 Review
Review

Affiliation:
1OMICS Centre of Excellence, G42 Healthcare, Abu Dhabi, UAE
2Victoria University, Footscray, Melbourne 3011, VIC, Australia
Email: nitin.saksena@bigpond.com; Nitin.saksena@G42.ai
ORCID: https://orcid.org/0000-0002-2431-9871
Affiliation:
3Institut National de la Santé et de la Recherche Médicale; Centre de Recherche des Cordeliers, Equipe-Immuno-pathologie et Immuno-intervention Thérapeutique, Sorbonne Université, Université de Paris, 75006 Paris, France
ORCID: https://orcid.org/0000-0002-2888-2418
Affiliation:
4Herpes Neuropathogenesis Research Group, The Westmead Institute for Medical Research and The University of Sydney, Westmead NSW 2145, Sydney, Australia
ORCID: https://orcid.org/0000-0002-4868-9789
Explor Immunol. 2021;1:61–79 DOI: https://doi.org/10.37349/ei.2021.00007
Received: March 03, 2021 Accepted: May 18, 2021 Published: June 30, 2021
Academic Editor: Jorge Kalil, National Institutes Science Technology, Brazil
As the severe acute respiratory syndrome coronavirus (SARS-CoV)-2 is a new virus, the current knowledge on the immunopathogenesis of this newly emerged SARS-CoV-2 is beginning to unravel with intensive ongoing global research efforts. Although a plethora of new studies have been published in a short space of time describing how the virus causes disease and incurs insults on the host immune system and the underlying immunopathogenic mechanisms remain to be elucidated. Thus, the discussion in this review is based on the most current knowledge on the immunopathogenesis of SARS-CoV-2 that has emerged in the past 12 months. The main objective is to shed light on the most current concepts in immunopathological aspects of the lung, bloodstream, and brain caused by the SARS-CoV-2, which has led to the current pandemic resulting in > 100 million infections and > 2 million deaths, and ongoing.
Coronaviruses belong to a group of enveloped RNA viruses that commonly infect human hosts during every flu season. They commonly result in the low-grade upper respiratory tract infection, akin to the common cold virus showing an overlap in symptoms, thereby going largely unnoticed potential [1]. The severe acute respiratory syndrome coronavirus (SARS-CoV)-2 is one of the exceptions that have led to a global pandemic and are defined by the more severe respiratory illness, reminiscent of its predecessor [SARS-CoV-1 and Middle East respiratory syndrome (MERS)], and resulting in more deaths globally in comparison to its earlier counterparts in 2002 and 2012, respectively [2]. The China Health Authority alerted the World Health Organization (WHO) about the number of cases of pneumonia of unknown etiology that was affecting the Wuhan City in Hubei Province in Central China in December 2019 [3]. The global pandemic [called coronavirus disease 2019 (COVID-19)] was declared related to the new etiology of the SARS-CoV-2, which now has spread to every part of the globe [2].
During the last 12 months, we have witnessed rapid transmission of the SARS-CoV-2 across the globe with > 3 million deaths worldwide, and still ongoing [4], in addition to the unprecedented magnitude of economic and livelihood loss globally. As of May 23, 2021, the global infections with SARS-CoV-2 stand at 167,073,071 with 3,469,517 deaths, and 148,019,244 recoveries globally [2]. Notable is the recovery rate at 85.7%, which varies depending on the demographics [2]. The global death rate stands at 2.1%, with the highest mortality noted in the USA. Of 33,882,333 infected, 603,876 deaths were reported, with 27,471,242 recoveries from SARS-CoV-2 infection as of May 23, 2021 [2]. The underlying reason for such high pathogenic potential in the USA remains poorly understood, apart from the contribution of comorbidities to the mortality rate [2].
Although the highly pathogenic nature of this coronavirus (SARS-CoV-2) has resulted in large outbreaks and rapid transmission, the underlying immunopathogenesis of the virus remains incompletely understood. Thus, the main objective of this review is to provide the latest snapshot of the progress on understanding immunopathogenesis caused by SARS-CoV-2 and its pathogenic consequences at the level of immunity, lungs, and the brain.
The three severe acute respiratory syndrome (SARS) viruses discovered, including the SARS-CoV-2, share the same mechanism of pathogenesis, although they differ in entry receptor, they use to gain access to the host cell. For instance, the Spike (S) protein (S2) of the MERS uses carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM-1) receptor protein, which is expressed on epithelial cells [5]. The host cell entry of both SARS-CoV and SARS-CoV-2 virus is mediated via angiotensin-converting enzyme (ACE) 2 receptor-mediated endocytosis, followed by uncoating, and the RNA genome release into the cytoplasm [6]. The binding of S protein to ACE2 results in down-regulation, and this is thought to be the primary cause leading to lung injury [7]. ACE2 can simultaneously upregulate ACE1 via the negative feedback [8] leading to excessive angiotensin-II. Excessive angiotensin II, in turn, binds to the angiotensin type 1a receptors (AGTR1a) receptors, leading to excessive vascular pulmonary activity consistent with lung pathology that is observed in pulmonary destruction [8, 9].
