 Review
Review

Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
†These authors contributed equally to the work.
ORCID: https://orcid.org/0000-0002-9870-7893
Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
†These authors contributed equally to the work.
ORCID: https://orcid.org/0000-0003-4793-6378
Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
ORCID: https://orcid.org/0000-0003-3492-4415
Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
ORCID: https://orcid.org/0000-0001-8014-8205
Affiliation:
1Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
2Hematology, “Azienda Ospedaliero-Universitaria di Parma”, 43126 Parma, Italy
Email: nicola.giuliani@unipr.it
ORCID: https://orcid.org/0000-0003-3457-3774
Explor Target Antitumor Ther. 2022;3:463–479 DOI: https://doi.org/10.37349/etat.2022.00095
Received: March 15, 2022 Accepted: July 08, 2022 Published: August 31, 2022
Academic Editor: Giancarlo Pruneri, Fondazione IRCCS Istituto Nazionale dei Tumori, Italy
The article belongs to the special issue Off-Label Drugs and -Omics Data in Cancer Treatment
Multiple myeloma (MM) is a blood cancer that derives from plasma cells (PCs), which will accumulate in the bone marrow (BM). Over time, several drugs have been developed to treat this disease that is still uncurable. The therapies used to treat the disease target immune activity, inhibit proteasome activity, and involve the use of monoclonal antibodies. However, MM is a highly heterogeneous disease, in fact, there are several mutations in signaling pathways that are particularly important for MM cell biology and that are possible therapeutic targets. Indeed, some studies suggest that MM is driven by mutations within the rat sarcoma virus (RAS) signaling cascade, which regulates cell survival and proliferation. The RAS/proto-oncogene, serine/threonine kinase (RAF)/mitogen-activated extracellular signal-regulated kinase (ERK) kinase (MEK)/ERK signaling pathway is deregulated in several cancers, for which drugs have been developed to inhibit these pathways. In addition to the signaling pathways, the disease implements mechanisms to ensure the survival and consequently a high replicative capacity. This strategy consists in the deregulation of apoptosis. In particular, some cases of MM show overexpression of anti-apoptotic proteins belonging to the B cell lymphoma 2 (BCL-2) family that represent a possible druggable target. Venetoclax is an anti-BCL-2 molecule used in hematological malignancies that may be used in selected MM patients based on their molecular profile. We focused on the possible effects in MM of off-label drugs that are currently used for other cancers with the same molecular characteristics. Their use, combined with the current treatments, could be a good strategy against MM.
Multiple myeloma (MM) is a hematological malignancy, with an annual incidence of approximately 6.6 cases/100,000 people [1]. It is a plasma cells (PCs) neoplasm, which provides the function of secreting antibodies in the bone marrow (BM). This tumor is usually diagnosed by serum protein electrophoresis (SPEP), in which a distinctive M peak is evident, or by estimation of free light chains in urine [2]. MM cells are the malignant counterparts of long-lived PCs in the post-germinal center (GC), characterized by strong BM dependence and somatic hypermutation (SHM) of immunoglobulin (Ig) genes [3]. MM is affected by in-depth genetic alterations that occur during the progression of the disease, which is why it is generally recognized as a highly heterogeneous disease. Its evolutionary course starts with a pre-malignant status noted as monoclonal gammopathy of undetermined significance (MGUS) [4], a posterior stage termed smoldering MM (SMM) [5], and may ultimately escalate into symptomatic myeloma [2]. The hallmark genetic events (or aberrations) are commonly classified as primary and secondary events. Primary aberrations are grouped into two subgroups: non-hyperdiploidy, comprised of primary Ig (t) translocations involving the Ig heavy chain (IGH) in the 14q32 region including t(4;14), t(11;14), t(14;20), and the hyperdiploidy group comprised of trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19 [6]. Multi-copy chromosomes carry genes whose overexpression may be responsible for the replicative potential of cells. The MGUS clone is subsequently affected by secondary abnormalities that may or may not lead to the transition to the MM [7]. Secondary abnormalities, also genetic, include MYC proto-oncogene (MYC) translocation, 17p or 1p32 chromosome deletion, 1q chromosome amplification or mutations of rat sarcoma virus (RAS) gene [NRAS proto-oncogene (NRAS) and KRAS proto-oncogene (KRAS)], B-Raf proto-oncogene (BRAF), mitogen-activated protein kinase (MAPK), and nuclear factor kappa-light chain enhancer of activated B cells (NF-κB) pathways or overexpression of the anti-apoptotic protein B cell lymphoma 2 (BCL-2) [8] (Figure 1).
