 Review
Review

Affiliation:
Alayev Lab, Stern College for Women, Biology Department, Yeshiva University, New York, NY 10174, USA
Email: david.musheyev@yu.edu
ORCID: https://orcid.org/0000-0002-7490-6492
Affiliation:
Alayev Lab, Stern College for Women, Biology Department, Yeshiva University, New York, NY 10174, USA
Explor Target Antitumor Ther. 2022;3:480–496 DOI: https://doi.org/10.37349/etat.2022.00096
Received: November 04, 2021 Accepted: May 24, 2022 Published: August 31, 2022
Academic Editor: Valerie Speirs, University of Aberdeen, UK
The article belongs to the special issue Endocrine Resistant Breast Cancer
Endocrine resistance is a major hurdle in the treatment of estrogen receptor (ER)-positive breast cancer. When abnormally regulated, molecular signals responsible for cellular proliferation, as well as ER itself, allow for cellular evasion of ER-dependent treatments. Therefore, pharmacological treatments that target these evasion mechanisms are beneficial for the treatment of endocrine-resistant breast cancers. This review summarizes currently understood molecular signals that contribute to endocrine resistance and their crosstalk that stem from mitogen-activated protein kinase (MAPK), phosphoinositol-3 kinase/protein kinase B (PI3K/AKT), mechanistic target of rapamycin (mTOR), cyclin-dependent kinases 4 and 6 (CDK4/6) and aberrant ER function. Recent clinical trials that target these molecular signals as a treatment strategy for endocrine-resistant breast cancer are also highlighted.
The connection between the function of endocrine organs and breast cancer was described as early as 1896 in a case series by Beatson [1], who described remission of breast cancer after a bilateral oophorectomy, an excision of ovaries, in premenopausal women. Because the women described were premenopausal, their ovaries were likely still producing estrogen until their removal. The removal of the estrogen-producing organs could be correlated with the remission of cancer implying cancer’s sensitivity to steroid hormones such as estrogen. Beatson’s observations came 62 years before the discovery of the estrogen receptor (ER) by Fuentes and Silveyra [2] and its eventual correlation with breast cancer in future studies. Known to be sensitive to estrogen signaling due to its expression of the ER, ER-positive breast cancer accounts for 75% of all breast cancer [3]. Discoveries in the signaling of ER and its effects on breast cancer led to different therapeutic approaches to treating ER-positive breast cancer by targeting ER. These treatments include selective ER modulators (SERMs), selective ER down-regulators (SERDs), and aromatase inhibitors (AIs), all of which have been shown to reduce mortality and disease recurrence, and were followed by the development of combination therapies [4]. However, resistance to these therapies poses a challenge and requires alternative treatments. Mechanisms of resistance stem from two major sources, direct effects on ER—such as mutations and alterations in functions—or through activation of parallel molecular signals leading to ER independent cellular proliferation.
The ER is a transcription factor that contains 6 functional domains termed by letters A–F that represent relate to specific functions of ER. Most notably, the C domain contains the DNA binding domain (DBD) that allows for ER’s nuclear action, the D domain contains nuclear localization signal (NLS) that allows translocation of ER to the nucleus, and the E domain contains the ligand binding domain (LBD) that allows interaction of ER with its ligand—estrogen (Figure 1) [5]. There are two ERs, ERα and ERβ, that share homology at the protein level [5], however, they differ in their cellular actions and are variably expressed in different tissue types [6]. Because it is the ERα subtype that is predominately expressed in breast cancers and has been targeted by endocrine therapies in the treatment of breast cancer, our discussion will focus on targeting Erα [6–8].
