 Review
Review

Affiliation:
1Oncology Division, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte, 1649-028 Lisboa, Portugal
†These authors share the first authorship.
Email: inessasso.pinho@gmail.com
ORCID: https://orcid.org/0000-0002-8377-0179
Affiliation:
1Oncology Division, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte, 1649-028 Lisboa, Portugal
2Luis Costa Laboratory, Instituto de Medicina Molecular-João Lobo Antunes, Faculdade de Medicina de Lisboa, 1649-028 Lisboa, Portugal
†These authors share the first authorship.
ORCID: https://orcid.org/0000-0003-3558-3966
Affiliation:
2Luis Costa Laboratory, Instituto de Medicina Molecular-João Lobo Antunes, Faculdade de Medicina de Lisboa, 1649-028 Lisboa, Portugal
ORCID: https://orcid.org/0000-0003-0985-0821
Affiliation:
2Luis Costa Laboratory, Instituto de Medicina Molecular-João Lobo Antunes, Faculdade de Medicina de Lisboa, 1649-028 Lisboa, Portugal
ORCID: https://orcid.org/0000-0002-6917-4477
Affiliation:
1Oncology Division, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte, 1649-028 Lisboa, Portugal
2Luis Costa Laboratory, Instituto de Medicina Molecular-João Lobo Antunes, Faculdade de Medicina de Lisboa, 1649-028 Lisboa, Portugal
ORCID: https://orcid.org/0000-0002-5506-0233
Affiliation:
1Oncology Division, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte, 1649-028 Lisboa, Portugal
ORCID: https://orcid.org/0000-0002-9147-7896
Affiliation:
1Oncology Division, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte, 1649-028 Lisboa, Portugal
2Luis Costa Laboratory, Instituto de Medicina Molecular-João Lobo Antunes, Faculdade de Medicina de Lisboa, 1649-028 Lisboa, Portugal
Email: luiscosta.oncology@gmail.com
ORCID: https://orcid.org/0000-0002-4782-7318
Explor Target Antitumor Ther. 2022;3:337–361 DOI: https://doi.org/10.37349/etat.2022.00086
Received: February 04, 2022 Accepted: March 24, 2022 Published: June 20, 2022
Academic Editor: Simon Langdon, University of Edinburgh, UK
The article belongs to the special issue Endocrine Resistant Breast Cancer
The most common breast cancer (BC) subtypes are hormone-dependent, being either estrogen receptor-positive (ER+), progesterone receptor-positive (PR+), or both, and altogether comprise the luminal subtype. The mainstay of treatment for luminal BC is endocrine therapy (ET), which includes several agents that act either directly targeting ER action or suppressing estrogen production. Over the years, ET has proven efficacy in reducing mortality and improving clinical outcomes in metastatic and nonmetastatic BC. However, the development of ET resistance promotes cancer survival and progression and hinders the use of endocrine agents. Several mechanisms implicated in endocrine resistance have now been extensively studied. Based on the current clinical and pre-clinical data, the present article briefly reviews the well-established pathways of ET resistance and continues by focusing on the three most recently uncovered pathways, which may mediate resistance to ET, namely receptor activator of nuclear factor kappa B ligand (RANKL)/receptor activator of nuclear factor kappa B (RANK), nuclear factor kappa B (NFκB), and Notch. It additionally overviews the evidence underlying the approval of combined therapies to overcome ET resistance in BC, while highlighting the relevance of future studies focusing on putative mediators of ET resistance to uncover new therapeutic options for the disease.
Breast cancer (BC) is the most common solid malignancy in females. Accounting for nearly one in four newly diagnosed cancer cases, BC is currently the leading cause of death in women worldwide [1].
The disease is characterized by remarkable clinical, morphological, and molecular heterogeneity. At the molecular level, BC is mainly classified as hormone receptor-positive (HR+), according to the expression of estrogen receptor (ER) and progesterone receptors (PRs), and/or human epidermal growth factor receptor 2-positive (HER2+; HER2/ERBB2), if HER2 oncogene is amplified. Tumors lacking expression of all three receptors are classified as triple-negative BCs (TNBCs). These distinct molecular subtypes have different clinical outcomes and therapeutic options [2].
Approximately 70% of all BCs are ER+, with proliferation tightly linked to the ER signaling pathway [3]. The ER is an intracellular receptor activated by estrogen that acts as a transcription factor, regulating the expression of genes related to BC tumorigenesis, proliferation, and survival [4]. ER exists in two isoforms, ERα and ERβ, which can homo- or hetero-dimerize to mediate transcriptional activity. ERα, encoded by the gene estrogen receptor 1 (ESR1) on chromosome 6, is ubiquitously expressed in multiple organs, including the mammary gland, acts as a promoter of tumorigenesis, and is the main predictive biomarker of endocrine therapy (ET) efficacy [5, 6].
Previous studies in ERα knockout mice have shown that ERα is essential for the onset of BC development and progression [7]. Consequently, inhibition of ERα through ET has become one of the major strategies for the prevention and treatment of BC.
ERβ, encoded by ESR2 on chromosome 14, is highly expressed in the prostate and ovaries and appears to have the opposite effect of ERα, restricting estrogen-dependent cell proliferation. In fact, some reports correlated ERβ higher expression with better survival and response to tamoxifen (TAM), irrespective of the ERα status, with lower levels of ERβ potentially contributing to endocrine resistance [8, 9]. This view of ERβ as purely suppressive in BC is defective, with a growing body of evidence reporting elevated ERβ expression as a predictor of poor prognosis and reduced disease-free survival (DFS) in women with ERα+ BC who underwent ET [10, 11].
Alternative messenger RNA (mRNA) splicing affects the expression of ERα and ERβ, leading to distinctive cellular localization, ligand-binding properties, and post-translational modifications of both receptors. ERα and ERβ have five structural and functional domains, namely, the amino-terminal domain (A/B domain), DNA binding domain (DBD/C-domain), hinge region domain (D-domain), ligand-binding domain (LBD/E-domain), and the carboxyl-terminal domain (F-domain) whose function remains unclear. ERα and ERβ share 96% homology in the DBD region, however, the A/B domain, D-domain, and F-domain are divergent (Figure 1) [12]. At the present moment, the function of ERβ in BC cancer remains a matter of debate and its role in the pathophysiology of estrogen signaling and endocrine resistance is still not fully understood.
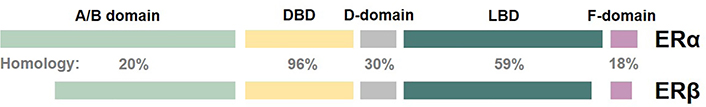
Schematic representation of the structural domains and percentage of homology of ERα and ERβ
For newly diagnosed BC, the pathological determination of positive ER status is an essential criterion for ET treatment. ERα status is determined by immunohistochemistry (IHC) staining on formalin-fixed paraffin-embedded (FFPE) tumor tissue sections, with positivity if ER is detected in ≥ 1% tumor cells, as this predicts responsiveness to ET [13].
