Review
Review

Affiliation:
Department of Neurosurgery, College of Medicine, University of Florida, Gainesville, Florida 32610, USA
Email: squintin@ufl.edu
ORCID: https://orcid.org/0000-0002-5880-8470
Affiliation:
Department of Neurosurgery, College of Medicine, University of Florida, Gainesville, Florida 32610, USA
Affiliation:
Department of Neurosurgery, College of Medicine, University of Florida, Gainesville, Florida 32610, USA
Affiliation:
Department of Neurosurgery, College of Medicine, University of Florida, Gainesville, Florida 32610, USA
Affiliation:
Department of Neurosurgery, College of Medicine, University of Florida, Gainesville, Florida 32610, USA
Email: Brandon.Lucke-Wold@neurosurgery.ufl.edu
ORCID: https://orcid.org/0000-0001-6577-4080
Explor Neuroprot Ther. 2022;2:118–130 DOI: https://doi.org/10.37349/ent.2022.00023
Received: March 29, 2022 Accepted: May 09, 2022 Published: June 21, 2022
Academic Editor: Rafael Franco, Universidad de Barcelona, Spain
The article belongs to the special issue Emerging Concepts in Subarachnoid Hemorrhage
The glymphatic system, or glial-lymphatic system, is a waste clearance system composed of perivascular channels formed by astrocytes that mediate the clearance of proteins and metabolites from the brain. These channels facilitate the movement of cerebrospinal fluid throughout brain parenchyma and are critical for homeostasis. Disruption of the glymphatic system leads to an accumulation of these waste products as well as increased interstitial fluid in the brain. These phenomena are also seen during and after subarachnoid hemorrhages (SAH), contributing to the brain damage seen after rupture of a major blood vessel. Herein this review provides an overview of the glymphatic system, its disruption during SAH, and its function in recovery following SAH. The review also outlines drugs which target the glymphatic system and may have therapeutic applications following SAH.
The central nervous system (CNS) is unique in that it is the only organ system which lacks well defined lymphatic vessels to aid in the elimination of interstitial metabolic waste products [1]. Approximately 60–68% of total water content in the CNS lies within intracellular spaces, while the remaining water content occupies extracellular spaces [1]. Extracellular fluid can further be divided into interstitial fluid (ISF), cerebrospinal fluid (CSF), and blood compartments, each roughly comprising 10% of the intracranial water volume [2]. In other organs, products of cellular metabolism released into the ISF are cleared into the venous blood through systems of lymphatic vessels [3]. Venous and lymphatic outflow in the CNS is not as well defined as in peripheral systems. However, the approximate 500 mL of CSF secreted by the choroid plexus has been shown to find reentry through arachnoid granulations (AG) and through parenchymal glymphatic channels [4, 5]. AG are protrusions of the arachnoid mater into the dural venous sinus that facilitate CSF reabsorption via vesicular transport [6]. Subsets of AG have also been implicated in lymphatic outflow of CSF via dural intravascular channels [7]. While these findings imply an important role for AG mediated CSF turn-over, studies also have found that patients with AG agenesis have seemingly normal CSF flow, suggesting bulk flow may occur through perivascular routes via the glymphatic system (GS) [8]. In mediating this flow, the GS allows for distribution of CSF in the parenchyma and clearance of soluble proteins and metabolites from the CNS [2]. In addition to waste elimination, it has also been shown that the GS is responsible for regulating brain-wide distribution of other compounds including amino acids, lipids, glucose, and neuromodulators [2].
Previous studies have attempted to map brain ISF drainage from the CNS by injecting and monitoring the temporal distribution of traceable solutes in the brain parenchyma [9]. The ISF efflux has been monitored in rodent models via striatal horse radish peroxidase (HRP) injections, showing considerable amounts of HRP in the perivascular space (PVS) of rodents sacrificed at later time points [10]. This experiment demonstrated that the perivascular drainage of ISF was, in part, directed to the subarachnoid CSF. Rennels et al. [11] concluded that CSF could penetrate the brain parenchyma using the same perivascular conduits that ISF employs for drainage. Additionally, studies have demonstrated that CSF and ISF continuously interchange. This exchange of fluids is facilitated by the convective influx of CSF along the periarterial space [12]. From the subarachnoid space, CSF is propagated into the PVSs through a combination of factors including arterial pulsatility, CSF pressure gradients, and respiration. Then the transport of CSF into the brain parenchyma is facilitated by aquaporin-4 (AQP4) water channels [12, 13]. CSF movement into the parenchyma pushes convective ISF within the tissue towards the perivenous spaces surrounding the large deep veins [2]. The ISF is then collected in the perivenous space from where it is then drained out of the CNS and into the cervical lymphatic system [14, 15].