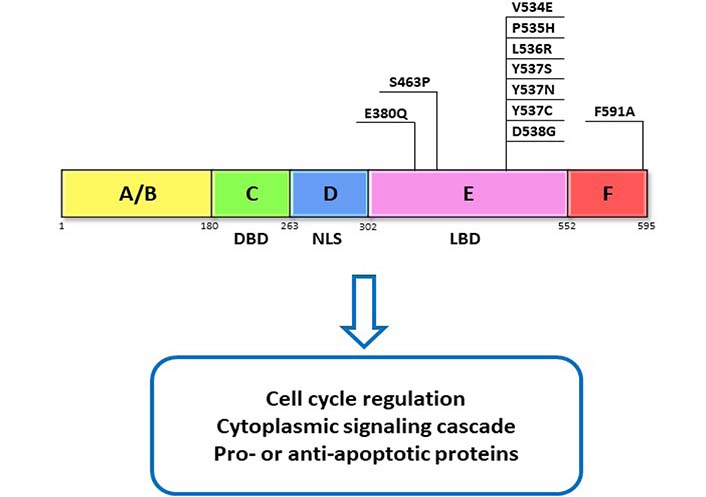
ER domains and function. ERα functional domains and functions are represented as A–F. DBD, NLS, and LBD domain as well as their commonly reported mutations in the LBD. These mutations have been found to contribute to resistance through activation of ERα which is independent of its ligand
Effects from ERα signaling can follow canonical pathways, whereby the ERα effect is nuclear and acts either directly or through intermediate activators and promoters on genetic response elements. Alternatively, the effect can be non-canonical, and exhibit immediate cytoplasmic response in cellular signaling cascades such as through phosphorylation of extracellular signal-regulated kinase (ERK) or protein kinase B (AKT) [9]. Transcriptional effects of ERα may be manifested either through canonical or non-canonical pathways, which is known as “nuclear-initiated steroid signaling” (NISS) [10]. Such nuclear functions through direct ER actions involve the cytoplasmic binding of estrogen to ER and the translocation of ER into the nucleus to act on enhancers and promoters that contain particular palindromic sequences known as estrogen response elements (EREs) [11–13]. Such actions depend on transcription and translation and therefore tend to be slower than the direct-acting membrane-initiated steroid signaling (MISS), which is activated by membrane-bound ER and leads to the activation of kinase signaling cascades [7]. Signaling pathways just downstream of MISS include a variety of signaling cascades including mitogen-activated protein kinase (MAPK), AKT, and Src [7, 14]. Furthermore, ER activation can occur independently of its known ligand, estrogen, through the ligand-independent mechanism, whereby ER is phosphorylated directly by other signaling cascades such as the MAPK/ERK pathway [15] or the phosphoinositol-3 kinase (PI3K)/AKT pathway [16, 17]. Responses to both nuclear and membrane-initiated signaling, as well as ligand-independent signaling, have been implicated in tumorigenesis and discoveries in the signaling pathways have allowed for the development of combination therapies and more targeted treatment [18]. Convergence between canonical and non-canonical ER activation as well as crosstalk between nuclear initiated and membrane initiated signaling has been implicated in treatment resistance and has been a topic of investigation in clinical and pre-clinical studies [19–21].
Treatments targeting estrogen signaling either through a reduction in estrogen production or direct action on the receptor are known as endocrine therapies and have been a part of the core strategy of treating ER-positive breast cancer. SERDs and SERMs both act directly on ER and reduce signaling with slight variations in their mechanisms of action. While SERMs compete with estrogen for receptor binding, SERDs bind with ER and mark ER for degradation [22]. Despite also being an endocrine therapy that affects ER, AI action does not involve direct action with ER, however, it reduces estrogen production by inhibiting a key step in the conversion of testosterone to estradiol [23].
The first Food and Drug Administration (FDA)-approved endocrine therapy was in the 1970s when tamoxifen, a SERM, has become the mainstay therapy in the treatment of ER-positive breast cancer and has been shown to reduce mortality and recurrence risk for 15 years after 5 years of treatment [24]. Despite being relatively well tolerated, some patients experienced adverse side effects and lowering the dose and duration of tamoxifen in patients with ER-positive breast cancer in an attempt to reduce side effects still yielded benefits in overall survival and a reduced recurrence rate [25]. In the ATLAS trial, in patients who tolerated treatment, intervention with tamoxifen monotherapy in early-stage breast cancer for 10 years improved overall survival and late recurrence when compared to 5 years of therapy, further highlighting the impactful utility of SERMs in endocrine sensitive tumors [26].
Despite having similar mechanisms of action to SERMs, SERDs bind to ERα with a stronger affinity than tamoxifen [27] and were shown to be effective in both tamoxifen-resistant and tamoxifen-sensitive cell lines [28]. Unlike SERMs, they are primarily approved for use in postmenopausal patients [29].
Similar to SERDs, AIs were found to provide a better clinical response rate in postmenopausal women when compared to women treated with tamoxifen [30]. In particular, the third generation AI, letrozole, has been shown to increase the time to disease progression when compared to tamoxifen [31].