Due to the pivotal role of ER signaling in ER+ BC, ET constitutes the treatment backbone in this subtype. ET is associated with a mortality reduction of 25–30% in the palliative setting [7] and a nearly 40% reduction in the relative risk of recurrence in the adjuvant setting [8]. Several ET options with different mechanisms of action have been developed over the past decades and are currently available in clinical practice, including inhibitors of ER activity, modulators of ER half-life, and regulators of estrogen availability [3, 14].
ET agents are mainly divided into three classes: estrogen synthesis inhibitors [e.g., aromatase inhibitors (AIs), as letrozole, anastrozole, and exemestane], selective ER modulators (SERMs; e.g., TAM), and selective ER downregulators [SERDs; e.g., fulvestrant (FULV)]. AIs suppress estrogen production through the inhibition of the enzyme aromatase. Aromatase is responsible for the conversion of androgens produced by different tissues (e.g., ovary, adrenal gland, adipose tissue, breast) into estrogen. By decreasing the levels of estrogen available to bind and activate ER, the ER signaling is impaired. SERMs competitively bind to both ER isoforms, causing conformational changes in the receptor. These changes result in the formation of an inactive ER complex, partially impairing the ER transcriptional activity. SERDs also competitively bind to ER, but the conformational changes induced in the ER complex lead to its degradation, thereby inhibiting its translocation to the nucleus and abolishing ER transcription activity (Figure 2) [3, 14, 15].
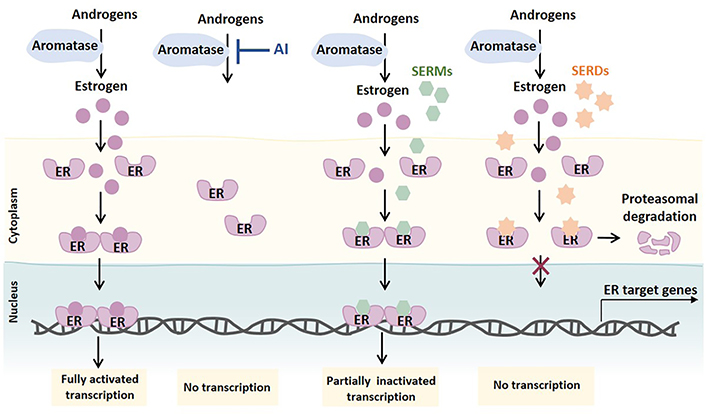
Mechanism of action of ETs primarily used to treat ER+ BC. Androgens produced by different tissues are converted into estrogens by aromatase. Upon estrogen binding, ER dimers translocate into the nucleus as transcriptionally active ER complexes. AIs block the synthesis of estrogen by blocking androgen aromatization. SERMs competitively bind to ER and partially impair ER signaling by forming an inactive ER complex. SERDs, considered pure ER antagonists, also competitively bind to ER, but inhibit ER transcription by causing ER complex changes that lead to ER proteasome-dependent degradation
Moreover, breast carcinomas that co-express ER and PR tend to have a better response to ET [16, 17]. PR signaling is dependent on ERα, therefore tumors with negative PR status may present altered ERα signaling and therefore an ineffective ET response [18].
However, 40–50% of patients treated with ET are at risk of cancer recurrence or progression due to intrinsic or acquired resistance [19]. Intrinsic or de novo resistance—defined as existing prior to treatment start or developing early on the course of treatment—is present in around 50% of patients with metastatic ER+ BC [20]. The primary mechanism of intrinsic resistance is the loss of expression of ERα [4]. However, it has been recently shown that patients carrying inactive cytochrome P450 2D6 (CYP2D6) alleles— approximately 8% of Caucasian women—are unable to convert TAM into its active metabolite, endoxifen, being thus less responsive to TAM [21]. Acquired resistance occurs in a significant percentage of patients after initial response during prolonged exposure to ET. The mechanisms responsible for acquired resistance are multiple and include the activation of several signaling pathways. Importantly, some mechanisms involved in intrinsic and acquired resistance overlap.
In this review, the already well-established mechanisms of ET resistance will be revisited, and the evidence underlying three pathways recently reported as putative mediators of ET resistance in BC—receptor activator of nuclear factor kappa B ligand (RANKL)/receptor activator of nuclear factor kappa B (RANK), nuclear factor kappa B (NFκB), and Notch—will be addressed.
In recent years, several molecular mechanisms of ET resistance have been the subject of comprehensive reviews, with the complex crosstalk between ER signaling and other signaling pathways responsible for orchestrating the phenomenon now intelligible [12, 22, 23–31]. Since a thorough description of ET resistance mechanisms is beyond the scope of this review, the main mechanisms will be hereinafter briefly summarized (Table 1).
Mechanisms of endocrine resistance [3]
| Resistance pathway | Mechanism | Reference(s) |
|---|---|---|
| ER expression and activity loss | Mutations | [32] |
| Gene regulation | [33] | |
| Post-transcriptional modifications (e.g., splice variants, mRNA stability) | [34, 35] | |
| Post-translational modifications | [36] | |
| Transcriptional machinery of ER | Down-regulation of co-repressors (e.g., NCoR) | [37] |
| Over-expression of co-activators (e.g., AIB1) | [38, 39] | |
| Increased expression of transcriptional factors (e.g., AP-1, SP-1, NFκB) | [40, 41] | |
| Cross-talk between ER and RTKs | EGF/EGFR | [42–44] |
| HER2 | [44–46] | |
| IGF1R | [47, 48] | |
| PI3Ks/Akt | [48–52] | |
| p44/42 MAPK | [53, 54] | |
| Stress-induced kinases (JNK, p38 MAPK) | [55] | |
| Cell cycle regulators | Over-expression of positive regulators (e.g., MYC and cyclins E1 and D1) | [56] |
| Reduced expression of negative regulators (e.g., p21 and p27) | [57, 58] | |
| Over-expression of anti-apoptotic molecules (e.g., BCL-XL) | [59] | |
| Reduced expression of pro-apoptotic molecules (e.g., BCL-2-interacting killer and caspase 9) | [59] |
AIB1: amplified in breast 1; Akt: protein kinase B; AP-1: activator protein 1; BCL-2: B-cell lymphoma 2; BCL-XL: B-cell lymphoma-extra large; EGF: epidermal growth factor; EGFR: EGF receptor; IGF1R: insulin growth factor 1 receptor; JNK: c-Jun N-terminal kinase; MAPK: mitogen-activated protein kinase; NCoR: nuclear receptor corepressor; PI3Ks: phosphatidylinositol 3-kinases; SP-1: specificity protein 1
Note. Reprinted from “Biological mechanisms and clinical implications of endocrine resistance in breast cancer,” by Giuliano M, Schifp R, Osborne CK, Trivedi MV. Breast. 2011;20 Suppl 3:S42–9 (https://linkinghub.elsevier.com/retrieve/pii/S0960977611702934). CC BY-NC-ND.