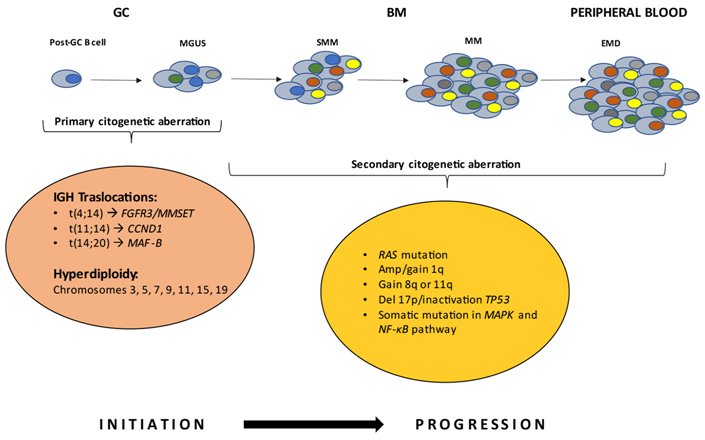
The basis of neoplastic transformation and development of MM are mutational events at the PC level. Early genetic events drive the MGUS stage. Further mutations induce deregulations in the PCs, leading to the transition to the SMM and MM stages. The latter steps are characterized by the proliferation of monoclonal PCs in the BM microenvironment. In rare cases, it can evolve into extramedullary disease (EMD). FGFR3: fibroblast growth factor receptor 3; MMSET: multiple myeloma SET domain; CCND1: cyclin D1; MAF-B: musculoaponeurotic fibrosarcoma oncogene homolog B; Amp: amplification; Del 17p: deletion 17p; TP53: tumor protein p53
The most common translocation in MM, impacting 15% of patients, is t(4;14)(p16;q32), and is associated with a very unfavorable prognosis [9]. Translocation t(4;14) results in the overexpression of two key genes, FGFR3 and MMSET, also known as nuclear receptor-binding SET domain 2 (NSD2) or Wolf-Hirschhorn syndrome candidate 1 (WHSC1). Overexpression of MMSET isoforms is a universal feature of t(4; 14) [10–12]; whereas the same is not true for FGFR3, as approximately 30% of patients affected by MM with t(4;14) do not express FGFR3. Transforming activity of this gene has been reported both in vitro and in vivo.
The t(11;14)(q13;q32), is detected in 15% of MM patients. Translocation can be observed in the condition of MGUS. This genetic event results in the over-regulation of nuclear CCND1 [13]. An intriguing aspect is that patients diagnosed with this translocation can remain stable in the MGUS status with no disease progression for decades. On this basis, the outcome can be assumed to be therefore favorable.
Translocation t(14;20) is diagnosed in 3% of patients with MM [14]. It is associated with a poor prognosis and, as a result of chromosome aberration, there is an overexpression of the MAF-B gene [15, 16].
The disease evolves along with secondary genetic disorders, including MYC translocation, which can occur in ~15% of MM at diagnosis and 50% of more severe stages [17]. The MYC gene codifies an extremely important transcription factor engaged in various cellular functions such as cell growth [18], proliferation [19], and protein synthesis [20]. MYC is implicated in the enumerated biological processes and plays a cardinal role in tumor advancement. In MM patients, its overexpression is characteristically linked to a poor prognosis.
As a secondary genetic event, it is also good to focus on copy number variations (CNVs), which are a hallmark of MM progression. This genetic event consists of DNA gain or loss at a whole chromosome arm, a whole chromosome, or a focal region [21]. It is a disorder typically known as aneuploidy and discriminates cancer cells from their benign counterparts. CNVs are detected in approximately 70% of MM cases [22].
CNV-containing PCs have a selective advantage in promoting disease progression because the amplified or deleted regions contain significant genes involved in the development and progression of MM [23]. The gold standard technique to detect selected CNVs is fluorescence in situ hybridization (FISH), but its results are limited to the probes used in each analysis. Some centers also use the single nucleotide polymorphism (SNP) microarray to reveal copy number alterations at a higher resolution.
Yellapantula et al. [24] used a custom next-generation sequencing (NGS) acquisition panel specifically designed to help the identification of rearrangements in MM. In their study, they reported high sensitivity (> 99%) and specificity (> 99%) for revealing chromosomal gains and losses [24].
The CNVs most commonly found in MM are on chromosomes 1, 13, 17. On chromosome 17, it is important to evaluate the role of TP53 gene, which has cancer suppressor function. In particular, chromosome 17p can be deleted in MM. This peculiar genetic aberration is also claimed to be a secondary event in disease progression [25]. Biallelic TP53 inactivation is accepted as an advanced driver in MM and has been most closely associated with low overall survival and resistance to standard treatments. It is found in 3.7% of patients [26]. In MM, TP53 mutations pose late events in disease progression; in fact, this mutation is rarely observed at diagnosis, demonstrating a central role in the progression of MM [27].
Another chromosome susceptible to CNVs is chromosome 13, which undergoes a deletion at arm q. The deletion is found in 50% of newly diagnosed MM patients.