Several circulating lymphocytes, monocytes, macrophages and lymphoid tissues, epithelial cells of the respiratory tract, epithelium of the renal distal tubules, mucosa of the intestine, neurons of the brain, have shown the presence of SARS-CoV-2 viral particles and viral DNA. The pneumocytes and enterocytes are the primary target cells as they express a copious amount of ACE2-through which they enter the host cell. The epithelial cells of cerebral neurons, immune cells, and renal tubules are among the other cell types infected by SARS-CoV-2, and the pathology in these cell types could result from direct infection of the target cells by the virus or indirect injury, resulting from factors such as immune responses, cellular activation, circulatory dysfunction, and hypoxia [10, 11]. Severe SARS-CoV-2 infection is typified by viral pneumonia, which culminates into acute respiratory distress syndrome (ARDS) [12, 13]. ARDS is a severe hypoxemic respiratory failure, characterized by inflammatory injury to the alveolar-capillary barrier (ACB) and protein-rich oedema fluid into the pulmonary airspaces [14].
As discussed in the preceding section that the cells of the respiratory system from the upper airway to the lungs are the cells virus has the strongest affinity with and the damage to these cells is central to SARS-CoV-2 pathogenesis [11]. The development of bronchial organoids made from four different cell types from the outer bronchial layer (or epithelium) by Takayama’s group showed that upon infection with SARS-CoV-2 the virus predominantly infects stem cells in the epithelium, also called basal cells, but did not enter the club cells that are protective in nature [15]. It remains to be established whether the virus spread can occur from these basal cells to other cells.
Similarly, in a parallel study, Chen’s group used mini lung organoids and has shown that some cells die upon infection with SARS-CoV-2, which could be attributed to the induction of chemokines and cytokines mediated by viral infection, thereby triggering a vigorous immune response, which in most cases prove fatal [16]. These observations are also consistent with the cytokine storm that SARS-CoV-2 incurs in the infected host (Figure 1). As a consequence of this hyper-immune response, it suddenly culminates into ARDS- a significant feature of SARS-CoV-2 infection (Figure 1) in the older adult group and is associated with mortality [17], but the presence of comorbidities, such as cardiovascular disease, hypertension, lower lymphocyte counts, diabetes mellitus; kidney disease, etc., are associated with poor outcomes in SARS-CoV-2-induced ARDS. ARDS results in causing diffused alveolar damage in the lung tissue, with hyaline membrane formation in the alveoli at the acute stage. This is followed by the organizing state where interstitial widening, oedema, and fibroblast proliferation occur. In an independent study on SARS-CoV-2-infected of a patient by Xu et al. [18] on a lung biopsy from a patient with SARS-CoV-2 pneumonia, there was a marked absence of lymphopenia and neutropenia, with selective infiltration of mononuclear cells. In contrast, the peripheral blood showed downregulation classical activation markers human leukocyte antigen-DR isotype (HLA-DR) and CD38 and these cells were highly positive for both perforin and granulysin CD38+ cells or were mainly the inflammatory T helper 17 (TH17) cells. This vigorous immune response could attribute to lung damage indicated by diffused alveolar damage- a feature characteristic of ARDS. This acute lung infection is characterized by ARDS in concomitance with high-level surface expression of ACE2 on pneumocytes [19, 20] (Figure 1). Notable is the levels of C-reactive protein and counts of blood neutrophil remain elevated in concomitance with lymphopenia and are frequently observed uniquely in SARS-CoV-2-ARDS patients on invasive ventilation [21].