The glymphatic pathway functions by redirecting the flow of CSF into the brain along arterial perivascular structures into the brain interstitium [16] (Figure 1). CSF from the subarachnoid space flows into the brain via PVSs of the leptomeningeal arteries [17, 18]. The endfeet of astroglial cells densely express AQP4 water channels which assist the flow of CSF into the brain parenchyma [16]. Within the brain interstitium, the CSF is dispersed by a net fluid movement directed towards the venous perivascular and perineuronal spaces [7]. Ultimately, CSF exits along the meningeal lymphatic vessels and AG [9, 12, 19]. This pathway continues to direct the flow towards venous perivascular and perineuronal spaces, clearing solutes from the neuropil into meningeal and cervical lymphatic drainage vessels [11].
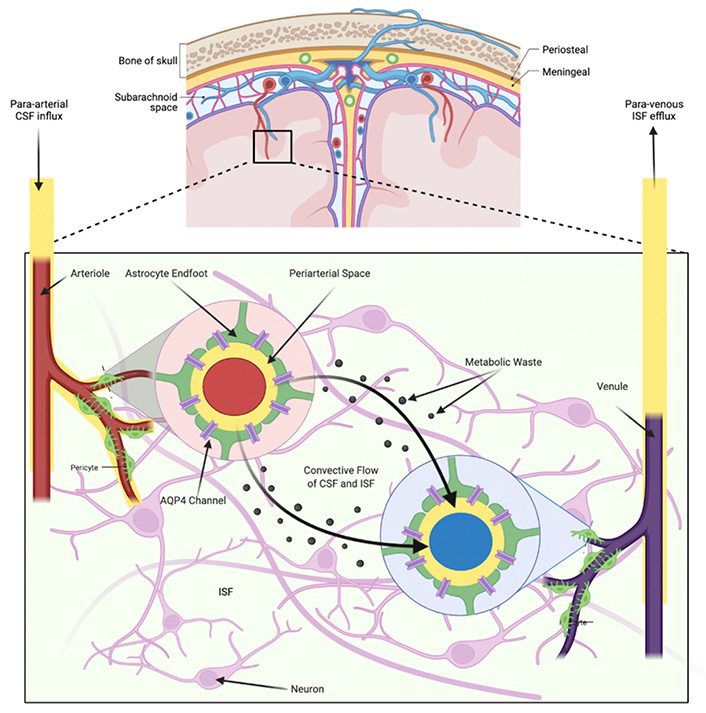
Overview of the circulation of CSF and ISF through the glymphatic pathway in brain parenchyma
The flow of CSF into the brain via PVSs of major cerebral arteries drives interstitial metabolites towards venous PVSs. The polarized nature of AQP4 water channels that are expressed on the astrocytic end feet bounded to both arteries and veins allow for CSF movement into the interstitium from periarterial spaces, and from the interstitium into perivenous spaces. This flow of CSF removes metabolites from the brain parenchyma that are generated during neural activity.
The dynamics of the GS continue to be elucidated as it is studied in the context of CNS and neurovascular diseases. Specifically, maintenance of glymphatic flow is shown to be important in recovery from subarachnoid hemorrhage (SAH). Liu et al. [20] showed that AQP4 plays a key role in the ISF drainage in a rat SAH model. In their experiment, SAH outcomes on AQP4 knockout rats were compared to controls who underwent sham surgery (a surgical procedure similar to the one used to induce SAH). AQP4 deficient rats displayed no improvement in neurological function or neuroinflammation seven days post SAH in addition to decreased CSF flow through the cerebral interstitium. These findings emphasized the important role that the GS plays in the clearance of excess fluid and harmful metabolites, especially following brain injury such as SAH.