In an attempt to identify alternative targets for treating breast cancers, exploratory studies looked for hormone receptors beyond the classic three targets in breast cancer. A receptor that was identified to be present in at least half of their samples was luteinizing hormone-releasing hormone (LHRH) and was even identified in triple-negative breast cancer samples, that are notoriously resistant to traditional targeted hormone treatments [32, 33]. Leuprolide is an LHRH agonist that, when given continuously inhibits pituitary hormone release (“chemical oophorectomy”) and leads to a subsequent reduction of estrogen production—similar to that described in Beatson’s oophorectomies. For this reason, it is primarily used to treat premenopausal women with limited use in hormone-resistant cancers [34]. Younger patients tend to have a higher risk of recurrence, therefore ovarian function suppression with gonadotrophin-releasing hormone (GnRH) agonists such as leuprolide. Treatment with LHRH agonists can preserve ovarian function by reversibly suppressing ovarian function and can rescue function post-chemotherapy, even in hormone receptor-negative tumors [35]. This strategy of preserving ovarian function is especially important to younger patients who have cancers dependent on chemotherapy, which can have detrimental effects on fertility [36]. It is important to note, that this restoration of ovarian function in some instances can even occur post-menopause. In a case report, a patient was described to have a resumption of menses post-AI treatment despite having previously been determined to be postmenopausal, suggesting a rescue of ovarian function by AI and necessitating closer monitoring of patient status and fertility and appropriate patient education [37].
Despite the three classes of endocrine therapies’ action on estrogen signaling, the difference between the therapies in the patient population they benefit emphasizes the importance of tailoring treatments to the disease. In the case of hormone therapy treatment resistance, switching to different hormone therapies is a strategy that is implemented in an attempt to overcome resistance. Unfortunately, response rates to treatment after switching over are not always as impressive as they are with initial hormone treatments, and require different treatment strategies [38]. For example, in patients who developed resistance to tamoxifen therapy, crossover to AI therapy reduced the risk of recurrence, however, did not provide a survival benefit [24]. Indicating that we still need a better understanding of the mechanisms that contribute to endocrine resistance and the development of better treatment strategies to overcome this resistance.
Resistance to endocrine therapies can be acquired as a result of long-term endocrine therapy or be a result of an intrinsic mechanism at play before the initiation of treatment. Both of these mechanisms can stem from direct actions on ER, such as the case of estrogen receptor 1 (ESR1) mutations leading to ER hyperactivation or to activation of ER-independent pathways responsible for cell growth and survival.
About 20% of ER-positive breast cancers lose ER expression and switch over to ER-independent mechanisms for cell proliferation after treatment with endocrine therapies [39, 40]. Examples of the loss of ER expression can stem from histone deacetylase inhibition [41] or methylation at the ESR1 gene promoter [42]. Direct mutations in the ESR1 gene can also lead to resistance. For instance, mutations in the LBD (Figure 1), which are found in 20% of metastatic ER-positive cancers and are often acquired following hormone treatment [43–48]. Such mutations cause ER to take on a conformation similar to the estrogen-bound receptor and therefore lead to constitutive activation of ER signaling independent of any ligands [49]. Some mutations can alter the transcriptional profile of genes activated in response to ER signaling leading to activation of pathways involved in growth, invasion, and metastasis, thereby creating a more aggressive tumor that is less responsive to conventional therapies [50, 51]. An even rarer mutation results in the fusion of ESR1 to different genes leading to constitutive activation of ER target genes and estrogen-independent growth [52]. Identification of specific mutations might offer avenues for creating treatments based on the specific mutations, as was identified in the case of ESR1-fusions, which were highly sensitive to treatment with a SERM/SERD and human epidermal growth factor receptor 2 (HER2) or Src inhibitor and cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitors [53, 54]. All of these changes to ER expression in one way or another lead to hyperactivation of ER that is independent of estrogen. This makes endocrine therapy a less useful intervention in the treatment of these tumors as reducing the production of estrogen, in the case of AIs, or competing for ER binding sites, with SERMs, would not change the signaling outcome. Though next-generation SERMS and SERDs are able to act on ER despite certain mutations [55] and have shown to be relatively tolerable in phase 1 studies [56, 57], these treatments do not address the activation of other pro- survival signaling mechanisms.