ET resistance has been associated with alterations in the ER expression at gene and protein levels or in the expression of CYP19A1 (or ARO1) gene encoding for aromatase. Pre-clinical and clinical evidence supports that ER expression may change during the natural history of disease and under ET exposure, and that, a switch to an ER-negative phenotype results in ET inefficacy. An estimated 20% of ER+ BC patients treated with ET lose ER expression over time [3, 12, 60], mainly due to epigenetic and post-transcriptional mechanisms [3, 4, 15, 61]. The first point mutations in the ESR1 gene, which encodes for ERα, were reported two decades ago [62]. However, it was only more recently that these mutations were found to enable hormone-independent ER transcriptional activity, resulting in constitutive ER activity and ET resistance [4, 60, 61, 63–65]. Such mutations mostly occur in the LBD of ESR1 [4, 64, 65] and are detected in 20% of recurrent BCs following long-term treatment with AIs or TAM [63]. Although at a much lower frequency than point mutations, ESR1 amplification and gene fusions have also been reported [63–65].
Next-generation oral SERMs or SERDs (e.g., elacestrant, SAR43985, rintodestrant, ZN-c5, H3B-6545, and ARV-471) are currently in clinical development to target both wild-type and mutant ER as a way to overcome ER mutation-driven resistance to ET [63].
Additionally, ER transcriptional activity can be modulated by different coregulatory proteins and/or transcriptional factors. Coregulatory proteins can be coactivators [e.g., nuclear receptor coactivator 1 (NCoA1), NCoA2, NCoA3] or corepressors [e.g., NCoR1, nuclear receptor subfamily 2 group F member 2 (NR2F2)] and are recruited to bind to ER and respectively enhance or repress the transcriptional activity of specific DNA elements [estrogen response elements (EREs)] located in the promoter regions of different ER target genes. Moreover, transcriptional factors, like AP-1 and SP-1, regulate the transcription of genes that do not contain EREs. Changes in the expression of coregulatory proteins or transcriptional factors critically influence the effectiveness of ET and are also associated with endocrine resistance [3, 6, 12].
Amplification of the CYP19A1 gene is also found in AI-resistant breast tumors (21.5% of AI-treated and < 2% of primary tumors), leading to increased aromatase activity, estrogen availability, and consequent ER signaling [66]. Although irreversibly resistant to AIs, these tumors remain sensitive to FULV, being strong candidates for treatment with SERDs [27, 63].
Overall, any variation in ER expression or signaling may contribute to endocrine resistance and more aggressive phenotypes. The regulation of ER expression is complex and not totally understood as reviewed by Hua et al. [66], requiring further investigation.
Cell cycle and apoptosis regulators play a key role in the proliferation of normal and BC cells. Therefore, it comes with no surprise that deregulation of these proteins is associated with endocrine resistance.
Several studies have shown that the activity of both positive and negative cell cycle regulators can have an impact on BC sensitivity to ET [3, 67, 68]. Increased expression of positive regulators, like c-MYC and cyclins D1 and E1, can activate cyclin-dependent kinases (CDKs), contributing to aberrant cell cycle progression and resistance to ET. Reduced activity of negative cell cycle regulators, like p21 and p27, and inactivation of retinoblastoma (RB) tumor suppressor also enable aberrant cell cycle progression, contributing to endocrine resistance. Additionally, increased expression of anti-apoptotic molecules like BCL-XL and reduced expression of pro-apoptotic molecules like BCL-2-interacting killer and caspase 9 also play a role in ET resistance [3, 4, 12].
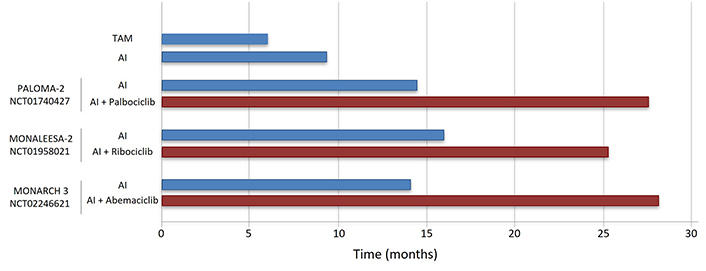
Three CDK4/6 inhibitors (CDK4/6i; palbociclib, ribociclib, abemaciclib) are currently approved in combination with standard ET to treat metastatic ER+ BC. Clinical studies showed that combined CDK4/6i and letrozole significantly improve progression-free survival (PFS) compared to letrozole alone in advanced ER+ BC resulted in the approval of CDK4/6i in combination with an AI in first line treatment of this disease [69–71]. In addition, the combination of a CDK4/6i and FULV has been approved for use following progression on initial AI monotherapy (Table 2, Figures 3 and 4) [20, 72, 73].
Summary of clinical trials leading to the approval of new drugs to overcome endocrine resistance in HR+/HER2– BC
| Drug | Target | Study | Phase | Population | Therapy line | Treatment arms | Outcome | Reference(s) |
|---|---|---|---|---|---|---|---|---|
| CDK4/6i | ||||||||
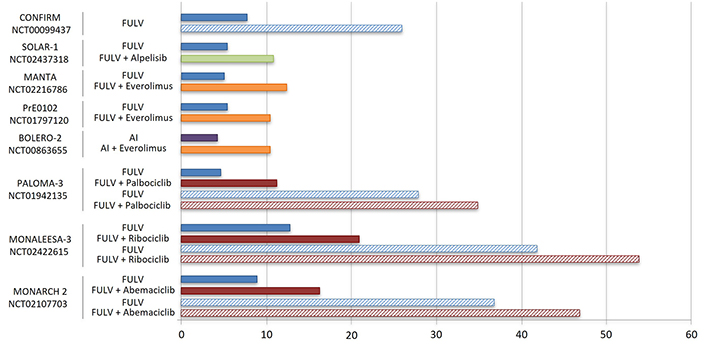
| Palbociclib | CDK4/6 | PALOMA-3 NCT01942135 | III | HR+/HER2– postmenopausal advanced BC | Second or later lines | Palbociclib + FULV vs. placebo + FULV | mPFS: 11.2 mo vs. 4.6 mo; hazard ratio: 0.46; P < 0.0001mOS: 34.9 mo vs. 28 mo; hazard ratio: 0.81; P = 0.09 | [72, 74] |
| Ribociclib | CDK4/6 | MONALEESA-3 NCT02422615 | III | HR+/HER2– postmenopausal advanced BC | First or second line | Ribociclib + FULV vs. placebo + FULV | mPFS: 21.0 mo vs. 13 mo; hazard ratio: 0.59; P < 0.001mOS: 54 mo vs. 42 mo; hazard ratio: 0.73; P < 0.01 | [75, 76] |
| Abemaciclib | CDK4/6 | MONARCH 2 NCT02107703 | III | HR+/HER2– postmenopausal advanced BC | Second or later lines | Abemaciclib + FULV vs. placebo + FULV | mPFS: 16.4 mo vs. 9.3 mo; hazard ratio: 0.55; P < 0.001mOS: 46.7 mo vs. 37.3 mo; hazard ratio: 0.75; P = 0.01 | [73, 77] |