Initially, this genetic event was associated with a poor prognosis. Over time, however, this view has changed, as recent studies have shown that the deletion coincides with other remarkable genetic events such as t(4;14). Thus, establishing the correct prognosis associated with this deletion remains a challenge. There are about 68 genes influenced by the deletion of the region 13q, including RB transcriptional corepressor 1 (RB1), EBP like (EBPL), ribonuclease H2 subunit B (RNASEH2B), RCC1 and BTB domain containing protein 2 (RCBTB2), and the microRNAs microRNA-16-1 (miR-16-1) and miR-15a. The region containing these genes is 13q14.11-13q14.3 [28]. Among these genes, one with a special role in cell cycle regulation is RB1. The miRNA cluster containing miR-16-1 and miR-15a, instead, seems to be deleted in chronic lymphocytic leukemia and regulates the expression of several cell cycle genes [29].
An incisive role in the progression of MM is assumed by chromosome 1q, particularly the region 1q21. In 40% of de novo cases and 70% relapsed-refractory MM (RRMM) patients, gain and/or amplification of chromosome arm 1q21 (1q21+) is detected [30, 31]. The presence of 3 copies of this region is defined as gain, while ≥ 4 copies characterize amplification. Chromosome amplification 1q (1q21 amp) is one of the most common secondary cytogenetic abnormalities in patients with MM. Moreover, not all the genes contained in this region have been identified, but those that are thought to be determinants in the disease development include BCL-9, cyclin-dependent kinase regulatory subunit 1B (CKS1B), interleukin-6 receptor (IL6R), interleukin-2 enhancer binding factor 2 (ILF2), and myeloid cell leukemia 1 (MCL-1). 1q21 amp is often associated with drug resistance, disease progression, and poor prognosis [32].
The pathogenesis of MM also highlights changes occurring in the BM microenvironment [33]. Genetic defects identified in the tumor and interactions between MM and BM microenvironment cells lead to the activation of signaling pathways that enhance the expansion of malignant clones and stimulate two phenomena essential for MM progression, namely neo-angiogenesis and osteoclastogenesis [34, 35]. Over-regulated signaling pathways are represented by the RAS/RAF/MEK/ERK-(also known as MAPK-) pathway, the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)- and NF-κB-pathways, but also the Janus kinase (JAK)/signal transducers and activators of transcription (STAT)-, Hedgehog-, Notch-, transforming growth factor beta (TGFβ)-, and Wingless-related integration site (WNT)- pathways. The RAS/RAF/MEK/ERK pathway consists of a cascade of intracellular proteins with kinase activity involved in cell proliferation, growth, adhesion, and apoptosis [36]. RAS [NRAS, KRAS, and HRAS proto-oncogene (HRAS)] activates RAF-kinases, which, in turn, phosphorylate MEK and finally ERK-kinase. Many cancers share mutations in protein kinases involved in this pathway. Therefore, one strategy implemented in oncology has been to target regulators of this pathway, as in colorectal cancer, hairy cell leukemia, and melanoma. The percentage of MM patients presenting with mutations in the MAPK pathway are 43–53%, thus resulting in a fairly common gene event in this disease [37–39].
Thus, in this review we will focus on the RAS/RAF/MEK/ERK signaling pathway and the role of the anti-apoptotic protein BCL-2, both of whom are engaged in the pathogenesis of MM and discuss the implementation of related targeting therapies. In fact, to survive the numerous genetic insults and pro-apoptotic changes, malignant cells must upregulate anti-apoptotic BCL-2 proteins and thus become highly dependent on the activity of these proteins [40]. Therefore, BCL-2 may also represent a therapeutic target in the struggle against MM.
Off-label drug use refers to the use of drugs under conditions other than those for which they were approved, such as dose, age of the patient, administration route, and contraindications [41]. This strategy is implemented to treat health problems for which there are no other currently approved drugs, for example in the case of uncommon diseases or specific subset of patients [42]. Off-label uses may prove particularly valuable in treating patients with an orphan disease, for which it may be the only available treatment [43].
The purpose of off-label use is to aid in the recovery of an individual patient. In oncology, pediatrics, geriatrics, and obstetrics, patient care can be difficult if off-label use is not employed [44, 45]. An estimate claims that off-label prescribing reaches 90% in the pediatric population or 40% in adults [46]. In oncology it achieves up to 50% of patients, and is widely applied in pediatric oncology [47]. The use of off-label drugs is believed to have contributed to an overall cure rate of more than 70% in pediatric malignancies [48]. Underlining, consequently, that cancer patients can benefit greatly from this phenomenon. One issue raised, however, is that the use of off-label drugs is not always supported by solid scientific evidence [41]. Scientific review and investigation of medications, that are tested and approved for a given condition, help protect the patient as much as possible from side effects. By using off-label drugs, this protection may be compromised [49], so they should be used in a precise and controlled manner.
Several patients with different tumors expressing the same target therefore may benefit from off-label prescription [50]. This concept is carefully translated into MM. Accordingly, it is possible to employ a variety of off-label drugs that can block the de-regulated molecular pathways in this cancer.
