 Review
Review

Affiliation:
Oncology Department, Geneva University Hospital (HUG), 1205 Geneva, Switzerland
ORCID: https://orcid.org/0009-0006-9050-2267
Affiliation:
Oncology Department, Geneva University Hospital (HUG), 1205 Geneva, Switzerland
Email: hiba.mechahougui@hug.ch
ORCID: https://orcid.org/0009-0005-9784-0333
Explor Target Antitumor Ther. 2025;6:1002328 DOI: https://doi.org/10.37349/etat.2025.1002328
Received: January 31, 2025 Accepted: May 28, 2025 Published: June 23, 2025
Academic Editor: Giulia Martini, University of Campania “Luigi Vanvitelli”, Italy
The article belongs to the special issue Liquid Biopsy: Has Already Changed the Clinical Decision-Making in Solid Tumors Treatment?
Biliary tract cancers (BTCs) are aggressive malignancies associated with poor prognosis and limited treatment options. Advances in precision oncology, notably the identification of recurrent molecular alterations such as fibroblast growth factor receptor 2 (FGFR2) fusions, isocitrate dehydrogenase 1 (IDH1) mutations, ERBB2 amplifications, and v-Raf murine sarcoma viral oncogene homolog B (BRAF) V600E mutations, have introduced new therapeutic avenues and modest survival benefits for patients with advanced disease. However, the practical implementation of targeted therapies remains hampered by challenges in tumor tissue acquisition and molecular testing, highlighting the need for alternative genomic profiling strategies. This comprehensive review examines the role of liquid biopsy as a non-invasive strategy for molecular profiling in BTCs, with a focus on the clinical applications of plasma and bile-derived circulating tumor DNA (ctDNA). We synthesized findings from recent clinical studies evaluating mutation detection rates, concordance between liquid biopsy and tissue-based assays, and the comparative performance of plasma versus bile ctDNA. Liquid biopsy demonstrates high rates of mutation detection and good concordance with tissue analyses. Bile-derived ctDNA, owing to its proximity to the tumor, consistently shows higher sensitivity and mutant allele frequencies (MAFs) than plasma ctDNA. Nevertheless, challenges remain, including lower sensitivity for detecting structural alterations (e.g., gene fusions), variability in ctDNA yield depending on disease status, and a lack of assay standardization across platforms. Liquid biopsy, particularly through bile ctDNA analysis, emerges as a promising adjunct to tissue biopsy for molecular profiling in BTCs. It offers opportunities for earlier, less invasive, and more personalized treatment decisions. Future directions should aim at developing tumor-informed liquid biopsy strategies that increase precision, reduce costs, and ultimately improve patient outcomes. Prospective studies are needed to confirm its clinical utility and survival impact.
Biliary tract cancers (BTCs) are aggressive malignancies with distinct epidemiological and molecular features. It includes intrahepatic cholangiocarcinoma (iCCA), perihilar cholangiocarcinoma (pCCA), distal cholangiocarcinoma (dCCA), and gallbladder cancer (GBC). iCCA originates above the second-order bile ducts, while pCCA and dCCA [collectively called extrahepatic cholangiocarcinoma (eCCA)] are anatomically divided at the cystic duct.
Globally, pCCA represents the most common subtype, followed by dCCA and iCCA [1]. CCA incidence and mortality vary widely across regions and between subtypes. iCCA rates are highest in Southeast Asia, particularly Thailand, where incidence reaches 85 per 100,000, significantly outpacing Western countries, which report rates below 3.5 per 100,000, making it a rare cancer in this population [2]. Mortality from iCCA has risen globally over the last decade, with sharp increases in Eastern Europe (e.g., Latvia and Lithuania with annual percentage changes exceeding 18%) and moderate rises in North America and Oceania. Conversely, eCCA mortality is generally lower, with only a few countries exceeding 1 per 100,000, such as Hungary and Germany. Trends in eCCA are more variable, with increases in some regions, such as Central Europe, and declines in others, including parts of North America and Australia. These variations reflect differences in risk factors, diagnostic practices, healthcare access, and disease classification [3]. Early-stage diagnosis is rare, with most patients presenting with advanced disease, resulting in a poor 5-year survival rate of 7–20% [1].
The ABC-02 trial [4] established gemcitabine-cisplatin as the first-line standard for unresectable BTCs. Recently, the therapeutic landscape has evolved with the addition of immune checkpoint inhibitors to traditional chemotherapy, offering a new standard of care in first-line treatment [5].