| Palbociclib | CDK4/6 | PALOMA-2NCT01740427 | III | HR+/HER2– postmenopausal advanced BC | First line | Palbociclib + letrozole vs. placebo + letrozole | mPFS: 27.6 mo vs. 14.5 mo; hazard ratio 0.56, P < 0.001ORR: 42% vs. 35% | [69, 78] |
| Ribociclib | CDK4/6 | MONALEESA-2NCT01958021 | III | HR+/HER2– postmenopausal advanced BC | First line | Ribociclib + letrozole vs. placebo + letrozole | mPFS: 25.3 mo vs. 16 mo; hazard ratio: 0.57; P < 0.001ORR: 43% vs. 29% | [79] |
| CDK4/6 | MONALEESA-7 NCT02278120 | III | HR+/HER2– premenopausal advanced BC | First line | Ribociclib + letrozole/anastrozole/TAM + goserelin vs. placebo + letrozole/anastrozole/TAM + goserelin | mPFS: 24 mo vs. 13 mo; hazard ratio: 0.55, P < 0.0001mOS: n.r. vs. 40.7 mo; hazard ratio: 0.71; P = 0.00973 | [70, 80] | |
| Abemaciclib | CDK4/6 | MONARCH 3NCT02246621 | III | HR+/HER2– postmenopausal advanced BC | First line | Abemaciclib + letrozole vs. placebo + letrozole | mPFS: 28.2 mo vs. 14.2 mo; hazard ratio: 0.54; P < 0.001ORR: 59% vs. 44% | [81] |
| PI3K/AKT/mTOR inhibitors | ||||||||
| Everolimus | mTOR1 | BOLERO-2NCT00863655 | III | HR+/HER2– postmenopausal advanced BC | Second or later lines | Everolimus + exemestane vs. placebo + exemestane | mPFS: 10.6 mo vs. 4.1 mo; hazard ratio: 0.36, P < 0.001ORR: 7% vs. 0.4% | [82] |
| MANTANCT02216786 | II | HR+/HER2– postmenopausal advanced BC | Second or later lines | Everolimus + FULV vs. placebo + FULV | mPFS: 12.3 mo vs. 5.4 mo; hazard ratio: 0.63, P = 0.01 | [83] | ||
| PrE0102NCT01797120 | II | HR+/HER2– postmenopausal advanced BC | Second or later lines | Everolimus + FULV vs. placebo + FULV | mPFS: 10.3 mo vs. 5.1 mo; hazard ratio: 0.61, P = 0.02 | [84] | ||
| Alpelisib | Class I PI3K p110α | SOLAR-1NCT02437318 | III | HR+/HER2– postmenopausal advanced BC | First or second line | Alpelisib + FULV vs. placebo + FULV | mPFS: 11.0 mo vs. 5.7 mo; hazard ratio: 0.65, P < 0.001ORR: 26.6% vs. 12.8%; PIK3CA- mutant subsetmOS: 39.3 mo vs. 31.4 mo; hazard ratio: 0.86, P = 0.15 | [71, 85] |
mo: months; mOS: median overall survival; mPFS: median PFS; mTOR: mammalian target of rapamycin; n.r.: not reached; ORR: overall response rate; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
The activity of cell cycle and apoptosis regulators can be modulated by tyrosine kinase receptors (TKRs) and transcription factor signaling pathways, with evidence of crosstalk between ER signaling, TKR pathways, cell cycle regulators, and apoptotic molecules towards ET escape.
Comprehensive evidence suggests that endocrine resistance can be driven by increased expression and activity of TKRs and subsequent activation of downstream signaling pathways, such as PI3K/AKT/mTOR and MAPK. Upon aberrant activation of TKRs (most commonly by amplification or mutation), downstream intracellular signal transduction pathways are activated, inducing ER phosphorylation and activation in the absence of estradiol [3, 4, 60, 63].
TKRs are a family of cell membrane receptors with a tyrosine kinase intracellular domain that phosphorylates tyrosine residues of target proteins. They include HER2, EGFR, FGFRs, and IGFR. HER2/ERBB2 amplification has long been known to reduce sensitivity to anti-estrogens in ER+/HER2+ tumors, with the current standard of care for this BC subtype being a combination of ET and HER2 inhibitors. Treatment of HER2-amplified BC will not be explored, as it is out of the scope of this review but has been recently reviewed by Chow et al. [87]. More recently, HER2-activating mutations have been associated with both intrinsic and acquired ET resistance [88, 89]. Somatic mutations in HER2 are observed in approximately 5% of endocrine-resistant, non-HER2-amplified BCs [90]. Tumors with an incidence of HER2 mutations as high as 10% are further enriched in lobular histology [91]. Combined blockade of HER2 mutations and ER results in synergistic antitumor activity in vitro and in vivo [88, 89], and the combination of neratinib (a tyrosine kinase inhibitor of HER2, EGFR, HER4, and HER3 heterodimers) and FULV has shown promising activity in metastatic ER+/HER2-mutated BC [91].
FGFR1–4 constitute a family of four highly conserved TKRs. Deregulation of FGFR has been reported in a variety of human cancers, including BC [92]. FGFR1 amplification is found in approximately 14% of metastatic BCs and is associated with de novo endocrine resistance [93]. FGFR2 genomic and expression alterations can be found in approximately 6% of BCs [94], with FGFR2 associated with endocrine resistance in vitro [95]. Clinical trials combining pan-FGFR inhibitors and ET are ongoing in FGFR1/2-amplified ER+/HER2– metastatic BC. FOENIX-MBC2 TAS-120-201 (NCT04024436) is a phase II trial evaluating the combination of FULV with futibatinib (TAS-120). The triple combination of ET, CDK4/6i, and FGFR inhibitor is also being explored in a phase Ib trial (VICC BRE 16126; NCT03238196). FGFR4 genomic and expression alterations can be found in approximately 7% of BC patients [94]. Recently, FGFR4 overexpression and hotspot mutations have been associated with metastases and endocrine resistance in lobular metastatic BC [96], and a recent study suggested that FGFR4 promotes the transition from a more differentiated luminal phenotype to a highly proliferative and metastatic, endocrine-resistant, HER2-enriched subtype [97]. The combination of ET with an FGFR4-selective inhibitor seems to be an attractive strategy in tumors exhibiting FGFR4 genomic signatures.