Studies characterizing molecular mechanisms responsible for tumorigenesis in breast cancer highlight the vast heterogeneity in breast cancers and elucidate potential mechanisms that are responsible for treatment resistance as well as offer treatment strategies to overcome resistance [58, 59]. Such studies identified clusters of changes in cells including mutations in ERα, mutations in the rat sarcoma virus/Raf/extracellular-signal regulated kinase kinases/MAPK (Ras/Raf/MEK/MAPK) pathway, as well as other molecular pathways and transcription factors [4]. More frequently occurring mutations that lead to the loss of function in the corepressors of ER, and subsequent endocrine resistance, occur in 13–55% of breast cancers and lead to disinhibition of ER expression. However, preclinical studies show an acquired sensitivity to combination treatment with histone deacetylase inhibitors, thus further highlighting the importance of tumor characterization [17]. The above-mentioned mutations lead to a shift towards ER-independent signaling, essentially making the tumor behave as if it were ER-negative. Treatments targeting these alternatively activated pathways would be more appropriate in these cases than traditional endocrine therapy.
Parallel signaling pathways that regulate cell proliferation, protein synthesis, and metabolism, may interact with ER signaling and with other pathways and may contribute to the progression of cancer, despite endocrine therapy. This is because an alternative mechanism is available for estrogen signaling that is independent of direct ligand binding with ER as a result of cytoplasmic crosstalk.
Both independent of estrogen signaling and through the reciprocal activation of ER bidirectional crosstalk between growth factor signaling cascades and ER lead to the activation of downstream pro-survival signaling [60, 61]. Examples of such estrogen-independent signaling include ER activation through ERK1/2 and PI3K/AKT, which phosphorylate ER on serines 118 and 167 causing resistance to endocrine therapy [15, 62]. Often, such tumors would exhibit an increase in the expression and activity of growth factors driven by several causes including activating mutations [4] and loss of ER signaling [63]. Such growth factor-induced ER signaling, interestingly, exhibits a distinct expression profile that differs from estrogen-dependent ER signaling [64]. Such a shift in growth factor overactivation, particularly fibroblast growth factor receptor (FGFR), was observed in 40% of metastatic ER-positive breast cancer after acquiring resistance to endocrine therapy and is associated with a worse prognosis [65, 66]. The estrogen-independent ER activation resulting from signal crosstalk underscores the importance of targeting multiple pathways, including the ER, in the case of treatment resistance and the importance of tumor categorization to identify the appropriate polytherapy.
Studies showing an improvement in breast cancer survival in response to implementing community cancer screening and subsequent earlier detection and treatment of breast cancer suggest a correlation between treatment success and relative time of treatment in the disease course [67]. The earlier treatment is less likely to be endocrine-resistant, and it is important to not only treat early in the disease course but also treat appropriately to the tumor type and molecular profile. Due to the crosstalk between ER and other signaling pathways, novel treatment strategies are aimed at inhibiting signaling pathways related to receptor tyrosine kinases, including MAPK and PI3K/AKT, and CDK4/6. In the case of hyperactivation of growth factor signaling, treatments that target tyrosine kinases and other downstream effectors of the growth factors were shown to improve the sensitivity to endocrine therapy [68, 69], and offer an avenue for polytherapy approaches in targeting multiple pathways simultaneously in response to acquired resistance. For example, in ER-positive cells with HER2 activating mutations, targeting HER2 or downstream effectors such as PI3Kα and target of rapamycin complex 1 (TORC1) sensitized the cells to fulvestrant treatment [70]. Clinically, combination treatment with endocrine therapy and HER2 inhibitors in patients with ER-positive breast cancer and HER2 activating mutations led to improved progression-free survival and duration of response when compared to endocrine therapy alone, thus showing benefit in combination approaches based on tumor characteristics [71].