The RAS/RAF/MEK/ERK pathway is a critical intermediary of many essential biological processes. It is involved in cell proliferation, migration, survival, and angiogenesis. Members of the RAS protein subfamily, NRAS, KRAS, and HRAS, function as molecular switches in cellular signal transduction. These small guanosine triphosphate hydrolases (GTPases) activate RAF-kinases, which in turn phosphorylate MEKs and finally ERK-kinases.
MAPK-pathway mutations are one of the most common mutations found in MM [37]. It is also intriguing to note that the number of patients with mutations appears to be higher in relapsed disease [39]. The literature data on the prognostic significance of mutations on the MAPK pathway do not follow a univocal trend but, on the contrary, seem to be conflicting. While some research groups have found negative effects of NRAS mutations [51], others have observed a negative effect in KRAS mutations [52], and still others have found no prognostic effect [53]. Despite the high prevalence of mutations activating the MAPK pathway, these controversial results can be explained by considering that only some of the mutations effectively appear to activate it.
It has been previously demonstrated that only KRAS G12D and BRAF V600E consistently resulted in phosphorylation of the ERK downstream target (Figure 2). Other mutations were associated with increased phospho-ERK levels in only a small percentage of cases.
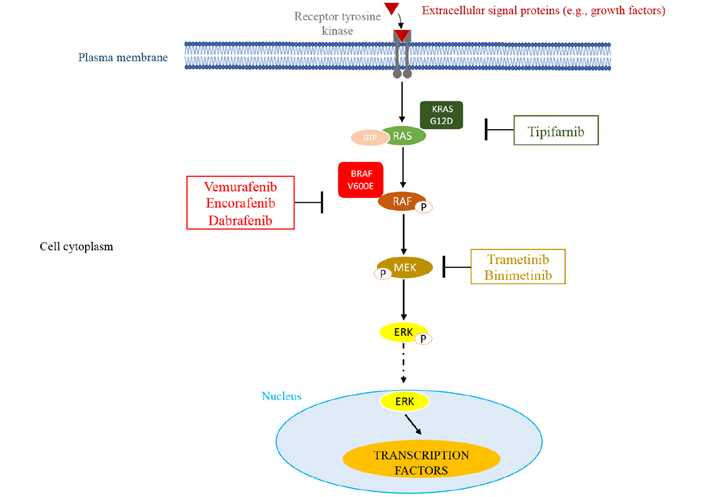
MAPK pathway and potential sites of therapeutic intervention with inhibitors. This molecular pathway is triggered by various extracellular signals. The main components of this pathway are RAS, RAF, and MEK leading to the activation of ERK through its phosphorylation (P). Once activated, ERK migrates into the nucleus where it activates transcription factors that affect cell proliferation and survival. In myeloma, overactive RAS/RAF/MEK/ERK signaling resulting from genetic mutations in the RAS and BRAF GTPases can be targeted by small molecule inhibitors of RAS G12D (tipifarnib), BRAF V600E (vemurafenib, encorafenib, dabrafenib), or MEK (trametinib, binimetinib)
As a consequence of the difficulty in targeting RAS, to date only one specific inhibitor has been identified for the KRAS G12C mutation, but this is rare in MM [54]. An investigation was conducted using the inhibitor tipifarnib, which was found to inhibit RAS and to have limited activity in MM patients. It also does not diminish, significantly, the activation of the MAPK pathway [55]. Along the lines of what was observed, the emphasis shifted to MAPK pathway inhibitors having downstream targets of RAS, which include BRAF and MEK.
Due to several pitfalls, it is necessary to accomplish patients screening. In a number of malignancies, it has been noted how, in the absence of the mutation involving BRAF, treatment of patients with inhibitors of this mutation can drive an increase in RAS signaling those results in activation of the pathway. This effect would appear to be triggered by a decrease in negative feedback at the level of RAS [56], binding of wild-type BRAF to CRAF, and subsequent MAPK signaling through CRAF [57, 58].
Attention must also be paid to how to identify patients who benefit from inhibition of the MAPK pathway [59]. A retrospective study that examined the effects of treatment with trametinib (MEK 1/2 inhibitor) illustrates this problem well. In that study, patients with mutations determining MAPK pathway triggering or carrying oncogenic mutations in NRAS, KRAS, and BRAF were selected. Of this cohort of patients, only 40% achieved at least a partial response (PR) when treated with trametinib coupled with other drugs, whereas only 10% exhibited at least a PR when treated solely with trametinib [60].
This underscores the critical importance, in terms of therapeutic efficacy, of precisely identifying patients who might be eligible for MAPK pathway inhibition. Several recent studies have therefore included only patients with the BRAF V600E/K mutation, which has been shown to consistently activate ERK and has also been closely examined in a variety of other cancers. In addition, several potent BRAF V600E mutation-specific inhibitors are available, such as vemurafenib, encorafenib, and dabrafenib. Approximately 5% of patients harbor a BRAF V600E clone or subclone. Early data on targeting the BRAF V600E mutation in patients with MM have been contradictory: some studies have shown treatment efficacy in RRMM patients with the mutation [61, 62], while others did not detect any response [63, 64].