The PI3K/AKT/mTOR pathway is one of the most frequently activated pathways in several types of cancer [98] and the link between PI3K/AKT/mTOR pathway deregulation and endocrine resistance in BC is well established [31, 99]. A number of drugs targeting PI3K/AKT/mTOR are currently being investigated in clinical trials in combination with standard therapies as a strategy to overcome acquired resistance in BC. Somatic mutations in PIK3CA are the most common cause of aberrant PI3K/AKT/mTOR pathway activation, prompting the development of various PI3K inhibitors (PI3Ki) with a different selectivity against the four PI3K catalytic subunit isoforms [100]. The selective alpha isoform inhibitor alpelisib (BYL719) was the first oral PI3Ki developed and was recently approved for the treatment of advanced PIK3CA-mutated ER+/HER2– BC progressing on previous ET (Table 2, Figure 4) [85]. AKT is another potential therapeutic target to overcome endocrine resistance, with several AKT inhibitors (AKTi) currently under clinical investigation. The combination of the pan-AKT inhibitor capivasertib (AZD5363) with FULV has been shown to improve PFS in metastatic ER+ BC progressing on AIs [101]. This combination may be particularly effective in ER+ BC with AKT1 mutations [102, 103]. Notably, it has been recently shown that triple inhibition by FULV, CDK4/6i, and AKTi durably impairs BC cell growth, prevents progression, and reduces metastases of tumor xenografts resistant to the combination of CDK4/6i and FULV or FULV alone [104]. A phase III trial (CAPItello-291; NCT04305496) is currently ongoing to evaluate capivasertib in combination with FULV in metastatic ER+ BC following progression on AIs. Furthermore, a phase Ib/III trial is evaluating capivasertib plus palbociclib and FULV in locally advanced, unresectable, or metastatic ER+ BC (CAPItello-292; NCT0486266).
Inhibition of mTOR, a downstream target of the PI3K pathway, has also been shown to reduce endocrine resistance. The mTOR complex-1 inhibitor everolimus in combination with exemestane was shown to prolong PFS in advanced ER+/HER2– BC after progression on non-steroidal AIs [82], independently of PIK3CA mutational status [105]. Consistently, patients with AI-resistant metastatic BC have also been shown to obtain clinical benefits from everolimus combined with FULV (Table 2, Figure 4) [83, 84].
RANKL and its receptor RANK belong to the tumor necrosis factor (TNF) superfamily and were identified in the late 1990s as key regulators of osteoclastogenesis [106, 107]. The pivotal role of the RANKL-RANK pathway in bone physiology and pathology led to the development of denosumab, a fully human anti-RANKL monoclonal antibody currently approved for the treatment of bone remodeling diseases, including cancer-induced bone metastases [108, 109].
In the last decade, extensive research stemming from the observation of RANK-expressing RANKL-sensitive cancer cell lines from different tumor types, including BC [110–113], clearly corroborated that the RANKL-RANK pathway is a major mediator of breast physiology and carcinogenesis [107, 114–116] as recently reviewed by our study group [117]. In view of this evidence, inhibition of the RANK pathway emerged as a promising strategy in BC prevention and treatment.
The fact that expansion of RANK-positive mammary basal stem cells is mediated by paracrine RANKL signaling [115] and that RANK-positive BC cases are more common among TNBC subtypes [118–123] established the relevance of the RANKL-RANK pathway mainly in this BC subtype. Accordingly, numerous studies have shown that RANK-expressing TNBC is a more aggressive subtype, as RANKL activates a signaling cascade involving NFκB, AKT (PKB), JNK, extracellular signal-regulated kinase (ERK), proto-oncogene tyrosine-protein kinase Src (Src), and MAPK, increasing migration, invasion, stemness, transformation, epithelial-mesenchymal transition (EMT), anchorage-independent growth, and metastatic ability [117]. It was only recently that our own research [124] and that of Benítez et al. [125] disclosed a link between RANK expression and luminal BC phenotype and carcinogenesis, respectively. In a recent study, our group characterized the phenotype of RANK-overexpressing (RANK OE) luminal BC cell lines, showing that RANK OE cells have a staminal and mesenchymal phenotype, with decreased proliferation rate and decreased susceptibility to chemotherapy, but are more invasive in vivo [124]. In silico analysis of the transcriptome of human breast tumors confirmed the association between RANK expression and stem cell and mesenchymal markers in ER+/HER2– tumors, leading to the hypothesis that luminal RANK-positive cells may constitute an important reservoir of slow cycling, therapy-resistant cancer cells [124].
Prior studies have shown that multiparous mouse mammary tumor virus (MMTV)-RANK mice, with RANK OE in the mammary gland under MMTV, develop spontaneous breast tumors with long latency and only after multiple pregnancies [126], although tumor latency decreases and tumor incidence increases compared to wild-type after carcinogenic protocols [22, 115]. Genetic or pharmacological inhibition of RANK signaling abrogated carcinogenesis in this model and also delayed tumor onset and decreased tumor and metastases incidence in HER2+ or polyomavirus-induced BC, MMTV-Neu, and MMTV-polyomavirus middle T antigen (PyMT) mice [127, 128]. In these models, RANK was focally expressed in non-transformed mammary glands but increased in mammary pre-neoplastic lesions and invasive adenocarcinomas. The long latency of MMTV-RANK carcinogenesis was recently found to be due to RANK-driven senescence, which initially delays tumor onset in oncogene-driven models but promotes stemness, luminal-like tumor growth, and metastases in later stages of tumor progression [125, 128]. In accordance with these findings, transcriptomic analysis of human breast tumors from luminal subtype linked RANK expression to senescence [125].
The work of our group has also disclosed that RANK OE cells are more resistant to FULV compared to RANK-low counterparts [124]. This suggests that the RANK pathway may be implicated in intrinsic ET resistance. Interestingly, continuous pathway activation by prolonged RANKL exposure led to a decrease in proliferation and down-regulated ER and PR, with an increase in FULV resistance. This suggests that RANK signaling may also be associated with the induction of acquired resistance to ET due to ER loss. Activation of the RANK signaling pathway leads to an increase in noncanonical NFκB signaling, which plays a key role in the above-mentioned regulation of proliferation of mammary epithelial cells—through the RANKL–RANK–NFκB inhibitor (IκB) kinase α (KKα; IKKα)–IκBα–p50/p65–cyclin D1 axis [129]. Unlike the canonical NFκB pathway that responds rapidly to signals from different receptors, the noncanonical NFκB pathway specifically responds to a small group of receptors, namely lymphotoxin-β receptor (LTβR), B-cell-activating factor receptor (BAFFR) belonging to the TNF family receptor, CD40, and RANK [130].
The RANK–NFκB axis has been associated with increased resistance to the anti-HER2 therapy lapatinib in RANK-positive HER2+ BC cells [131]. Moreover, although several signaling pathways crosstalk due to activation of different receptors, lapatinib was not able to counteract RANK-induced NFκB activation in this study. Conversely, it was previously reported that NFκB inhibitors abrogate RANKL-induced EMT, cell migration, and invasion [132].
It is widely accepted and clearly demonstrated that multiple mechanisms are involved in the crosstalk between NFκB and ER. Accordingly, there is compelling evidence of a role for the NFκB pathway in ET resistance, indicating that RANK-mediated NFκB signaling may contribute to ET resistance. This suggests that it may be of particular interest to further study the mechanism of RANK-mediated ET resistance and investigate the efficacy of RANK pathway inhibition—either at the RANKL level or via inhibition of downstream mediators like NFκB—as a mechanism to overcome it (Figure 5).