The TOPAZ-1 trial, a phase III study, tested the efficacy of the addition of durvalumab, a programmed cell death ligand 1 (PD-L1) inhibitor to standard chemotherapy in advanced BTCs [5]. Patients with newly diagnosed, inoperable, or metastatic BTCs were randomized to receive either durvalumab or placebo with gemcitabine and cisplatin for up to 8 cycles, followed by durvalumab or placebo maintenance therapy every 28 days until disease progression or withdrawal. Durvalumab significantly improved median overall survival (mOS) (12.9 vs. 11.5 months) and the two-year survival rate was 23.6% for durvalumab versus 11.5% for placebo. These findings represent the most significant advance in first-line BTCs treatment since the ABC-02 trial and led to the Food and Drug Administration (FDA) approval of the durvalumab, gemcitabine, and cisplatin combination in September 2022. The KEYNOTE-966 trial followed TOPAZ-1, evaluating pembrolizumab combined with gemcitabine and cisplatin in treatment-naive metastatic or inoperable BTCs patients [6]. Involving 1,069 patients, this trial showed a median OS of 12.7 months for pembrolizumab versus 10.8 months for placebo. Subgroup analysis revealed that iCCA patients benefited most, compared to eCCA or GBC.
Despite recent therapeutic advances, precision diagnostics remain underexploited in BTCs. This review highlights the emerging role of molecular tools, such as liquid biopsy, in refining patient management, addresses the current gap between this technological innovation and a still limited clinical application, and discusses the persistent technical limitations that must be understood to better inform clinical decision-making.
Recent advancements in genetic screening have revealed distinct molecular profiles across CCA subtypes.
iCCA is characterized by frequent mutations in isocitrate dehydrogenase 1 (IDH1) (≈ 15–25% [7, 8]) and fibroblast growth factor receptor 2 (FGFR2) fusions or rearrangements (≈ 10–45% [7, 8]), which are among the most well-characterized alterations and appear to be mutually exclusive [7]. Less common mutations in iCCA include neurotrophic tyrosine receptor kinase (NTRK) gene fusions (4% [9]) and BRAF V600E mutations (5% [8]).
pCCAs and dCCAs predominantly harbor ERBB2 amplifications, KRAS, TP53, and SMAD4 mutations, with KRAS mutations occurring more frequently than in iCCA [10]. GBC shows amplification of human epidermal growth factor receptor 2 (HER2) in approximately 10–20% of cases [11].
Although rare (< 2%), DNA mismatch repair deficiency (dMMR) can be observed across all BTCs subtypes [12]. In the TOPAZ-1 trial, dMMR was observed in only 1.5% of cases. Microsatellite instability (MSI) status can be assessed either by immunohistochemistry (IHC) targeting mismatch repair proteins (MLH1, MSH2, MSH6, and PMS2) or by DNA-based assays analyzing microsatellite sequences. The choice between technologies such as next-generation sequencing (NGS), RNA sequencing, or IHC depends on the target alteration and the type of material available, whether tumor tissue or circulating tumor DNA (ctDNA) [11]. Deficiency in any of the four major mismatch repair genes leads to high MSI, a hypermutator phenotype, and increased neoantigen production, making dMMR tumors strong candidates for immune checkpoint blockade [13] (Figure 1).
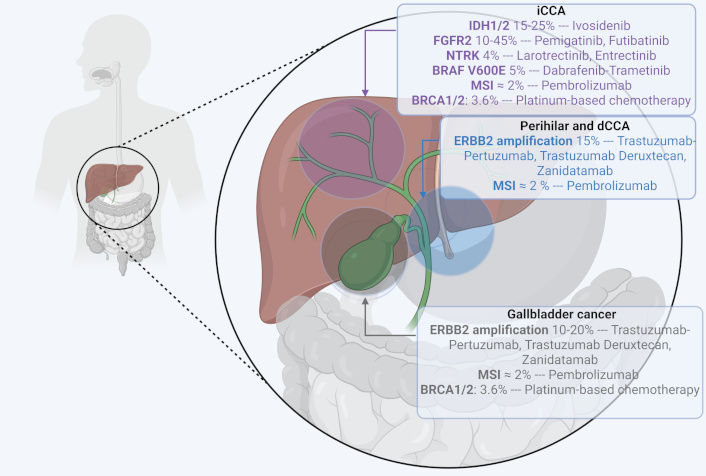
Molecular alterations in biliary tract cancers and matched targeted therapy. BRAF: v-Raf murine sarcoma viral oncogene homolog B; BRCA1/2: breast cancer 1/2; dCCA: distal cholangiocarcinoma; FGFR2: fibroblast growth factor receptor 2; iCCA: intrahepatic cholangiocarcinoma; IDH1/2: isocitrate dehydrogenase 1/2; MSI: microsatellite instability; NTRK: neurotrophic tyrosine receptor kinase. Created in BioRender. Mechahougui, H. (2025) https://BioRender.com/t3pmut8
FGFR alterations are critical drivers of oncogenesis in CCA and include mutations and rearrangements. FGFR2 functions as a receptor for FGFs and is part of the FGFR1–4 receptor tyrosine kinases family [14]. Under physiological conditions, the FGF/FGFR2 signaling pathway plays important roles in embryonic development, tissue repair, tumor angiogenesis, and proliferation [15] but FGFR2 fusions or rearrangements can lead to constitutive activation of the receptor, driving tumorigenesis and progression [16]. These alterations are present in approximately 10–15% of patients with iCCA but are almost absent in eCCA and GBC [7]. The identification of FGFR2 alterations has led to the development of FGFR inhibitors, which have shown clinical benefit in molecularly selected populations.