Although BRAF targeting is effective in tumor types carrying mutant BRAF, however, there is rapid resistance against BRAF inhibitors. This phenomenon can be explained by several mechanisms among which one of the most frequent involves the acquisition of activating mutations upstream or downstream of BRAF in NRAS or MEK, leading to alternative signaling and bypass of BRAF [65, 66]. In addition, MEK inhibition has been seen to induce therapeutic resistance through upregulation of other signaling pathways, such as the PI3K/AKT pathway [67]. To circumvent these resistance mechanisms, it has been suggested to combine BRAF inhibition with MEK inhibition in a way that acts on two of the interactors involved in the signaling pathway. These approaches have been found to be very efficient in melanoma cases and, notably, dual inhibition has been more effective than BRAF inhibition alone [68]. Currently, dual inhibition is the most widely used strategy in MM studies.
The BIRMA trial examined the inhibition of BRAF and MEK in MM patients carrying the BRAF V600E/K mutation using a combination of encorafenib and binimetinib (a selective inhibitor of MEK). Preliminary results from this study showed an overall response rate (ORR) of 82% with 9 of 11 patients having at least one PR. This study demonstrated that, at least for some patients with MM, pathway inhibition leads to clinically meaningful responses [69].
Regarding the BRAF/MEK co-inhibitor, according to a study by Haertle et al. [70], proteosome inhibitors were ineffective with RAF/NRAS/KRAS activating mutations. In contrast, co-treatment of bortezomib and BRAF/MEK co-inhibitor had a synergistic action in the presence of activating mutations.
Another trial (NCT03091257), tests dabrafenib and/or trametinib in patients with RRMM. The purpose of the study is to examine the efficacy of BRAF inhibition in mutation-positive patients and the effects of MEK inhibition in patients with only RAS mutations.
Thus, we can state that inhibition of the MAPK pathway shows promising activity in a subset of patients. Therefore, a preliminary assessment of the presence of mutations involved in this pathway could guide decision-making on therapeutic strategies.
The BCL-2 gene family encodes more than 20 proteins that regulate the intrinsic apoptosis pathway and are fundamental to the balance between cell survival and death [71].
The BCL-2 family proteins can be categorized into three groups, namely antiapoptotic multidomain proteins (such as BCL-2, BCL-XL, and MCL-1), proapoptotic multidomain proteins [like BCL-2-associated X (BAX), BCL-2 antagonist/killer (BAK), and BCL-2-related ovarian killer (BOK)], and proapoptotic BCL-2 homology domain 3 (BH3)-only members [e.g., P53 up-regulated modulator of apoptosis (PUMA), NOXA, BH3 interacting-domain death agonist (BID), and BLC-2 interacting mediator of cell death (BIM)] [71].
BCL-2 family proteins are capable of giving rise to different interactions; however, their corresponding interaction turns out to be selective and specific. For example, BIM and PUMA can bind all members of the anti-apoptotic multidomain BCL-2 family, whereas NOXA binds only to MCL-1 [72, 73]. In contrast, BCL-2 associated agonist of cell death (BAD) interacts with BCL-XL and BCL-2, but not with MCL-1 [72, 74].
The intrinsic pathway involves the mitochondria and after receiving the stimulus, the pro-apoptotic BH3-only members bind and neutralize the antiapoptotic proteins. This leads to oligomerization of multi-domain pro-apoptotic member (BAX/BAK) present on mitochondrial membrane surface whose activation leads to permeabilization and formation of pores in outer mitochondrial membrane, releasing various apoptotic mediators [high-temperature requirement protein A2 (HtrA2), also called Omi, second mitochondria-derived activator of caspase (Smac)/direct inhibitor of apoptosis (IAP)-binding protein with low pI (DIABLO), cytochrome c, endonuclease G (Endo G) and apoptosis-inducing factor (AIF)] [75]. BAK may contribute to early mitochondrial fragmentation while BAX is probably more important for subsequent pores development and degeneration in the outer membrane [75]. The release of cytochrome c in cytosol causes the association of apoptosis protease-activating factor 1 (APAF-1) and ATP/dATP to form intracellular apoptosome that activates caspase-9 [76]. Disrupted mitochondria also produce Smac/DIABLO, which releases caspase-3 from X-linked IAP (XIAP)-mediated inhibition. The role of IAP is to act a guardian inside a cell to defend against the mediator of apoptosis (HtrA2/Omi, Smac/DIABLO) by binding to caspase-3/-9 whereas Endo G and AIF operate independently of caspase causing chromatin condensation and fragmentation. Anti-apoptotic members of the BCL-2 family, through direct protein interactions involving binding to their BH3 motifs, inhibit the activity of BAK/BAX. Inhibition of the anti-apoptotic members of this protein family has been accepted in clinical practice being that BCL-2 family members are key regulators of common apoptotic pathways [77].