Dysregulation in receptor tyrosine kinase signaling has long been implicated in the development of cancer due to its involvement in a variety of key cellular functions including growth, differentiation, and motility [72]. Particularly in breast cancer, upregulation of tyrosine kinase receptors has been correlated with a poor prognosis [73–75]. Tyrosine kinase receptor signaling starts at the cellular membrane where the receptor is activated through an auto-phosphorylation event, commonly through direct extracellular ligand-binding by growth factors which leads to further downstream activation of kinase cascades including PI3K/AKT and MAPK [76, 77]. These cascades then activate cell growth and survival pathways [78, 79]. Dysregulation of elements along these signaling pathways, either through activating mutations, upregulation of activators, or pathway crosstalk, drives the pathogenesis of cancer by promoting cell proliferation [78, 80]. Dysregulation of receptor tyrosine kinase signaling has been correlated with endocrine resistance and combination therapies with endocrine therapy and receptor tyrosine kinase inhibitors have yielded promising results in preclinical studies by reversing endocrine resistance and inducing apoptosis [81]. Pathways downstream of receptor tyrosine kinases that are often implicated in endocrine resistance and have been investigated for the treatment of endocrine-resistant breast cancer include the MAPK signaling pathway and the PI3K/AKT pathways as well as their shared downstream mechanistic target of rapamycin (mTOR) complex and CDK4/6.
MAPK signaling pathway is activated by growth factors and regulates cellular processes such as growth, proliferation, development, and differentiation. The MAPK pathway includes cascades of serine-threonine kinases with three major pathways: ERK, p38, and c-Jun [82]. Upon growth factor binding to the growth factor receptor, the Ras/Raf/MEK1/2 activation leads to activation of ERK1/2 in the cytoplasm. In turn, ERK1/2 can have cytoplasm action via activation of downstream targets such as MAPK interacting protein kinases (MNKs) and ribosomal S6 kinases (S6Ks, RSKs) or nuclear action via activation of transcription factors such as c-FOS, c-Myc, and activating protein-1 (AP-1) (Figure 2). Overactivation of the MAPK signaling pathway has been shown to be a driver of tamoxifen resistance and treatments with drugs targeting the signaling cascade were successful in reversing the resistance [65, 83]. Of these pathways, the ERK pathway is known to act on ERα by phosphorylating it on serine 118 leading to estrogen-independent ER activation [84]. Inhibition of the ERK pathway has been shown to upregulate ERα and sensitize cancer cells to tamoxifen [85]. Similarly, another experiment in MCF-7 cells that overexpress epidermal growth factor receptor (EGFR) showed that ERK inhibition sensitized the cells to SERD treatment [43]. This evidence highlights the crosstalk between ER signaling and ERK signaling and suggests the potential for a self-perpetuating positive loop between the two pathways [86]. Another way that estrogen signaling affects the MAPK pathway is through G protein-coupled ER (GPER)-dependent EGFR activation and related pathways [87, 88]. GPER is thought to be an ER-related protein that is membrane-bound and is similarly activated by estrogen [89, 90]. In an ER-negative cell line estrogen treatment after transfection with GPER led to an increase in MAPK phosphorylation, an effect that was not observed in cells that were not expressing GPER [91]. Furthermore, extended treatment with tamoxifen led to an increase in membrane-bound GPER, suggesting a potential alternative ER-independent mechanism that can be activated with estrogen [92].
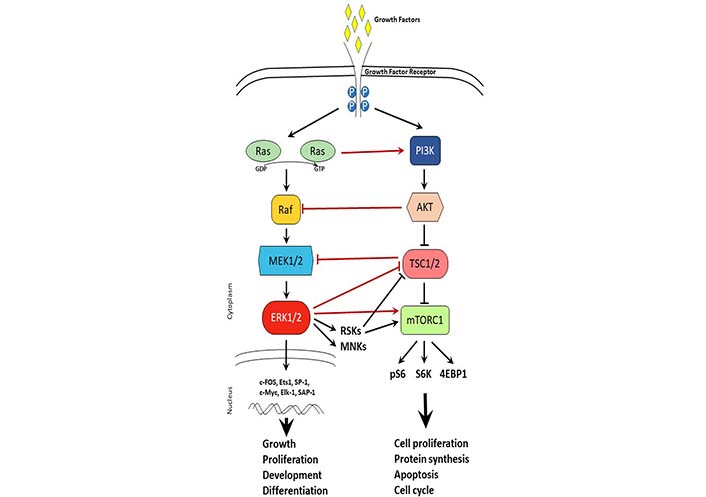
Cross-talk between MAPK and mTOR signaling pathways. Schematic representation of MAPK and mTOR signaling pathways upon growth factor activation. Arrows represent activation, while t-shaped lines represent inhibition. Black lines depict signaling within a pathway. Red lines depict crosstalk between signaling pathways. TSC1/2: tuberous sclerosis 1/2; 4EBP1: eukaryotic translation initiation factor 4E-binding protein 1; P: phosphorylation; pS6: phosphorylated S6; GDP: guanosine diphosphate; GTP: guanosine triphosphate