Pemigatinib, an oral selective and reversible inhibitor of FGFR1–3, demonstrated clinical activity in the phase II FIGHT-202 trial, achieving an objective response rate (ORR) of 35.5% and a mOS of 21.1 months [17]. Based on these results, the U.S. FDA and the European Medicines Agency (EMA) approved pemigatinib as a second-line therapy for advanced BTCs harboring FGFR2 fusions or rearrangements following progression on systemic treatment [17]. Infigratinib, another selective FGFR1–3 inhibitor, demonstrated an ORR of 23.1% in a phase II trial [18] involving patients with advanced CCA harboring FGFR2 fusions or rearrangements. The highest response rate (34%) was observed in those who had received only one prior line of therapy. Although approved by the FDA, the application for EMA approval was withdrawn for strategic economic reasons of the sponsor, limiting its availability in Europe [19]. Futibatinib, a highly selective and irreversible FGFR1–4 inhibitor, has demonstrated efficacy even in tumors resistant to other FGFR inhibitors, owing to its irreversible binding. In the phase II FOENIX-CCA1 trial [20], patients with FGFR2 fusions achieved an ORR of 42% and a mOS of 21.7 months. Futibatinib is now approved by both the FDA and EMA for advanced BTCs with FGFR2 fusions or rearrangements after progression on systemic therapy. Other FGFR inhibitors, such as lirafugratinib (RLY-4008) [21], derazantinib [22], and erdafitinib [23], have also shown promising early results.
FGFR inhibitors are now being evaluated in first-line settings for iCCA with FGFR2 fusions. The FIGHT-302 trial (NCT03656536 ) is comparing pemigatinib to gemcitabine-cisplatin in advanced iCCA, while the FOENIX-CCA3 trial (NCT04093362) is evaluating futibatinib in a similar patient population.
IDH1 encodes an enzyme, IDH1, that plays a critical role in cellular metabolism, DNA transcription, and repair by converting isocitrate to α-ketoglutarate. Mutations in the IDH1 gene lead to the production of the oncometabolite R-2-hydroxyglutarate (R-2HG), which disrupts epigenetic processes, causes DNA damage, and alters histone methylation, thereby driving tumorigenesis [24].
Ivosidenib, an oral inhibitor of IDH1, was tested in the phase III ClarIDHy trial [25], with an improved mOS (10.3 months with ivosidenib vs. 7.5 months with placebo). After adjusting for crossover, the control group’s mOS was estimated at 5.1 months. Based on these findings, the FDA approved ivosidenib in 2021 for pretreated advanced or metastatic BTCs with proven IDH1 mutations, and EMA approval followed in 2023. Other IDH inhibitors are under development with CCA cohorts, including olutasidenib [26] and LY3410738 [27]. Resistance mechanisms include secondary mutations, isoform switching, or persistently elevated R-2HG levels [28]. More recently, the tyrosine kinase inhibitor dasatinib is being explored in clinical trials like the phase II trial NCT02428855, that evaluates dasatinib in IDH-mutant iCCA.
The RAS-RAF-MEK-ERK pathway, frequently activated by KRAS mutations across all CCA subtypes, plays a central role in cell proliferation and survival and is linked to poor prognosis [29]. The BRAF V600E mutation, a downstream component, is found in 5% of CCA cases [8], predominantly in iCCA, and is associated with advanced disease stages, resistance to chemotherapy, and lower survival rates [30]. The V600E mutation leads to constitutive activation of kinase activity, escaping physiological control and promoting unchecked cell proliferation [31]. These mutations can be detected through NGS, polymerase chain reaction (PCR), or Sanger sequencing, which are superior to IHC analysis for therapy decisions [32].
Due to the rarity of this alteration, the combination of the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib was evaluated in the Rare Oncology Agnostic Research (ROAR) trial, a basket study investigating dabrafenib plus trametinib in BRAF V600E-mutated rare cancers [33]. Among 43 BTC patients, the ORR was 47% and the mOS was 14.0 months. Other molecules have been tested, like ulixertinib [34], an ERK 1/2 inhibitor, and selumetinib a MEK inhibitor [35]. Ongoing trials are exploring novel combinations, like PD-L1 inhibitor atezolizumab with the MEK inhibitor cobimetinib (NCT03201458), and the BRAF inhibitor ABM-1310 (NCT05501912 and NCT04190628).
HER2, encoded by the ERBB2 gene, is a receptor tyrosine kinase that plays a critical role in tumorigenesis by activating downstream signaling pathways like RAS/MAPK, PI3K/Akt, and JAK/STAT, which contribute to its role in oncogenesis [36]. In eCCA and GBC, ERBB2 amplification occurs in 10-20% [11].