Several BH3 mimetics have entered clinical trials [78], although, due to the absence of a trustworthy validation assay to directly test the mitochondrial activity of new candidate BH3 mimetics, there have been many erroneous reports of agents advertised as BH3 mimetics despite their off-target mechanisms of action. BH3 profiling assesses the activity of a compound at the mitochondrial level by measuring cytochrome c release as a marker for mitochondrial outer membrane permeabilization.
Villalobos-Ortiz et al. [79] proposed a comprehensive biochemical toolkit consisting of BH3 profiling in conjunction with the high-throughput Annexin V/Hoechst viability assay for validation of BH3 mimetic candidates. In order to accredit mitochondrial assays of BH3 mimetics, they performed a BH3 profiling method (iBH3) that consists of exposing mitochondria to standardized concentrations of BH3 peptides and measuring outer mitochondrial membrane permeabilization by cytochrome c release using flow cytometry [79].
Another approach was proposed by Bhola et al. [80] who developed a high-throughput method to assess the sensitivity to BH3 mimetics of tumors within 24 h of excision, providing data demonstrating that the very process of prolonged ex vivo propagation rapidly alters chemical vulnerabilities relative to the primary tumor [80].
The same authors nicely demonstrated a relationship between BCL-2 pathway and RAS signaling in cancer stem cell providing the rational to design new strategy to treat resistant cancers [81].
Drugs mimicking the action of BH3-only proteins indirectly lead to BAX/BAK activation. This allows permeabilization of the mitochondrial outer membrane, apoptosome formation and subsequent caspase activation and apoptosis. This is particularly true in the treatment of chronic lymphatic leukemia, typified by the approval of the BCL-2 inhibitor venetoclax as a particularly effective rescue therapy for this disease [70]. The ability of venetoclax is to bind BCL-2, through high-affinity binding, causing blockade of its signaling in the cell; this is the mechanism of action that is responsible for the TP53-independent apoptotic event [82] (Figure 3).
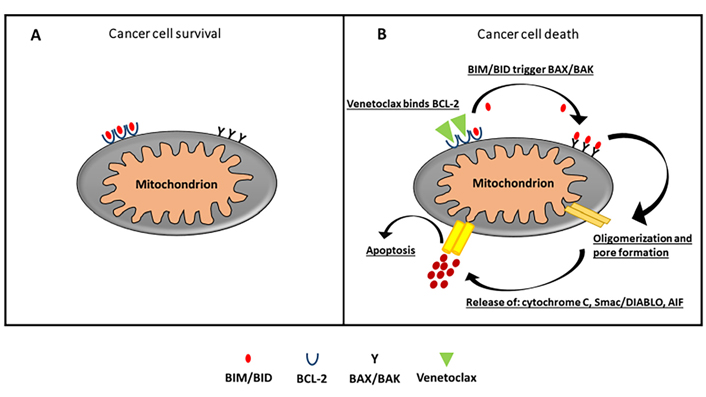
A. In cancer cells, excessive production of BCL-2 sequesters and blocks the function of pro-apoptotic protein and evades apoptosis. B. Venetoclax, a selective BCL-2 inhibitor, displaces and reactivates pro-apoptotic proteins bound to the BCL-2 binding groove. This prompts to oligomerization of BAX/BAK whose activation leads to permeabilization and formation of pores in outer mitochondrial membrane, releasing cytochrome c, Smac/DIABLO, and AIF
Chronic lymphocytic leukemia cells can effectively evade apoptosis due to their marked dependence on BCL-2 activity; in other malignancies, distinct members of the BCL-2 family by contrast may have a greater influence. This is the situation in MM. Indeed, in PC and MM, previous preclinical studies have indicated that MCL-1 may be the major anti-apoptotic analog of BCL-2 [8]. Based on this, venetoclax being specific for BCL-2, its clinical trial in MM patients has begun with moderate enthusiasm.
However, it has been demonstrated in MM cell lines and in primary patient samples that venetoclax is highly effective in a specific subgroup of MM with t(11;14), which is present in approximately 20% of MM, mainly because of the higher BCL-2/MCL-1 messenger RNA (mRNA) ratio [83].
Nevertheless, even among patients with t(11;14) myeloma, the response rate is only 40–60% [84]. Knockdown of CCND1 has been shown to fail to induce resistance to venetoclax, suggesting that t(11;14) and CCND1 have no direct roles in response to venetoclax [85].