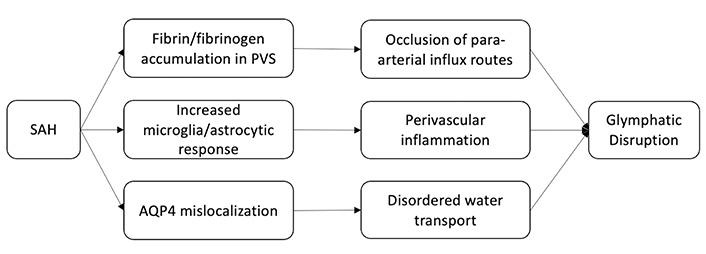
Although important for neurologic recovery following brain injury, the GS itself has been shown to be disrupted by SAH. Gaberel et al. [21] showed this while evaluating glymphatic function in mice following various stroke subtypes. Using contrast-enhanced magnetic resonance imaging (MRI), they found evidence of severe glymphatic impairment after SAH and in the acute phase of ischemic stroke, but not following carotid ligature or intracerebral hemorrhage. More specifically, in the SAH model, they observed occlusion of para-arterial influx routes that persisted after bilateral decompressive craniectomy, as well as the accumulation of fibrin/fibrinogen in PVSs causing diminished waste clearance. Luo et al. [22] observed a similar phenomenon coinciding with increased inflammatory markers and perivascular fibrinogen deposition one hour after SAH and reduced microcirculation within six hours. Immunohistochemistry (IHC) of the perivascular tissue showed increased microglia and astrocyte activation with persistent elevation in inflammatory markers 7 days post SAH. Fibrinogen deposition and subsequent microvascular occlusion were ameliorated by tissue plasminogen activator improving recovery in wild type (WT) mice but not in AQP4 deficient mice.
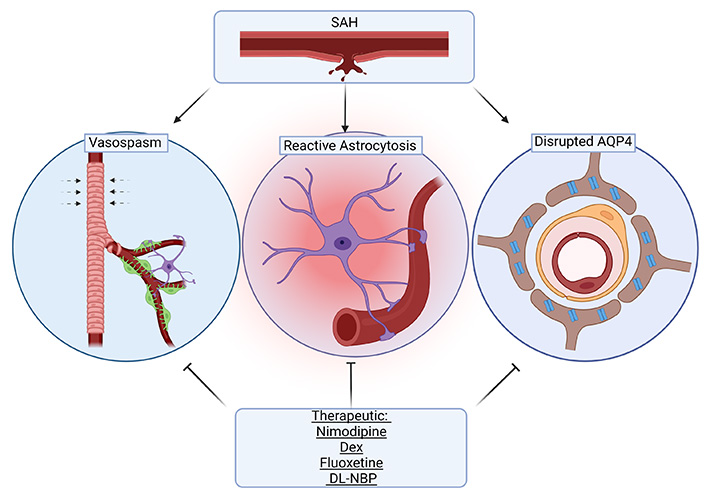
Following SAH, blood components can enter the PVS and result in coagulation and macrophage chemotaxis, both of which impair liquid exchange [23]. The entrance of peripheral immune cells into the CNS along with other blood components has been shown to be a driver of inflammation following SAH [24, 25, 26]. Elevated CSF proinflammatory monocytes and T-cells are found early on in SAH patients in association with elevated proinflammatory cytokines [27]. While peripheral immune cell penetration into the CNS may also occur in less severe contexts, in the context of SAH disruption of glymphatic flow is thought to lead to the accumulation of both cells and cytokines, potentiating further disruption [28]. Early on, astrocytes undergo a phenotypic shift in response to inflammatory insult that is commonly referred to as reactive astrogliosis [29]. These astrocytes perpetuate the inflammatory cascade via cytokine production [30]. Additionally, the expression of AQP4 in astrocytes is up-regulated and polarization is disrupted. As previously alluded to, the abundance and distribution of AQP regulates the efficiency and direction of water transport in the CNS. AQP subtypes are differentially expressed throughout the CNS, and this polarity allows for the directional transport of water [31]. In healthy tissue, AQP4 is localized at the perivascular endfeet and terminal pads of astrocyte processes [32, 33]. Following brain damage, such as SAH or cerebral ischemia, there is a distinct redistribution of AQP4 away from the endfeet, that has been tied to a transcriptional change in astrocyte expression of AQP4 isoforms (ratio of M23 and M1 isoforms) [34, 35]. Although ISF transport increases due to overexpression of AQP4, disordered transport due to mislocalization cannot compensate for the edema caused by the SAH [36, 37] (Figure 2).