The phase IIa MyPathway trial [37], a basket study, assessed the combination of pertuzumab and trastuzumab in 39 patients with HER2-positive metastatic BTCs. The ORR was 23%, and the mOS was 10.9 months. Although regulatory approval for BTCs is pending, these results support the potential utility of HER2-targeting monoclonal antibodies. Trastuzumab deruxtecan, an antibody-drug-conjugate (ADC) combining trastuzumab with the topoisomerase I inhibitor deruxtecan, has shown significant efficacy in HER2-positive BTCs in the HERB trial [38]. Patients with HER2-positive BTC (IHC3+ or IHC2+/ISH+) achieved an ORR of 36.4% and a mOS of 7.1 months. Patients with HER2-low BTCs (IHC/ISH < 2+) had lower response rates but still benefited from treatment. The DESTINY-PanTumor02 trial [39] further validated trastuzumab deruxtecan’s efficacy, with an ORR of 56.3% and mOS of 12.4 months in HER2 IHC3+ BTCs. Zanidatamab, a bispecific antibody targeting two HER2 epitopes, has demonstrated rapid and durable responses in HER2-positive BTCs. In the HERIZON-BTC-01 trial [40], patients with ERBB2-amplified BTCs achieved an ORR of 41% and a median duration of response of 12.9 months.
Other agents under investigation include TAS0728 (an oral HER2 inhibitor) [41], RC48-ADC [42], and HER2-targeted bispecific antibodies in combination with chemotherapy [43]. While no HER2-targeted therapy is yet FDA or EMA-approved for BTCs, guidelines recommend their use in HER2-expressing cases [11].
The KEYNOTE-158 trial [44] evaluated pembrolizumab in MSI-H/dMMR tumors across 27 cancer types, including 22 BTC patients. In this cohort, pembrolizumab achieved a median progression-free survival (mPFS) of 4.2 months, a mOS of 24.3 months, and an ORR of 40.9%. Based on these results, pembrolizumab monotherapy is now recommended in European guidelines and approved by the EMA for advanced BTCs with dMMR or MSI-H after prior systemic therapy [45]. The FDA has also approved pembrolizumab for all MSI-H/dMMR tumors regardless of the cancer type. Ongoing trials, such as the MOST-CIRCUIT trial (NCT04969887), are investigating combination regimens of nivolumab with ipilimumab for MSI-H/dMMR BTCs.
Neurotrophin receptors (TRK A, B, and C) are crucial for cell proliferation and neuronal development [46] through the activation of signaling pathways like PI3K and MAPK [47]. However, NTRK alterations, particularly gene fusions, can drive tumorigenesis by causing ligand-independent activation of these pathways [48]. NTRK fusions are rare in BTCs, occurring in approximately 4% of cases [9].
Larotrectinib, a first-generation pan-NTRK inhibitor has demonstrated robust efficacy in solid tumors with NTRK fusions, including BTCs, in a pooled analysis of 3 clinical trials [49]. In an integrated analysis of three phase I/II trials (ALKA-372-001 [EudraCT 2012-000148-88], STARTRK-1 [NCT02097810], and STARTRK-2 [NCT02568267]), entrectinib, a potent CNS-active TRK inhibitor, showed also durable systemic and intracranial responses in patients with NTRK-fusion-positive solid tumors. Entrectinib received FDA approval for use in NTRK fusion-positive solid tumors, including CCA, following progression on prior systemic therapy [50]. Both larotrectinib and entrectinib are approved in the United States and Europe for treating unresectable or metastatic solid tumors with NTRK gene fusions.
BRCA mutations are observed in 3.6% of BTCs, with a higher prevalence of BRCA2 over BRCA1 mutations in iCCA and GBC [51]. These mutations are frequently associated with alterations in TP53, ARID1A, and KRAS, among other genes, and are linked to higher rates of MSI and elevated tumor mutational burden, indicating a more immunogenic tumor profile. BRCA mutations are associated with improved PFS in patients receiving platinum-based chemotherapy. Furthermore, BRCA-mutant tumors exhibit unique genetic and immunogenic characteristics, supporting the rationale for exploring PARP inhibitors in combination with immunotherapy and targeted therapies in this subgroup.
Precise tissue sampling and molecular profiling are critical for the diagnosis and management of CCA. In patients ineligible for curative-intent surgery, core biopsy is recommended to obtain material for histopathological and molecular analyses. However, diagnosis remains challenging due to poor tumor accessibility, particularly in the perihilar region, and biliary cytology achieves a sensitivity of only 20–40% [52]. Differentiating malignant from benign lesions is especially difficult in conditions such as primary sclerosing cholangitis or IgG4-related disease, where inflammatory changes can mimic neoplasia [53]. Misclassification exposes patients to unnecessary major surgeries, with significant associated morbidity and mortality. These limitations have underscored the urgent need for noninvasive diagnostic alternatives. Liquid biopsy, in particular, has gained interest as a complementary tool, especially after procedural restrictions during the COVID-19 pandemic. With tissue biopsy failure rates reaching up to 27% in CCA [54], liquid biopsy now offers a valuable approach for detecting actionable molecular alterations in advanced BTCs.