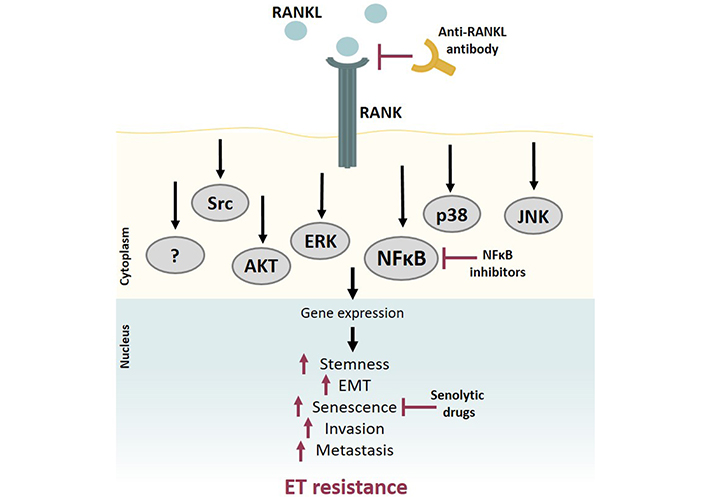
RANK pathway contributes to ET resistance. RANK activation by RANKL activates a signaling cascade that involves several downstream pathways, such as NFκB, AKT, ERK, and MAPK. RANK OE breast tumors are more aggressive, presenting a staminal and mesenchymal phenotype, with increased invasion and metastatic ability. RANKL-RANK pathway activation induces ET resistance through NFκB activation and/or other mechanisms yet to be clarified. Inhibition of RANK pathway signaling, either by blocking RANKL, inhibiting downstream mediators like NFκB, or using senolytic drugs to overcome RANK-induced senescence, may contribute to avoiding or circumventing ET resistance
The NFκB family comprises five inducible transcription factors p65(RelA), RelB, cRel, NFκB1 or p50, and NFκB2 or p52, all of which play critical roles in cell proliferation and survival and inflammatory and immune responses [133–136], being crucial for normal organ development, including of the mammary gland [137].
A growing body of evidence indicates abnormal activation of the NFκB pathway in multiple malignancies, suggesting a putative role for NFκB in tumorigenesis [134, 138–141] and chemotherapy resistance [142].
Members of the NFκB family have a conserved N-terminal Rel homology domain (RHD) [143] that is responsible for dimerization, nuclear translocation, DNA binding, and interaction with IκB. This family of molecules includes multiple proteins, among which IκBα, IκBβ, and IκBε, are the most important NFκB regulators. NFκB molecules exist either as homo or heterodimers, but the most abundant intracellular form is the p50(NFκB1)/p65(RelA) heterodimer. On unstimulated cells, NFκB homo or heterodimers are hijacked in the cell cytoplasm, binding to their IκB inhibitor [143]. Activation of the NFκB cascade occurs either through canonical or noncanonical pathways [144, 145].
The canonical pathway is activated following stimulation by inflammatory cytokines [interleukin-1 (IL-1), lipopolysaccharide (LPS), TNFα, T- and B-cell mitogens], growth factors, viral proteins, and extracellular physical and chemical stress [146, 147]. These ligands activate their receptors and initiate a downstream cascade that ultimately leads to phosphorylation and subsequent ubiquitination of IκBα by a trimeric IKK complex [IKKα, IKKβ, and IKKγ or NFκB essential modulator (NEMO)], enabling nuclear translocation of the p50/p65 heterodimer. Upon translocation, the complex interacts with κB sites to accelerate the transcription of target genes [144, 145, 148–150].
Conversely, in the noncanonical pathway, NFκB-inducing kinase (NIK) phosphorylates and activates IKKα in response to signals from a small group of receptors, including the RANK [151]. Activated KKα then phosphorylates p100, resulting in p52 liberation and promoting p52/RelB nuclear translocation [152, 153].
Contrarily to p50/RelA activation in the canonical pathway, the role of p52/RelB in therapy resistance remains poorly understood. Nevertheless, RelB expression was found to be high in ER+ BC cells [154], and inhibition of p52/RelB was shown to be able to reverse ER expression [155].
Regarding ET resistance, comprehensive evidence shows that NFκB regulates a series of genes relevant to endocrine resistance [156–158]. In fact, there seems to be a reciprocal interaction between endocrine treatments and the immune system. Inflammation cytokines like IL-1 and TNFα are higher in metastatic BC patients, where they seem to activate the NFκB pathway and lead to endocrine resistance. Antibodies directed against transcription factors upstream of this pathway have restored sensitivity in cell line models of endocrine-resistant BC [4]. There is also supporting evidence that a dysregulated immune response or excessive inflammation in the tumor microenvironment (TME) could be related to endocrine resistance and could promote BC progression and metastasis [159, 160]. Downstream NFκB-regulated proteins, like BCL-2, cyclin D1, and cytokines IL-6 and IL-8, have been shown to be major players in this process [148, 154]. Additionally, previous studies documented that estrogen withdrawal led to the increased transcriptional activity of p65(RelA) and sustained estrogen-independent tumor growth through upregulation of cyclin D1 and BCL-3 [157]. Oida et al. [161] focused on the role of NFκB in hormone dependency in BC and showed that NFκB inhibition enhanced ER expression and promoted recovery of TAM sensitivity in ER-reduced cell sublines. Another study by Nehra et al. [162] reported p65(RelA) inhibition, either by overexpression of mutant IκB or by synergistic restoration of sensitivity to TAM by the small-molecule NFκB inhibitor parthenolide in resistant MCF-7 cell lines, along with decreased BCL-2 expression and induced caspase-dependent apoptotic cell death in resistant cells. Notably, all effects could be reversed by caspase-8 specific inhibition. In TAM-resistant BC cells, Zhou et al. [134] described the increased transcriptional activity of NFκB and AP-1, which was suppressed after treatment with parthenolide or the proteasome inhibitor bortezomib. Additionally, increased p50/NFκB1, p52/NFκB2, and cRel expression were observed in breast tumors compared with normal surrounding tissues [163].
Owing to the possible putative role of NFκB in BC, targeting the NFκB pathway might be a successful therapeutic strategy. Several inhibitors have been reported to date, still in early developmental stages (Figure 6).