Interestingly, cerebral vasospasm following SAH is thought to be associated with glymphatic disruption. Cerebrovascular vasospasms are one of the key factors leading to poor prognosis and high mortality of SAH patients. Ecker and Riemenschneider [38] were the first to document vasospasms in SAH on angiography. Since this report, many studies have investigated therapeutics which may reverse vasospasms to prevent further neurological decline following SAH [39, 40]. Although vasospasms are a contributor to cerebral ischemia, there is limited evidence showing a strict causal relationship between the two. As previously discussed, SAH results in coagulation that impairs fluid exchange [23]. Recombinant tissue plasminogen activator (rt-PA) is used to clear coagulation in the PVS, which improves neurological function and reduces inflammation. rt-PA intrathecal injection after SAH can reduce the incidence of cerebral vasospasm and improve outcomes [41]. rt-PA treatment in rodent models of SAH has also been shown to enhance clearance of blood components including peripheral immune cells after SAH, also reducing vasospasm [22]. These findings support the central role of the GS in recovery from SAH. Although there is a lack of evidence establishing a direct link between vasospasm and glymphatic disruption, this is a current area of investigation.
As its internal regulation in humans is further elucidated, drugs that modulate the GS are being uncovered. Somewhat consistent with previously discussed mechanisms of regulation, existing drugs that affect the sleep-wake state, level of sedation, cerebral blood flow/pulse pressure, cerebrovascular tone, and AQP4 function, have been shown to affect the GS. Positive effects on glymphatic flow represent an additional therapeutic benefit that can be considered when selecting treatments or developing novel treatments for SAH. Below we explore select drugs which have been studied in the context of SAH, have been shown to enhance glymphatic flow, and the potential mechanisms underlying their effects.
Nimodipine is a second-generation antihypertensive drug that primarily sees use in managing vasospasm after SAH and is the only FDA-approved drug for managing outcomes in SAH [42]. Nimodipine has been widely shown to reduce mortality and improve neurological function after SAH and may serve as a prophylactic measure in aneurismal SAH [43, 44, 45]. Importantly, animal models have shown that nimodipine attenuates early brain injury, an important predictor of clinical outcomes in SAH patients [46, 47]. Recently it was shown that the drugs post-SAH neuroprotection, maybe due to its preservation of the GS [47]. In this study, SAH mice treated with nimodipine 2 h after SAH induction were shown to have reduced glial fibrillary acidic protein (GFAP) and AQP4 expression but reduced AQP4 perivascular polarization compared to SAH-vehicle controls. Parenchymal CSF flow was measured via intra-cisternal tracer, and a linear regression analysis confirmed that flow was inversely correlated with GFAP and AQP4 expression but positively correlated with AQP4 polarization [47]. SAH mice treated with nimodipine also had reduced cerebral edema, attenuation of neurological deficits, and improved cerebral blood flow 3 days following SAH compared to SAH-control groups, consistent with findings in other rodent studies [48]. Linear regression of these findings found a negative correlation between CSF tracer penetration and neurological deficits but no association between CSF tracer penetration and cerebral blood flow 3 days after SAH. One concern regarding nimodipine, as a calcium channel antagonist, is hypotension. Despite lowering cerebral pulse pressure, nimodipine penetrates the CSF and increases glymphatic flow [49]. This effect is somewhat paradoxical because pulse pressure is a driver of glymphatic flow [48]. While these preliminary studies show that nimodipine’s effect on the GS contributes to its neuroprotection post-SAH, more studies regarding the relationship between the GS, cerebral blood flow, blood pressure, and vascular possibility are needed. Additionally, the effect of typical doses of nimodipine on blood pressure may preclude its use in patients presenting with hypotension [50]. Further studies on the dose-dependent effect of nimodipine on pulse pressure, cerebral blood flow, and the GS may serve to maximize its benefits to SAH patients while minimizing its risks.