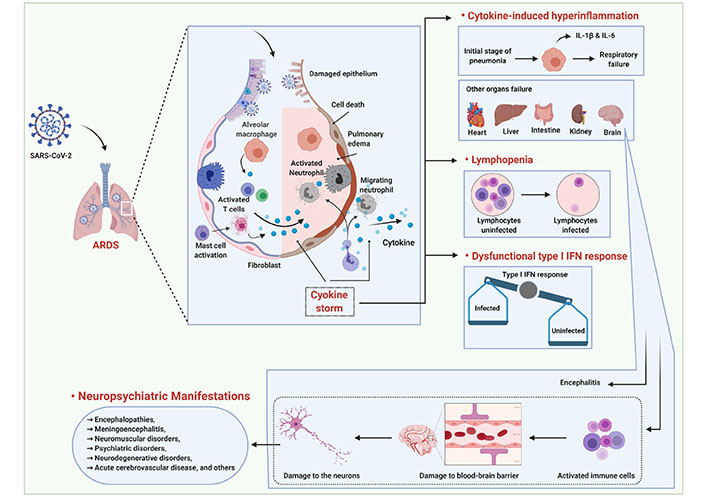
Schema showing the lung pathology caused by infection with SARS-CoV-2 during ARDS, cytokine-induced hyperinflammation. ARDS is a form of severe hypoxemic respiratory failure, which is mainly characterized by inflammatory injury to the alveolar capillary barrier with extravasation of protein-rich oedema fluid into the airspaces [14]. How ARDS leads to a cascade of events is shown in this schema. In severe infection, the SARS-CoV-2 is a systemic illness, which is typified by hyperinflammation, cytokine storm, and elevations injury biomarkers that damage other organs, which is a consequence of cellular activation. Late activation of Mast cells (MCs) by SARS-CoV-2 triggers the secretion of mediators stored in the granules, resulting in de novo synthesis of pro-inflammatory cytokines. SARS-CoV-2 induces IL-1 in macrophages and MCs, thereby causing the gene expression and activation of other pro-inflammatory cytokines. The stimulation and secretion of a pro-inflammatory complex leads to cytokine storm causing systemic and pulmonary damage [95, 96]. Together, the cellular activation contributes to hyperinflammation of lungs through cytokine storm or hypercytokinemia (IL-1β and IL-6) resulting in cell death, lymphopenia, pulmonary oedema and respiratory failure, followed by injury to heart, liver, intestine, kidney and the brain. Exaggerated systemic inflammation, or cytokine storm correlates with lymphocytopenia, which is a hallmark of severe disease. SARS-CoV-2 infection also hijacks the antiviral machinery and subverts it for its own propagation thereby creating an imbalance in Interferon responses within the host. Activated immune cells, especially the cells of macrophage lineage, can enter the blood-brain barrier and damage it to gain access into the CNS resulting in damaged neurons and consequently into a variety of neuropsychiatric and neuro-degenerative manifestations
Overall, it is apparent that the diffused alveolar damage in the lungs is the main unifying feature of the critically ill with SARS-CoV-2-induced ARDS [13]. Further, it is worth emphasizing that the innate immune responses play a vital role in the pathophysiology of ARDS. Apart from damaging lungs, the multiple immunologic processes that involve the activation of neutrophils, macrophages, mast cells, and dendritic cells also participate together in mediating collateral tissue injury to a variety of organs, such as the heart, liver kidneys, and brain. Vasodilation, endothelial permeability, leukocyte recruitment, which usually follow collateral tissue injury and inflammatory processes subsequently lead to pulmonary damage, organ stress due to hypoxemia, in particular cardiovascular stress.
In critically ill patients, a subset of patients, there is a continuous amplification of the host inflammatory response in the face of diminishing viral loads resulting in systemic inflammation thereby injuring the distal organs [22]. These inflammatory insults include are associated with mitochondrial damage-associated molecular patterns (DAMPs), which, through Toll-like receptors (TLRs) and Nod-like receptors (NLRs) signaling pathway leads to the activation of alveolar macrophages. Notable is that the microvascular permeability enhanced by mitochondrial DAMPs is independent of leukocytes. Consequently, the activated alveolar macrophages, mast cells, and other cell types release massive amounts of pro-inflammatory cytokines (cytokine storm) (Figure 1), typified by copious amounts of interleukin (IL)-1β, IL-2, IL-6, IL-8, IL-10, IL-7, granulocyte colony-stimulating factor (G-CSF), monocyte chemoattractant protein (MCP)-1, and tumour necrosis factor (TNF)-α in plasma of SARS-CoV-2 patients, with ARDS. Moreover, there is a negative effect on T cell proliferation and/or their survival with the abnormal plasma concentrations of circulating cytokines in severely ill COVID-19 patients. Cytokine storm further assists in recruiting circulating macrophages, mast cells, and neutrophils to the site of injury (Figure 1) [23], causing extensive damage to both the lung epithelia and endothelia, in addition to impairing the ACB. Interstitial pneumonia is also frequently associated with the cytokine storm [17, 24, 25]. It is the disruption of the ACB that allows protein-rich fluid to enter the alveoli causing pulmonary oedema (fluid accumulation), which interferes with gas exchange (reviewed in [26]). Further, ubiquitination also plays a vital role in regulating the abundant secretion of key proteins involved in ARDS, as a consequence of which secretion of pro-inflammatory cytokines causes the decreased function of ion channels [sodium (Na) and- potassium (K)-ATPase and epithelial sodium channels (ENaC)] and depletion of surfactant proteins resulting in alterations in lung cell viability-a process also termed as NETosis [14, 27].