Cell-free DNA (cfDNA) consists of small DNA fragments, typically 40 to 200 base pairs in length [55] released into the circulation through cellular apoptosis or necrosis. A fraction of cfDNA derived from tumor cells, known as ctDNA, harbors cancer-specific genetic and epigenetic alterations. Detection of ctDNA can be achieved through techniques such as PCR or NGS [56]. While PCR remains cost-effective and suitable for targeted mutation analysis, NGS offers a comprehensive assessment of genomic alterations, an advantage when addressing the genetic heterogeneity characteristic of tumors (Table 1).
Pros and cons of liquid biopsy in biliary tract cancers
| Pros | Cons |
|---|---|
| Minimally invasive, lower risk of complicationsRequires only a blood sample, reducing the risk associated with invasive procedures, and the delay. | Dependency on DNA shedding and tumor burdenLimited efficacy in localized disease. |
| Real-time monitoring of secondary mutationsAllows for frequent testing to monitor treatment response and disease progression. | Technical challengesRequires highly sensitive and specific assays, which are still under development and standardization. |
| Captures tumor heterogeneityCan detect multiple genetic alterations from different tumor sites, providing a comprehensive genetic profile. | Limited comprehensive dataDoes not provide histological information, which is essential for certain diagnostic and treatment decisions. |
| Faster turnaround timeResults can often be obtained more quickly than traditional biopsies, facilitating timely clinical decisions. | Higher costs and limited availabilityAdvanced technologies required may be expensive and not widely accessible in all healthcare settings. |
Studies in other tumor types, such as non-small cell lung cancer [57], have demonstrated the potential of liquid biopsy in localized settings to assess recurrence risk, refine prognostication, and guide adjuvant chemotherapy decisions. However, in resected CCA, evidence supporting the utility of ctDNA remains limited. A sub-analysis of the phase II STAMP trial [58] evaluated the feasibility of ctDNA to predict recurrence risk during adjuvant therapy in CCA. In this study, ctDNA was analyzed at three time points, before initiation of cisplatin-gemcitabine adjuvant chemotherapy, after five cycles, and after eight cycles, using a tumor-informed assay (Signatera). No significant differences in recurrence-free survival (RFS) or OS were observed based on ctDNA status at these time points. Although patients with detectable ctDNA prior to adjuvant chemotherapy showed a trend toward shorter RFS compared to ctDNA-negative patients, the association did not reach statistical significance. Importantly, patients with persistently positive ctDNA during adjuvant treatment uniformly experienced clinical recurrence, with significantly shorter RFS.
In the curative setting, whether early intervention based on detectable ctDNA, rather than waiting for radiographic recurrence to initiate systemic therapy, can improve outcomes remains unclear. When ctDNA is strongly prognostic for eventual radiographic recurrence but no validated early intervention strategies are available, its detection may not alter management and could instead increase patient anxiety. Isolated case reports [59, 60] have described instances where adjuvant therapy escalation guided by positive ctDNA findings appeared beneficial. Nonetheless, prospective studies specifically designed to determine whether ctDNA-guided interventions translate into meaningful clinical benefits are critically needed.
Several retrospective studies have evaluated the utility of ctDNA for initial molecular profiling in advanced BTCs. Mody et al. [61] analyzed 124 patients using a 73-gene ctDNA panel, identifying actionable alterations in 55%, including FGFR2 fusions, IDH1/2 mutations, HER2 amplifications, and BRAF mutations. Similarly, Ettrich et al. [62] reported a tissue-blood concordance rate of 74% in therapy-naive CCA patients, rising to 92% in iCCA. Lamarca et al. [63] demonstrated complete concordance between tissue and plasma ctDNA in 112 paired samples from 104 patients, even among those receiving active therapy. Specific targets such as IDH1 mutations appear particularly well detected by ctDNA, as shown by Aguado et al. [64], who reported a 92% concordance with tissue and observed that clearance of IDH1 mutations correlated with prolonged PFS in patients treated with ivosidenib. Similarly, Chen et al. [65] detected genetic alterations in 94.8% of ctDNA samples from 154 Chinese patients, with frequencies of IDH1 mutations and FGFR2 fusions comparable to tissue results (7.4% vs. 6% and 4.8% vs. 2.7%, respectively). Real-world data support the feasibility of early ctDNA testing. In a 2024 analysis of 1,726 advanced CCA patients [66], actionable alterations were detected in 18% of cases, mainly IDH1 mutations (11%) and FGFR2 fusions (9%), with a significant proportion tested before first-line therapy. However, despite these promising findings, sensitivity for detecting structural variants remains suboptimal. Hwang et al. [67] observed an 84.8% sensitivity for ctDNA genomic profiling overall, but only 40% sensitivity for HER2 amplifications and acknowledged persistent challenges in fusion detection, particularly when ctDNA levels were low.