Sedative anesthetics were among the first drugs shown to enhance glymphatic flow in an attempt to replicate the effects of natural sleep on brain metabolite clearance [51]. Dexmedetomidine (Dex) is a newer selective alpha-2 agonist that is used as a sedative perioperatively. Dex has been shown to have broad anti-inflammatory and antioxidant effects [52, 53, 54]. In the context of SAH, Dex attenuates early brain injury and reduces neurological deficits in rodent models [55, 56]. These findings are supported by a limited retrospective study, which found that low dose Dex within 24 h of SAH improved neurological outcomes [54]. Recent studies have shown that when Dex is given intrathecally or intravenously it enhances glymphatic delivery of coadministered anesthetics and opioids in rodent models, without affecting serum drug levels [57, 58]. Glymphatic flow is a mediator of drug delivery from the CSF to the parenchymal ISF thus, enhanced glymphatic efflux increases drug exposure in the brain [59]. Lilius et al. [57] showed that drugs such as naloxone and oxycodone coadministered with Dex had higher peak concentrations throughout the brain ISF in mice. In this study, hippocampal, thalamic, hypothalamic, and spinal concentrations of naloxone in control groups peaked at administration and decreased within 30 min whereas naloxone concentrations in Dex sedated mice were increased at thirty minutes and remained significantly elevated at the 2-hour mark. Supporting this effect of Dex on the glymphatic flow, the same study showed that amyloid-β IgG antibody delivered intrathecally saw greater distribution in the brains of Dex sedated mice versus controls. Studies also showed increased glymphatic efflux in Dex versus isoflurane sedation, indicating that mechanisms outside of simply inducing unconscious state are contributing to the Dex’s effect on the GSs [58]. Glymphatic efflux is shown to be greatest during slow-wave (non-rapid eye movement, NREM) sleep [51] which Dex has shown to reliably induce [60, 61]. Additionally, reducing cerebrovascular adrenergic tone has been shown to increase ISF volume independent of the sleep-wake state. Alpha-1 adrenergic antagonist applied directly to the cortex of awake mice increases interstitial volume comparable to that of sleeping or anesthetized mice [51]. Despite increased interstitial volume in these mice, previous studies have shown that Dex reduces vasogenic edema post-surgical injury [62] whereas alpha-1 agonist exacerbates vasogenic edema [63]. These previous findings suggest that Dex increases glymphatic clearance via induction of deep wave sleep and reduced adrenergic tone. Along with Dex, ketamine/xylazine (K/X), a common anesthetic used for rodents, has been shown to be superior in achieving slow-wave sleep than other forms of anesthesia, also correlating to delta wave electroencephalogram (EEG) [64]. This study also showed that glymphatic flow was highest when K/X was supplemented with Dex and pentobarbital. Given that poor grade SAH patients are often sedated, the use of Dex as a primary sedative may confer additional benefit to recovery. Supporting the safety of Dex for sedation in these patients, a clinical trial examining the safety and efficacy of Dex in SAH patients saw no significant difference in cerebral autoregulation [65]. Side effects of Dex are related to its sympatholytic effects, including transient hypertension, hypotension, and bradycardia [66]. Unlike nimodipine, a reduction in cerebral blood flow has been observed at high doses of Dex, however, this is quickly reversed and not associated with risk of vasospasm [66]. Interestingly, a limited preliminary study showed that perioperative administration of Dex and nimodipine resulted in faster rehabilitation times, showing that co-administration may be feasible [67]. These combined animal and clinical data suggest that Dex may improve outcomes in SAH and enhance the pharmacokinetics of administered drugs to the brain via improved GS function. Pending future studies on safety for use in SAH, Dex may provide direct neuroprotection post SAH via the previously outlined mechanism as well as enhanced delivery of other therapeutics such as nimodipine.
Fluoxetine is one of the most widely used selective serotonin reuptake inhibitors (SSRIs) to treat depression. It also may improve cognitive ability after long-term use in patients with vascular dementia and Alzheimer’s disease (AD) and may attenuate early brain injury after SAH [68, 69]. Similar to other drugs covered here, fluoxetine’s attenuation of early brain injury in SAH rodent models is attributed to its anti-inflammatory and anti-apoptotic effect [69]. However, improved GS function has been observed and represents an overlapping neuroprotective mechanism in this context. Fluoxetine’s influence on glymphatic flow has recently been studied as a potential mechanism by which it may ameliorate neurological diseases including SAH, specifically its effect on astrocyte AQP4 function and cerebrovascular autoregulation. In mouse models of depression, chronic stress has been shown to reduce AQP4 expression [70]. Additionally, reduced coverage of cerebrovasculature by AQP4 positive astrocytic endfeet has been observed in patients with major depressive disorder [71]. In a dual mouse model of depression and AD, fluoxetine reversed the reduction of AQP4 in mice that underwent chronic unpredictable stress, which was reflected as improved glymphatic flow and greater clearance of amyloid-β [71]. Previous studies also show that fluoxetine selectively increases cerebral blood flow, negating the effects of co-administered norepinephrine [51, 72]. These findings indicate that fluoxetine may interfere with calcium signaling in the cerebrovascular, altering its sympathetic tone which is a key regulator of glymphatic flow [2, 73–75]. The mechanisms of fluoxetine’s effects on APQ4 expression in astrocytes and on the cerebrovascular are not well understood. Both fluoxetine and Dex have been shown to attenuate inflammation in models of SAH, which is a potential mechanism by which astrocyte and APQ4 function are preserved in various disease states [55, 76]. Fluoxetine lacks the potential adverse hemodynamic effects of Dex and nimodipine while being studied in terms of long-term use, improving the feasibility of its post-SAH use [77, 78]. Fluoxetine also has a favorable safety profile in treating depression post-hemorrhagic stroke but has limited human studies during SAH [79].