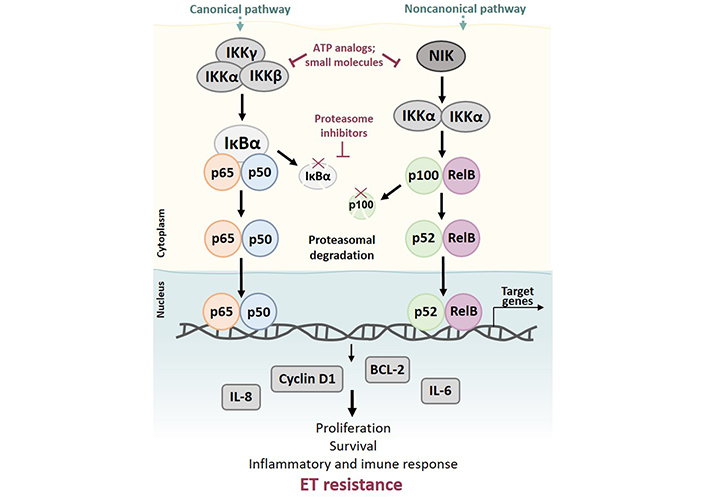
Activation of canonical and noncanonical NFκB pathways may lead to ET resistance. Activation of the canonical pathway can be induced by multiple stimuli, including inflammatory cytokines and growth factors, ultimately leading to phosphorylation and subsequent ubiquitination of IκBα by a trimeric IKK complex (IKKα, IKKβ, and IKKγ), allowing the nuclear translocation of p50/p65 heterodimer. Upon translocation, the complex accelerates the transcription of target genes. The noncanonical pathway is activated by TNF cytokine family members and results in phosphorylation of NIK and activation of IKKα. Activated IKKα then phosphorylates p100, resulting in the liberation of p52 and promoting p52/RelB nuclear translocation. Both canonical and noncanonical pathways play important roles in cell proliferation and survival and inflammatory and immune response. Deregulation of NFκB signaling can lead to drug resistance and expression of downstream NFκB-regulated proteins, like BCL-2, cyclin D1, and cytokines IL-6 and IL-8 described as contributors to ET resistance
Most of these inhibitors prevent proteasomal degradation of IκB proteins, resulting in NFκB hijacking in the cytoplasm. To accomplish this, the drug has either to inhibit IκB function (e.g., proteasome inhibitors), phosphorylation of IκB proteins (e.g., parthenolide), or IKKα and IKKβ. Selective IKKβ inhibitors in particular have shown exciting results in preclinical models, but also important toxicities, precluding their use in clinical practice [164]. Another drug class that can block NFκB activity is the class of nuclear translocation inhibitors. Regardless of the mechanism, the use of NFκB inhibitors in BC endocrine resistance may help elucidate target drivers of this phenomenon and contribute to restoring ET sensitivity.
It is acknowledged that BC subtypes have different clinical outcomes due to different biological and cellular mechanisms involved in tumor aggressiveness, metastases formation, and treatment response [165]. Increased Notch activity and/or Notch deregulation lead to the transformation of normal breast cells into cancer cells, with differential Notch ligand activation potentially associated with oncogenesis and progression of particular BC subtypes and poor clinical outcomes (Table 3) [166, 167].
Alterations in Notch pathway according to BC subtype [165]
| Subtype | BC subtype | |||
|---|---|---|---|---|
| HER2+ | Basal-like | Luminal A | Luminal B | |
| Notch activation (mRNA, protein) |
|
|
|
|
Note. Adapted from “Moving breast cancer therapy up a notch,” by Mollen EWJ, Ient J, Tjan-Heijnen VCG, Boersma LJ, Miele L, Smidt ML, et al. Front Oncol. 2018;8:518 (https://www.frontiersin.org/articles/10.3389/fonc.2018.00518/full). CC BY.
The Notch signaling pathway is mediated by one of four Notch receptors (Notch1–4). While Notch1 and Notch2 are ubiquitously expressed throughout development and in adult life, Notch3 and Notch4 are more abundant in vasculature cell subtypes. Notch1 and Notch2 knockouts are embryonically lethal due to multiple organ defects, while Notch3 and Notch4 knockouts are viable but display subtle vascular abnormalities [147]. Five Notch ligands are recognized in mammals: delta-like ligands 1 (DLL1), DLL3, DLL4, and Jagged 1 (JAG1) and JAG2 [166].
Depending on the cancer type, the Notch pathway can be hyper- or hypo-activated, which relies on specific Notch receptors. In some organs, both pathway activation and repression have been observed, depending on the tumor subtype or model system in question [168].
Overexpression of Notch1 and/or JAG1 have been associated with poor overall survival, supporting the role of Notch as a prognostic biomarker in BC [167]. While Notch gain-of-function mutations are present in a limited number of BCs, Notch is overexpressed, activated, and crosstalks with other oncogenic pathways in several breast tumors [165].
Some of the earliest known targets of Notch signaling include transcriptional repressors, such as the hairy/enhancer of split (HES) genes, the HES subfamily members HEY1, HEY2, and HEYL, c-Myc, and cyclin D1 (Figure 7) [167, 168].
The Notch pathway is involved in breast tumorigenesis, enhanced BC stem cell (BCSC) self-renewal and proliferation, promotion of EMT, angiogenesis, and immunomodulation [168, 169]. However, depending on the BC subtype and/or context, Notch can have an oncogenic or tumor suppressor role [166]. Dysregulation of Notch signaling, namely through activating Notch receptor activating mutations, overexpression of ligands and/or receptors, and/or overexpression of target genes, contributes to increased proliferation, cell transformation, and drug resistance in tumors such as breast, multiple myeloma, prostate, T-cell acute lymphoblastic leukemia, among others [167].
Cumulative evidence indicates that cancer stem cells (CSCs) are key drivers of acquired endocrine resistance in ERα+ breast tumors. Endocrine-resistant BC shows an increase in BCSCs with Notch3/4 expression [165].
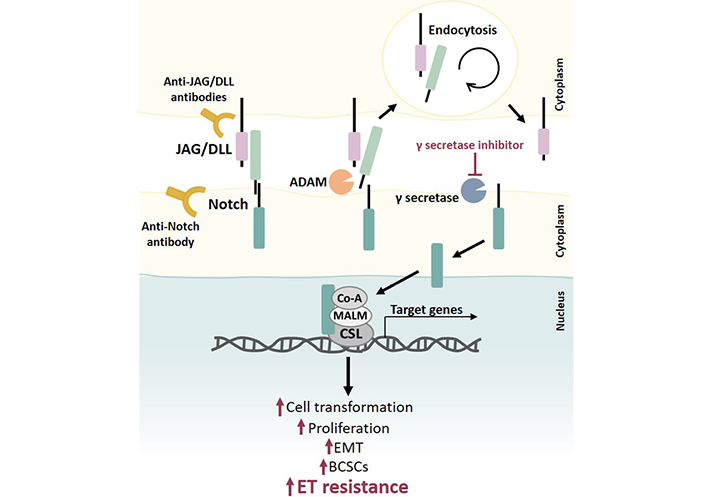
The role of Notch signaling in ET resistance. The Notch receptor is activated by binding to a ligand (JAG/DLL) presented in a neighboring cell. This interaction removes the extracellular portion of Notch from the transmembrane portion, resulting in its endocytosis followed by cleavage events by a disintegrin and metalloprotease (ADAM) and then by γ secretase, which allows the release of the intracellular Notch portion. This intracellular portion translocates to the nucleus, where it binds to DNA-binding protein CSL and recruits transcriptional coactivator Mastermind-like proteins (MALM) and other transcriptional coactivators to initiate transcription of Notch target genes. Notch regulates several cellular processes, and dysregulation of this pathway (e.g., through Notch receptor mutations, overexpression of ligands and/or receptors, and/or overexpression of target genes) contributes to increased cell transformation, proliferation, EMT, BCSCs population, and drug resistance, namely ET resistance. Notch signaling inhibition can be accomplished by using different molecules, such as γ-secretase inhibitors and antibodies anti-Notch ligands, and/or receptors. Co-A: coactivator
Estradiol has been shown to inhibit Notch activity by affecting the receptor cellular location, and TAM and raloxifene have been shown to block this effect, thereby activating the Notch pathway. Therefore, the Notch pathway can be hyperactivated in resistant BC cells, and this can be abrogated by blocking this pathway [166]. Taken together, this provides a strong rationale for studies combining Notch inhibitors with current BC treatment modalities [165].