However, important technical challenges persist, particularly in detecting structural alterations such as FGFR2 fusions. Berchuck et al. [68], in a large retrospective study of 2,068 ctDNA samples, identified molecular alterations in 84% of patients, with 44% carrying actionable targets. While high concordance rates were reported for IDH1 (87%) and BRAF V600E (100%) mutations, concordance for FGFR2 fusions was markedly low at 18%. This contrasts sharply with earlier reports from Ettrich et al. [62] and Lamarca et al. [63], suggesting variability in detection likely reflects both biological and technical factors. The Guardant360 assay, which relies on DNA hybrid capture, showed limited sensitivity, particularly for non-BICC1 fusion partners, due to narrow probe coverage and the challenge of detecting diverse rearrangements in cfDNA [69]. In contrast, assays like Illumina’s TruSight Oncology 500, which specifically targets known FGFR2 intronic breakpoints, have demonstrated significantly higher detection rates [70]. While cfDNA is a valuable tool for identifying truncal mutations and resistance mechanisms, tissue-based profiling remains essential when fusions are suspected. Advances such as anchored multiplex PCR, broader probe designs [68], and RNA-based methods may improve fusion detection in future practice [71].
In addition to these technical considerations, tumor biology and sampling context can also influence ctDNA accuracy. Okamura et al. [72] found higher concordance between ctDNA and metastatic lesions compared to primary tumors, suggesting that metastatic burden and anatomical site affect detectability.
Beyond detection, prognostic applications of ctDNA have been explored. Yang et al. [73] showed that blood-based copy number variation (CNV) analysis could stratify patients’ immunotherapy responses, with lower CNV risk scores correlating with improved disease control. Likewise, Berchuck et al. [68], showed that higher baseline ctDNA levels were associated with shorter OS, supporting ctDNA as a potential dynamic biomarker of disease burden and outcome (Table 2).
Selected trials evaluating liquid biopsy in blood in BTCs
| Year | Authors | Trial type | Population | Assay | Concordance rate liquid/tissue | Notable results |
|---|---|---|---|---|---|---|
| Localized BTCs | ||||||
| 2023 | Yoo et al. [58] | Randomized phase II |
| Signatera, tumor-informed assay | No comparison to tissue | Patients with positive ctDNA before adjuvant chemotherapy had shorter RFS than those with negative ctDNA |
| Metastatic BTCs | ||||||
| 2019 | Mody et al. [61] | Retrospective study |
| Guardant® | No comparison to tissue | Blood-based liquid biopsy can be used for molecular characterization and can identify clinically relevant alterations including 5% IDH1 and 7% FGFR2 mutations |
| 2019 | Ettrich et al. [62] | Retrospective study |
| QIAamp Circulating Nucleic Acid Kit for ctDNA extraction NGS of 15 gene panel, selected frequently mutated genes | No comparison to tissue |
|
| 2020 | Lamarca et al. [63] | Post hoc analysis of patient data collected as part of the prospective ABC-01, -02, and -03 |
| FoundationOne Liquid® Oncomine |
|
|
| 2020 | Aguado et al. [64] | ctDNA analysis of the randomized phase III trial ClarIDHy |
| ctDNA/digital PCR | IDH1: 92% concordance between plasma ctDNA and tissue samples |
|
| 2021 | Chen et al. [65] | Retrospective study |
| QIAamp Circulating Nucleic Acid Kit for cfDNA extraction |
|
|
| 2021 | Okamura et al. [72] | Observational genomic profiling study conducted under the UCSD-PREDICT prospective protocol (NCT02478931) |
| Guardant® |
|
|
| 2021 | Yang et al. [73] | Multicohort observational analysis |
| MagMAX cfDNA isolation Kit; TIANamp genomic DNA Kit | No comparison to tissue | CNV detection by liquid biopsy can predict response to immunotherapy
|
| 2022 | Berchuck et al. [68] | Retrospective, multi-institutional study |
| Guardant® |
| Targetable alterations detected in 44% of patients |
| 2025 | Hwang et al. [67] | Retrospective single-center study |
| Oncomine Comprehensive Assay and AlphaLiquid®100 panels |
|
|
| Evaluation of resistance mechanisms during treatment | ||||||
| 2017 | Goyal et al. [74] | Prospective translational analysis within the context of the BGJ398 phase II trial |
| Guardant® | No comparison to tissue |
|
BRAF: v-Raf murine sarcoma viral oncogene homolog B; BRCA1/2: breast cancer 1/2; BTCs: biliary tract cancers; cfDNA: cell-free DNA; CNV: copy number variation; ctDNA: circulating tumor DNA; eCCA: extrahepatic cholangiocarcinoma; FGFR2: fibroblast growth factor receptor 2; HR: hazard ratio; iCCA: intrahepatic cholangiocarcinoma; ICI: immune checkpoint inhibitor; IDH1: isocitrate dehydrogenase 1; MSI: microsatellite instability; NGS: next-generation sequencing; OS: overall survival; PCR: polymerase chain reaction; PD: progressive disease; PD-1: programmed cell death 1; PFS: progression-free survival; PPV: positive predictive value; PR: partial response; RFS: recurrence-free survival; SD: stable disease
Early in its development, liquid biopsy was already being explored as a tool for monitoring secondary resistance mutations during targeted therapy. In a phase II trial of FGFR2-targeted therapy with BGJ398, Goyal et al. [74] demonstrated the ability of cfDNA to detect acquired resistance alterations, including FGFR2 V564F and other kinase domain mutations. Similarly, Berchuck et al. [68] identified 31 FGFR mutations in plasma ctDNA that were undetectable in the corresponding tumor tissue, including several novel variants of uncertain clinical significance (Table 2).