Continuing the theme of drugs that may target inflammation, DL-3-n-butylphthalide (DL-NBP) is a drug that has been studied for its therapeutic effects on neurodegenerative diseases and diseases of the cerebrovascular including stroke, AD, and Parkinson’s disease [80–83]. DL-NBP targets multiple pathophysiological processes in the context of these diseases including inflammation, oxidative stress, thrombogenesis, neuronal apoptosis, and mitochondrial malfunction. Recently, it was shown that DL-NBP improves glymphatic clearance of amyloid-β and reduces deposition in transgenic mouse models of AD [80]. This study showed that DL-NBP increased the expression of perivascular AQP4. Interestingly DL-NBP has been shown to decrease AQP4 expression during the acute phase of an ischemic stroke while increasing and restoring AQP4 expression with long-term administration in models of AD, conferring vascular and neuroprotection in both contexts [80, 84]. The effect of long-term administration of DL-NBP on AQP4 may also contribute to its ability to promote revascularization and neuroregeneration in mouse models of SAH, in which AQP4 polarization is impaired [85, 86]. The purported disease modulating mechanism of DL-NBP is very broad and further research is needed to determine which of its targets influence the GS.
The drugs covered here represent treatments that have a history of clinical use and have also been found to influence glymphatic function in the context of SAH models. While many drugs have been explored to address the pathology seen in SAH, few have seen clinical use in this context and fewer have been explored for their influence on the GS [87]. As previously outlined, GS dysfunction seen in SAH is associated with subsequent neurological deficits and therefore is an important target in recovery from SAH. Overall, the findings suggest that these drugs function to improve glymphatic flow after SAH due to their effects on the cerebrovasculature to prevent vasospasm and due to reduced inflammatory/hypoxic changes in astrocytes, thus improving AQP4 function (Figure 3). These effects represent overlapping regulatory mechanisms which can be leveraged to overcome deleterious changes in the GS following SAH. Glymphatic studies in healthy versus disease populations are still in their early phases and more research is needed to understand the dynamics of the system in the acute versus later phases of the SAH prior to prescribing therapies for their apparent abilities to modulate the GS.
This work reviews our current understanding of the GS, its disruption in SAH, and its function in recovery from SAH. As we outlined, the GS represents an important target for ameliorating damage and post-acute sequelae of SAH. The previously outlined animal studies suggest that restoring or improving glymphatic function in the context of SAH reduces vasogenic cerebral edema, reduces tissue damage, and preserves long-term neurological function. While more advanced studies are needed, the fact that clinically approved drugs have been found to positively modulate the GS lowers the barrier to using them to improve patient outcomes in SAH. It is our hope that current evidence from animal studies and existing therapeutics will motivate further studies targeting the GS during acute and post-acute SAH.
AD: Alzheimer’s disease
AG: arachnoid granulations
AQP4: aquaporin-4
CNS: central nervous system
CSF: cerebrospinal fluid
Dex: dexmedetomidine
DL-NBP: DL-3-n-butylphthalide
GS: glymphatic system
ISF: interstitial fluid
PVS: perivascular space
rt-PA: recombinant tissue plasminogen activator
SAH: subarachnoid hemorrhage
SQ: primary author; AB, YM, JH: secondary authors, contributed to Figure 1 and 2 designs, as well as the “Introduction” and the “Disruption of the GS after subarachnoid hemorrhage” sections; BLW: project organizer, made final edits. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Brandon Lucke-Wold was funded by NIH R25 training grant. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Eric J. Panther, Brandon Lucke-Wold
Fettah Eren ... Sueda Ecem Yilmaz
Gonçalo Januário
Andrés Ricaurte-Fajardo ... Nathalia Melo Gonzalez
Mohammed A. Azab ... Brandon Lucke-Wold