The mTOR signaling pathway is a master sensor of nutrient availability in the cell that regulates processes such as cell proliferation, protein synthesis, apoptosis, and cell cycle. Just downstream of the PI3K/AKT pathway, mTOR can form two different complexes that are distinctly known as mTOR complex 1 (mTORC1) and mTORC2 [93, 94]. mTORC1 is composed of mTOR, raptor, deptor, and LST8 and is known to regulate cell growth, protein synthesis, autophagy, and metabolism [95, 96]. Activation of mTOR promotes cell proliferation which is seen in endocrine- resistant breast cancer [97]. mTOR is also upregulated by ERK and PI3K/AKT pathways, which poses an additional layer of signaling that is stimulating tumor survival and contributing to treatment resistance, however, offers an additional co-target to consider in combination therapies [98, 99]. Just downstream of mTORC1, S6K1 is often amplified in breast cancer and is correlated with a poorer prognosis [100]. Furthermore, S6K1 action on ERα also leads to a positive feedback loop just as observed with mTORC1, whereby S6K1 phosphorylation of ER leads to ER activation and, through mTOR, ER action activates S6K1 [48, 101]. Such positive feedback loops highlight the importance of combinatorial therapies to break the feedback signaling (Figure 2).
Evidence of crosstalk between the PI3K/AKT and MAPK and involvement of other tumorigenic pathways, as was seen with mTOR, has been identified in numerous studies. This crosstalk can sometimes be missed in patients as was seen with the case of acquired HER2 activation with endocrine resistance despite an initial negative HER2 status. Interestingly, HER2 activation was previously described in acquired endocrine resistance despite initial low expression and is understood to be a result of crosstalk between multiple pathways including MAPK and PI3K/AKT [102]. This crosstalk might explain the treatment response to trastuzumab, a HER2 inhibitor in patients whose tumors were initially expressing low levels of HER2 [103].
Further downstream of the receptor tyrosine kinase signaling and of ER, the activation of CDKs contributes to tumor proliferation and cell cycle progression as a result of transcriptional upregulation of cell cycle regulators, these include CDK2 and CDK4 [104]. Signaling responsible for CDK4/6 upregulation includes upstream mitogenic pathways such as PI3K/AKT, MAPK, mTOR, and ER signaling [105]. In the case of endocrine resistance, these CDKs are often found to be aberrantly hyperactivated [106]. Palbociclib, ribociclib, and abemaciclib are currently FDA-approved CDK4/6 inhibitors and are used in combination with endocrine therapies for the treatment of ER-positive/HER2-negative breast cancer [107, 108]. In the PALOMA-2 trial, combination therapy with palbociclib and letrozole led to improvements in progression-free survival compared to letrozole monotherapy in patients with untreated ER-positive/HER2-negative breast cancer [107]. Similar results were achieved in the PALOMA-3 trial in patients who had disease progression after initial treatment failure from endocrine therapy [108]. The discovery of crosstalk between CDK4/6 activity and signaling cascades that involve PI3K/mTOR [109] and FGFR [110] has sparked further studies that propose the use of triple therapy with CDK4/6 inhibitors, endocrine therapy, and drugs targeting the respective signaling cascades. In the TRINITI-1 trial combination treatment with a CDK4/6 inhibitor and everolimus in addition to endocrine therapy had the overall benefit and was relatively well tolerated [111].
Taking into account the molecular pathways involved in endocrine resistance and the dynamic nature of disease progression, clinical trials investigating treatment options in endocrine-resistant breast cancers turn to combinatory treatments and crossover to different therapies in response to shifts in molecular signaling. Combinatory treatments targeting pathways downstream of receptor tyrosine kinases, mTOR, and CDK4/6 have shown promise in clinical trials.