Follow-up data highlighted the effectiveness of TAS-120, an irreversible pan-FGFR inhibitor, in 4 patients with FGFR2 fusion-positive CCA who had developed resistance to prior FGFR inhibitors. These patients were selected for TAS-120 treatment based on serial biopsies, ctDNA analysis, and patient-derived tumor cell evaluation [75].
Recently, Goyal et al. [76] gave a new insight on how resistance to FGFR inhibitors emerges in FGFR2-altered CCA. Their study, which combined genomic analyses with in vitro and in vivo models, showed that more than 60% of patients who initially respond to treatment later develop secondary FGFR2 mutations. These include both “gatekeeper” mutations like V565F, which confer high-level resistance through marked impairment of drug binding, and “molecular brake” mutations affecting N550, which are more frequent in clinical samples. Notably, the latter do not substantially reduce inhibitor potency in biochemical or cellular assays, nor do they prevent drug binding. Structural studies suggest that N550 variants induce subtle conformational shifts that allow partial kinase reactivation while preserving some degree of drug interaction, an effect likely amplified in vivo, where FGFR inhibitor concentrations are limited by on-target toxicities and narrow therapeutic windows. These findings support a broader interpretation that resistance in FGFR2-altered tumors is shaped less by absolute half-maximal inhibitory concentration (IC50) shifts and more by a dynamic balance between residual signaling activity and pharmacokinetic constraints. Variants like N550K, though only modestly resistant in vitro, may be selectively favored in patients precisely because they retain this balance. This explains the emergence of diverse and often polyclonal resistance patterns, particularly under therapeutic pressure where drug exposure is suboptimal. On this basis, the authors provide a biological rationale for the use of tinengotinib, a multikinase inhibitor with broader target specificity, currently being evaluated in the randomized phase III trial FIRST-308 (NCT05948475).
Bile has emerged as a valuable alternative source of ctDNA in BTCs, particularly when tissue sampling is challenging due to biliary obstruction. Given its anatomical proximity to tumor tissue, bile-derived ctDNA offers a promising platform for somatic mutation detection, often surpassing plasma ctDNA in terms of sensitivity and accuracy [77]. For example, Shen et al. [78] investigated bile cfDNA in 6 patients with CCA and 4 with GBC, comparing findings to tumor DNA using a 150-gene panel. Bile cfDNA fragments were found to be longer, closely mirroring the fragment size of tumor DNA, and the assay demonstrated high sensitivity (94.7%) and specificity (99.9%) for detecting single nucleotide variants and indels [78].
Li et al. [79] further emphasized the advantages of bile-derived ctDNA, demonstrating consistently higher concentrations of cfDNA and a greater number of detectable genomic alterations in both bile supernatant and pellet compared to plasma. Mutant allele frequencie (MAF) was also significantly higher in bile samples, with bile-tumor tissue concordance ranging from 85% to 90%. These findings support bile ctDNA as a more reliable representation of tumor-derived genetic material, particularly in tumors with a high mutational burden.
Consistent evidence was provided by Han et al. [80], who reported an 80% concordance between bile ctDNA and tumor biopsy samples in a cohort of 42 BTC patients. Notably, bile ctDNA demonstrated superior sensitivity in detecting mutations in key oncogenes such as TP53 and KRAS, further highlighting its potential for improving molecular diagnostics in BTCs.
Expanding on these observations, Arechederra et al. [81] applied a bile-based NGS panel in 68 patients, achieving a sensitivity of 96.4% and a specificity of 69.2% for malignancy detection. These results collectively underscore the growing role of bile ctDNA as a powerful alternative for genomic profiling, particularly in settings where tissue sampling is limited or inconclusive.
Despite its advantages, logistical challenges, such as the need for bile sampling via endoscopic or surgical procedures, can limit its routine use. However, in cases of biliary obstruction, either at diagnosis or during local recurrence, bile ctDNA presents an attractive option when an endoscopic intervention is required. This approach complements plasma-based liquid biopsy, offering another tool for mutation detection and monitoring in BTCs (Table 3).