Interaction between cellular components of the TME and BC cells also regulates Notch signaling-driven therapeutic resistance in breast tumors. Different cellular components of the TME can induce CSC survival, stemness, and resistance through either transforming growth factor beta (TGF-β)-dependent mechanisms or by releasing soluble factors such as cytokines, chemokines, and growth factors that favor angiogenesis and an immunosuppressive environment. In turn, all these factors augment Notch ligand- and receptor-mediated chemoresistance, endocrine resistance, and radioresistance in breast tumors. Additionally, cancer-associated fibroblasts and tumor-associated macrophages can collaborate via cell-cell interaction to promote endocrine resistance, with Notch signaling potentially contributing to the crosstalk between these two cell types [166].
ET was the first targeted treatment developed in BC, and a long and thrilling journey has been traveled since the 1950s when the understanding of ER’s role in disease and its potential as a predictive biomarker began to change the treatment landscape of the disease. However, this remarkable treatment does not provide a cure for all patients, even in association with other types of treatment. Several mechanisms involving ERs, cell cycle and apoptosis regulators, and TKRs have been clearly implicated in endocrine resistance, with numerous drugs and combinations explored to overcome such resistance. Numerous clinical trials have been conducted in this setting, some of which with positive results that led to the approval of first and second line therapies to overcome resistance in BC.
It is clear that there are promising mechanisms and biomarkers of ET resistance that lack clinical validation. Deep analysis of clinical cohorts could expand the portfolio of potential biomarkers, a clear unmet need. However, the clinical and biological heterogeneity of large patient cohorts is challenging in terms of interpretation and clear identification of robust biomarkers. Technologies like next-generation sequencing (NGS) and digital droplet polymerase chain reaction (ddPCR), along with modern bioinformatics pipelines, are key for future research.
Moreover, the TME which includes a panoply of cellular and noncellular (matrix, cytokines, and physical) components affects the response to cancer therapy, namely ET. The communication between cells and the extracellular matrix, along with cell-to-cell communication, is critical for cellular transcriptomics and pathway activity. Therefore, pre-clinical research on the mechanism of resistance must evolve from two-dimensional (2D) cell culture models to heterocellular three-dimensional (3D) models. In this sense, the use of patient-derived organoids also represents an important tool.
Furthermore, the immune component of the TME has particular relevance in response to therapy. In addition, immunotherapy is gaining relevance in the context of ER+ BC. Unfortunately, patient-derived xenografts (PDXs), while undoubtedly useful for screening drug response, lack the immune component, unless conducted in humanized mice. The development of humanized mice at affordable costs will certainly accelerate research in this setting.
Overall, the discovery and understanding of these additional resistance mechanisms will predictably lead to even better outcomes for BC patients. Evidence of the role of new putative mediators of ET resistance, like RANKL-RANK, NFκB, and Notch pathways, supports further studies in the area.
A/B domain: amino-terminal domain
AIs: aromatase inhibitors
AKT: protein kinase B
AP-1: activator protein 1
BC: breast cancer
BCL-2: B-cell lymphoma 2
BCSC: breast cancer stem cell
CDK4/6i: cyclin-dependent kinase 4/6 inhibitors
CDKs: cyclin-dependent kinases
DBD: DNA binding domain
D-domain: hinge region domain
DLL1: delta-like ligands 1
EGFR: epidermal growth factor receptor
EMT: epithelial-mesenchymal transition
ER+: estrogen receptor-positive
ERK: extracellular signal-regulated kinase
ESR1: estrogen receptor 1
ET: endocrine therapy
F-domain: carboxyl-terminal domain
FGFRs: fibroblast growth factor receptors
FULV: fulvestrant
HER2+: human epidermal growth factor receptor 2-positive
HR+: hormone receptor-positive
IκB: nuclear factor kappa B inhibitor
IKKα: nuclear factor kappa B kinase α
IL-1: interleukin-1
JAG1: Jagged 1
JNK: c-jun N-terminal kinase
LBD: ligand-binding domain
MAPK: mitogen-activated protein kinase
MMTV: mouse mammary tumor virus
mOS: median overall survival
mPFS: median progression-free survival
mRNA: messenger RNA
mTOR: mammalian target of rapamycin
NCoA1: nuclear receptor coactivator 1
NFκB: nuclear factor kappa B
NIK: nuclear factor kappa B-inducing kinase
NR2F2: nuclear receptor subfamily 2 group F member 2
PFS: progression-free survival
PI3K: phosphatidylinositol 3-kinase
PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
PR+: progesterone receptor-positive
RANK: receptor activator of nuclear factor kappa B
RANK OE: receptor activator of nuclear factor kappa B-overexpressing
RANKL: receptor activator of nuclear factor kappa B ligand
SERDs: selective estrogen receptor downregulators
SERMs: selective estrogen receptor modulators
TAM: tamoxifen
TKRs: tyrosine kinase receptors
TME: tumor microenvironment
TNBCs: triple-negative breast cancers
TNF: tumor necrosis factor
The authors acknowledge Joana Cavaco Silva for assisting in manuscript revision.
ISP: contributed to the manuscript design and literature analysis, wrote part of the introduction and the NFκB pathway as well as global revision to the article.
CA: contributed to the initial design of the article, wrote Notch pathway and a substantial contribution and regulators of cell cycle apoptosis, as well as to global manuscript revision.
IG: contributed to the manuscript design and literature analysis, wrote part of the introduction and created some figures.
SC: wrote the RANK-RANKL pathway chapter, and full manuscript revision.
TRP: made the revision of TKRs and alternative pathways, made
RTS: specially contributed for the initial design of the article, contributed to the conclusion and review the final manuscript.
LC: contributed to conceptualization, design, direction and overall supervision of the manuscript.
All the authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
IG is supported by the FCT PhD grant SFRH/BD/139178/2018. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Kristin A. Altwegg, Ratna K. Vadlamudi
Alfredo Rossi ... Marta Carlesimo
Katarzyna Rygiel
Kate M. Moore ... Simon P. Langdon
Aglaia Skolariki ... Simon Lord
Samuel Jones ... Kathryn M. Taylor
Matthew Willman ... Brandon Lucke-Wold
Gary J. Cheng ... Dean C. Singleton
David Musheyev, Anya Alayev