To further explore the biology responsible for venetoclax sensitivity in myeloma and to potentially identify additional biomarkers, Gupta and colleagues [85] performed a data analysis based on assessment of RNA-sequencing and assay for transposase-accessible chromatin (ATAC)-sequencing from samples of patients screened for venetoclax sensitivity. The identical analysis was also conducted using cell lines. Venetoclax-sensitive myeloma is enriched in B-cell-associated genes that are not typically expressed in PCs [85, 86]. The expression of these genes could not be explained completely by t(11;14) because such genes were observed almost exclusively in venetoclax-sensitive t(11;14) and were mostly absent in venetoclax-resistant t(11;14). No single gene was consistently expressed in all sensitive cell lines or patient samples, suggesting that a panel of genes or cell surface markers may be required to distinguish sensitive and resistant myeloma.
Through ATAC-sequencing analysis, they also identified a pattern of B-cell-like chromatin accessibility in venetoclax-sensitive cell lines, suggesting increased binding of transcription factors at some of these accessible sites. Overexpression of one of these transcription factors, basic leucine zipper ATF-like transcription factor (BATF), led to increased sensitivity to venetoclax. BATF is an essential B-cell developmental transcription factor which is activated during the B-cell receptor signaling, with subsequent activation-induced deaminase expression and class change recombination. However, its expression and role in PCs are not understood [87]. BATF represses and promotes transcription depending on the presence of other transcription factors, including the PC transcription factor interferon regulatory factor 4 (IRF4), and may contribute to the venetoclax-sensitive transcription program in PCs through alterations in chromatin accessibility. Thus, despite possessing the characteristics of terminally differentiated PCs, venetoclax-sensitive myeloma aberrantly preserves or reactivates aspects of the B-cell program, including dependence on BCL-2 that is normally downregulated during differentiation.
Although both BCL-2 expression and the BCL-2/BCL-XL ratio are higher in venetoclax-sensitive cell lines and patients, they show a significant degree of overlap when comparing sensitive and resistant samples, thus making it difficult to select a specific cut-off that is useful for clinical decision-making [84, 88].
In the phase I clinical trial NCT01794520, venetoclax monotherapy was administered to patients with RRMM. Enrolled patients represented a heavily pre-treated group with a median of five prior lines of therapy. During the trial, the preclinical hypothesis [83] that myeloma patients with t(11;14) translocation responded more favorably to this therapy was confirmed. Therefore, this specific cohort was increased to include 30 patients. Compared with the entire study population (66 patients), the ORR was 21%; however, almost all of the patients who responded were from subgroup t(11;14). In this subgroup, the response rate amounted to 40%, which included 14% complete remissions (CRs). In contrast, only 2 of those patients without t(11;14) in the entire population of 66 individuals had a significant response to venetoclax [89].
When considering venetoclax as a targeted therapy for MM with t(11;14) and comparing it to other available treatment options for a population of heavily pre-treated RRMM patients, the use of venetoclax may emerge as an attractive salvage treatment option for these patients.
To expand the range of applicability of this approach the association between BCL-2 inhibition and other therapeutic modalities. Among its function, it is well established that bortezomib, a highly effective proteasome inhibitor for myeloma, can stabilize NOXA, an MCL-1 neutralizing protein [90].
Based on the above, being that the expression of MCL-1 may be a potential mechanism that triggers resistance to venetoclax activity, bortezomib has the potential to prove useful in combination with venetoclax. Apart from bortezomib, dexamethasone, a glucocorticoid used in therapeutic treatment in myeloma patients, has been shown to sensitize primary cells and myeloma lines to venetoclax activity. Based on this evidence, clinical trial M12-901 (NCT01794507) was initiated to test the triple combination venetoclax-bortezomib-dexamethasone in patients with RRMM [88]. Here, the response level reached 67% and was not limited to patients with t(11;14). The ORRs were similar in the different cytogenetic profiles; in particular, it was 47% in those patients who had the 17p deletion, a particularly high-risk subgroup with a severely impaired apoptosis process. It is important to note that higher levels of BCL-2 were observed in the subset of patients who had only 1–3 prior lines of therapy as compared to those with more than 4 lines of therapy [88].
These very promising early phase data prompted the initiation of clinical trial M14-031 (BELLINI), which aims to register the combination venetoclax-bortezomib-dexamethasone versus placebo-bortezomib- dexamethasone as a life-saving therapy for RRMM with 1–3 prior lines of therapy. Importantly, outcomes in patients with high-risk cytogenetics were distinct based on t(11;14) status and BCL-2 gene expression. Trends in progression-free survival and overall survival favored the venetoclax arm in patients with either t(11;14) or BCL-2 high gene expression regardless of cytogenetics status. In contrast, patients with high-risk cytogenetics and BCL-2 low gene expression in the absence of t(11;14) were most at risk when treated with venetoclax due to the higher risk of toxicity. The most common Grade 3/4 adverse events (AEs) were neutropenia (21% vs. 8%), thrombocytopenia (15% vs. 30%), anemia (16% vs. 15%), diarrhea (15% vs. 12%), and pneumonia (18% vs. 13%). There were 12 treatment-emergent AEs (TEAEs) leading to death in the venetoclax arm and 1 in the placebo arm. Deaths attributed to infections were more common in patients treated with the combination of venetoclax and bortezomib [91].