Selected trials evaluating liquid biopsy in bile in BTCs
| Bile cfDNA | ||||||
|---|---|---|---|---|---|---|
| Year | Authors | Trial type | Population | Assay | Concordance rate bile/tissue | Notable results |
| 2019 | Shen et al. [78] | Retrospective, single-center observational study |
| Customized panel of 150 tumor-related genes | Overall mutation concordance:
High mutational concordance:
| Bile-based liquid biopsy features high concordance with blood samples and tumor tissue:
|
| 2022 | Li et al. [79] | Retrospective single-center study |
| Customized xGen lockdown probe panel with 425 predefined cancer-related genes | Bile vs. plasma vs. tissue:
| Genomic profiling of bile (supernatant/pellet) showed significantly higher concordance with tumor tissue alterations than plasma |
| 2022 | Arechederra et al. [81] | Prospective cohort |
| Bilemut NGS assay |
| Superior performance demonstrated in 30 paired bile and tissue samples:
|
AJCC: American Joint Committee on Cancer; BTCs: biliary tract cancers; CCA: cholangiocarcinoma; cfDNA: cell-free DNA; CNV: copy number variation; MAF: mutant allele frequency; NGS: next-generation sequencing; SNV: single nucleotide variation
Targeted therapies have redefined the treatment landscape of BTCs, and as they expand into the first-line setting, ctDNA is emerging as a central tool in guiding therapeutic decisions. In real-world practice, early ctDNA testing can identify actionable alterations such as IDH1 mutations or FGFR2 fusions, facilitating timely initiation of matched treatments. This non-invasive approach is particularly valuable when tissue access is limited or biopsy material is insufficient, a common challenge in BTCs.
Beyond baseline profiling, serial ctDNA analysis offers a means to monitor tumor evolution and detect emerging resistance mechanisms. In other malignancies, such as EGFR T790M in non-small cell lung cancer or c-KIT secondary mutations in gastrointestinal stromal tumors, this has already translated into routine clinical practice. In BTCs, acquired mutations affecting the FGFR2 kinase domain have been identified through cfDNA analysis and are increasingly recognized as markers of therapeutic resistance. While the adaptation of treatment based on such resistance mutations is not yet standard care in BTCs, some next-generation FGFR inhibitors designed to overcome these alterations are currently under investigation [82]. Their clinical integration may soon enable a more dynamic, mutation-guided sequencing of therapies.
Nevertheless, analytical and biological limitations persist. Sensitivity for detecting amplifications and structural variants varies considerably across platforms; PCR-based assays may better detect predefined alterations, while NGS panels offer broader coverage but may struggle with complex rearrangements such as FGFR2 fusions, especially when involving rare partners [68]. RNA-based liquid biopsy approaches and anchored multiplex PCR are promising developments that may improve fusion detection. Moreover, ctDNA yield and interpretability depend on tumor biology, with metastatic lesions generally shedding more detectable DNA than primary or low-volume disease. This variability must be accounted for in clinical interpretation.
Looking ahead, ctDNA analysis may extend its utility beyond advanced disease. Several prospective trials, such as NCT05743959, NCT06171321, NCT04183712, and NCT06416397, are evaluating its role in detecting minimal residual disease (MRD) and anticipating recurrence in the adjuvant and surveillance settings. These applications are aligned with approaches being developed in colorectal and lung cancers, where MRD-guided interventions are under clinical validation.
To fully realize its potential in BTCs, ctDNA testing must be integrated into a framework that is both biologically informed and clinically actionable. The development of tumor-informed, stage-adapted, and alteration-specific strategies, coupled with prospective validation and standardized platforms, will be essential to move from technical feasibility to routine clinical impact.
ADC: antibody-drug-conjugate
BRAF: v-Raf murine sarcoma viral oncogene homolog B
BTCs: biliary tract cancers
CCA: cholangiocarcinoma
cfDNA: cell-free DNA
CNV: copy number variation
ctDNA: circulating tumor DNA
dCCA: distal cholangiocarcinoma
dMMR: DNA mismatch repair deficiency
eCCA: extrahepatic cholangiocarcinoma
EMA: European Medicines Agency
FDA: Food and Drug Administration
FGFR2: fibroblast growth factor receptor 2
FGFs: fibroblast growth factors
GBC: gallbladder cancer
HER2: human epidermal growth factor receptor 2
iCCA: intrahepatic cholangiocarcinoma
IDH1: isocitrate dehydrogenase 1
IHC: immunohistochemistry
mOS: median overall survival
MRD: minimal residual disease
MSI: microsatellite instability
NGS: next-generation sequencing
NTRK: neurotrophic tyrosine receptor kinase
ORR: objective response rate
OS: overall survival
pCCA: perihilar cholangiocarcinoma
PCR: polymerase chain reaction
PD-L1: programmed cell death ligand 1
PFS: progression-free survival
R-2HG: R-2-hydroxyglutarate
RFS: recurrence-free survival
JG: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. HM: Conceptualization, Investigation, Writing—original draft, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Jeanny B. Aragon-Ching
Zhuldyz Myrkhiyeva ... Aliya Bekmurzayeva
Ekaterina S. Kuligina ... Evgeny N. Imyanitov