Recent trials investigating the clinical benefit of inhibition of CDK4/6 yielded favorable results [112]. Results from the MONARCH 2 trial, a phase 3 randomized controlled trial (RCT) in which patients with ER-positive/HER2-negative breast cancer were treated with fulvestrant in combination with abemaciclib, CDK4/6 inhibitor, or with fulvesterant with placebo, resulted in improvements in overall survival in the abemaciclib treated group compared to the placebo-treated group [113]. Similarly the phase 2, LETPAL trial showed the benefit of the CDK4/6 inhibitor palbociclib with letrozole combination therapy when compared to chemotherapy in ER-positive/HER-negative breast cancer [114]. The results from these clinical trials further solidify the evidence found in pre-clinical investigations that were discussed earlier, showing that targeting CDK4/6 in conjunction with ER allows us to circumvent a cellular adaptation that causes upregulation in CDK4/6 and its resulting endocrine resistance. We now know that crosstalk among multiple pathways can contribute to treatment resistance, therefore studies are further investigating the use of polytherapies that target the known players in endocrine resistance. In a study investigating triple intervention with a CDK4/6 inhibitor, AKT inhibitor, and endocrine therapy in resistant tumors in postmenopausal ER-positive/HER2-negative breast tumors, initial results of the phase 1 trial showed that the triple therapy was well tolerated [115].
Evidence of the involvement of receptor tyrosine kinases and their downstream serine-threonine kinases in endocrine resistance prompted further investigations and eventually led to the characterization of the PIK3CA mutation which is found in nearly 40% of ER-positive breast cancers [116, 117]. The recent SOLAR-1 trial investigate a PI3K inhibitor, alpelisib, and found a benefit in median overall survival among patients with the PIK3CA mutation who were treated alpelisib and fulvesterant when compared to patients treated with fulvesterant and placebo [118]. This correlation with tumor characterization with its matching treatment allows for more effective treatment strategies than targeting ER alone.
Another receptor tyrosine kinase implicated in endocrine resistance that is currently investigated is the HER2 receptor. In endocrine-resistant breast cancer with low HER2 expression, treatment with an antibody-drug conjugate has shown promising results in clinical and preclinical studies [119]. Suggesting possible signaling overlap between HER2 and other members in the EGFR family of receptor tyrosine kinases. Trials investigating the transition to activating other molecular pathways give us a better understanding of the evolution of tumors as they progress through different treatments and offer insight into the value of retyping tumors as they progress.
The multitude of molecular subtypes of breast cancers highlights the heterogeneity of the disease and the need for disease classification and the development of tailored therapies. Furthermore, molecular changes throughout treatment as a result of genetic changes and crosstalk between multiple signaling cascades elucidates the dynamic nature of breast cancer progression and the value of polytherapy to simultaneously target multiple pathways to circumvent the effects of future shifts in molecular signaling. In response to this heterogeneity and dynamic molecular shifts technologies to assay tumor types have been developed to improve tumor classification and implement more targeted treatments [120]. The results of these studies identify specific targets in molecular signaling and mutations that may be used in the future to predict endocrine resistance prior to a trial of endocrine therapy, therefore allowing for a more aggressive treatment earlier in their disease course.
AIs: aromatase inhibitors
AKT: protein kinase B
CDK4/6: cyclin-dependent kinases 4 and 6
DBD: DNA binding domain
EGFR: epidermal growth factor receptor
ER: estrogen receptor
ERK: extracellular signal-regulated kinase
ESR1: estrogen receptor 1
GPER: G protein-coupled estrogen receptor
HER2: human epidermal growth factor receptor 2
LBD: ligand binding domain
LHRH: luteinizing hormone-releasing hormone
MAPK: mitogen-activated protein kinase
mTOR: mechanistic target of rapamycin
mTORC1: mechanistic target of rapamycin complex 1
NLS: nuclear localization signal
PI3K: phosphoinositol-3 kinase
S6Ks: S6 kinases
SERDs: selective estrogen receptor down-regulators
SERMs: selective estrogen receptor modulators
DM took the lead in writing the manuscript. AA provided intellectual support and edited the manuscript. Both of the authors contributed to manuscript revision, read and approved the submitted version.
The authors have no conflicts of interest to declare.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by NIH [CA220021] and the Appelbaum foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Kristin A. Altwegg, Ratna K. Vadlamudi
Alfredo Rossi ... Marta Carlesimo
Katarzyna Rygiel
Kate M. Moore ... Simon P. Langdon
Aglaia Skolariki ... Simon Lord
Samuel Jones ... Kathryn M. Taylor
Matthew Willman ... Brandon Lucke-Wold
Gary J. Cheng ... Dean C. Singleton
Inês Soares de Pinho ... Luís Costa