Therefore, ongoing venetoclax-based studies are focusing primarily on patients with t(11;14) and/or BCL-2 overexpression, marking the first biomarker-driven approach in MM. Based on these initial clinical data, it is conceivable that the inhibition of BCL-2 by venetoclax provided us with the entry point for individualized targeted therapy for a certain important subset of patients with MM. However, the combination bortezomib-venetoclax should be considered with caution in RRMM patients because of the high risk of infections and related toxic deaths.
The role of venetoclax will be further explored in the phase III CANOVA study (NCT03539744), which is evaluating the combination of venetoclax an dexamethasone versus pomalidomide and dexamethasone for t(11;14)-positive RRMM patients [92].
Importantly, as has already been established in another hematological malignancy (mutations in the BCL-2 family) [93], the use of venetoclax can lead to the development of acquired mutations and, consequently, to treatment resistance.
So, in fact, as with other therapies, most patients with MM who initially respond to venetoclax eventually relapse. Many of these acquired resistances may occur through changes in cellular dependence from BCL-2 to MCL-1 or BCL-XL [94].
MM is accompanied by high genomic instability reflected in multiple genetic abnormalities involving several cancer pathways; therefore, MM is typically characterized by high heterogeneity. The progress to discover sensitive genomic targets against which to develop novel agents with the potential to improve patient survival has made remarkable progress. However, these efforts have been severely challenged by the intricate biology of the disease and the molecular mechanisms of MM.
Despite all the progress, MM is still incurable in most patients. Future hopes are therefore directed toward identifying additional therapeutic strategies and new targets for its treatment.
In this review, we summarized the RAS/RAF/MEK/ERK and BCL-2 signaling pathway describing their involvement in the pathogenesis of MM and the therapeutic approaches developed on them.
The efficacy of these therapeutic agents was tested on different MM cell lines and in MM patients and produced encouraging results. MEK inhibitors in combination with other kinase or mutant gene inhibitors have shown promising results in patients with RRMM. The combination delays the onset of acquired resistance resulting in increased progression-free survival and overall patient survival.
Similarly, the BCL-2 inhibitor venetoclax as monotherapy and in combination with other anti-myeloma agents has demonstrated improved outcomes in early clinical trials in MM patients with BCL-2 overexpression regarding the cytogenetic status.
With the availability of different types of treatment, over time it will be more complex to choose which treatment approach is most suitable for the patient and, as a result, it becomes necessary to understand how to incorporate such strategies into existing treatment approaches. An appropriate option might be to use a different combination of pathway-specific drugs that, by interacting with each other, could reduce the likelihood of developing drug resistance.
This could be further aided through deep molecular analysis of MM cells profile of the single patient, which would allow the identification of patients who would benefit from these drugs.
AIF: apoptosis-inducing factor
BAK: B cell lymphoma 2 antagonist/killer
BATF: basic leucine zipper ATF-like transcription factor
BAX: B cell lymphoma 2-associated X
BCL-2: B cell lymphoma 2
BH3: B cell lymphoma 2 homology domain 3
BM: bone marrow
BRAF: B-Raf proto-oncogene
CCND1: cyclin D1
CNVs: copy number variations
DIABLO: direct inhibitor of apoptosis protein (IAP)-binding protein with low pI
ERK: extracellular signal-regulated kinase
FGFR3: fibroblast growth factor receptor 3
IAP: inhibitor of apoptosis protein
Ig: immunoglobulin
KRAS: KRAS proto-oncogene
MAPK: mitogen-activated protein kinase
MEK: mitogen-activated extracellular signal-regulated kinase kinase
MGUS: monoclonal gammopathy of undetermined significance
MM: multiple myeloma
miR-16-1: microRNA-16-1
MYC: MYC proto-oncogene
NRAS: NRAS proto-oncogene
ORR: overall response rate
PCs: plasma cells
PR: partial response
RAF: RAF proto-oncogene, serine/threonine kinase
RAS: rat sarcoma virus
RRMM: relapsed-refractory multiple myeloma
Smac: second mitochondria-derived activator of caspase
TP53: tumor protein p53
VR, NTI, and NG wrote the manuscript; VR and NTI designed the figures. All authors contributed to manuscript revision, read and approved the submitted version.
NG received research funding and honoraria from Amgen, Bristol Mayers Squibb, Celgene, Millenium Pharmaceutical, GSK, Takeda, and Janssen Pharmaceutical. The remaining authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by a grant from “Ricerca Finalizzata del Ministero della Salute Italiana” PE-2016-02361261. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Sara Ovejero, Jerome Moreaux
James H Stoeckle ... Gareth J Morgan
Iolanda Capone ... Giancarlo Pruneri
Christos Ladias ... Nikolaos A. Papanikolaou