SARS-CoV-2 infection can activate caspase-8, thereby triggering cellular apoptosis, in addition to the secretion of proinflammatory cytokines predominantly in the lung epithelial cells which are subsequently released through the virus-induced necroptosis pathway. This has been confirmed by the post-mortem lung sections derived from patients with fatal COVID-19, which is characterized by necrotic cell debris, inflammatory cell infiltration, and interstitial fibrosis in the lungs [28].
A recent comparison of the immune cell-subsets in hospitalized versus non-hospitalized individuals revealed significant subjugation of adaptive and innate immune cell frequency in hospitalized individuals when compared to the healthy and convalescent individuals. This was characterized by an increased frequency of T cell activation markers, in particular-CD69, OX40, HLA-DR, and CD154 (CD40L), respectively, uniquely in patients who were hospitalized, while programmed death-ligand 1 (PD-L1) and T cell immunoreceptor with Ig and ITIM domains (TIGIT)activation markers showed elevation found to be elevated in both categories of patients-hospitalized and non-hospitalized. Although an exception, the B cells also showed a similar pattern of activation/exhaustion, with increased frequency of CD69 and CD95 (Fas receptor) activation markers in hospitalized patients, whereas the increased programmed cell death protein 1 (PD-1) frequency was a feature of non-hospitalized individuals. Interestingly to note is that many of these changes also showed a sustained increase over time in longitudinal samples derived from non-hospitalized patients, suggesting a prolonged immune dysregulation much after the recovery from SARS-CoV-2 infection recovery. While these critical changes in T cell activation/exhaustion in non-hospitalized patients showed a positive correlation with age, the severely ill patients demonstrated elevated expression of both activation and exhaustion markers implying prolonged immune dysregulation after SARS-CoV-2 infection [29].
Another significant aspect of SARS-CoV-2 associated lung pathogenesis is the sepsis-induced ARDS, which is usually characterized by pulmonary thrombosis and, where the coagulation dysfunction appears to be commonly associated with elevated D-dimer (or fibrin degradation fragment) levels, which are the fragments of proteins usually produced upon the dissolution of a blood clot in the body, and D-dimer level of 500 nanograms per milliliter or higher is considered to be “abnormal” [13, 30].
Most deaths resulting from SARS-CoV-2-induced ARDS display evidence of thrombotic disseminated intravascular coagulation [13], shedding light on possible atypical manifestations observed in the lung, characterized by dilated pulmonary vessels on chest computed tomography (CT), and pleuritic pain. Vascular enlargement, which is rarely observed in typical ARDS, is a systematic feature of COVID-19 ARDS resulting in mortality. Thus, the COVID-19 ARDS appears to have the worst outcome than what is seen in typical ARDS by other causes and accounts for > 53% of total deaths in the COVID-19 pandemic [31, 32].
Considering this, it is important to discuss the role of the endothelium in SARS-CoV-2 infection. Both the arterial and venous endothelial cells and arterial smooth muscle have been shown to express ACE2 and other facilitators through which SARS-CoV-2 can gain access into these cells [33]. A universal attribute of these smooth muscle cells, endothelial cells, and perivascular pericytes across all organs is the expression of ACE2, implying that SARS-CoV-2, once in systemic circulation can rapidly disseminate throughout the body [33]. Interestingly, in a post-mortem lung tissues recovered from patients who died of SARS-CoV-2 infection revealed high numbers of ACE2-positive endothelial cells accompanied by critical alterations in the endothelial cell morphology typified disruption of intercellular junctions, cell swelling, and contact loss with the basal membrane [34]. Notable is that the co-morbidities such as hypertension, diabetes, and obesity, where endothelial dysfunction is a key determinant [35], exacerbate the risk of venous thromboembolic disease, apoptosis of endothelial cells, inflammation of organs, in addition to systemic vasculitis [36]. The inflammatory response in SARS-CoV-2 infected patients can activate coagulopathy. In SARS-CoV-2-associated coagulopathy [37], the underlying mechanisms that are involved in activating coagulation during the infectious process remain obscure, but appear to be a direct consequence of inflammatory response cascade induced by the virus, rather than any specific trait of the virus [38]. Klok et al. [39] have shown that in ICU-admitted SARS-CoV-2 patients, there was 31% more incidence of thrombotic complications. Already, based on the acute pulmonary oedema in critically ill COVID-19 patients with micro-thrombosis of small pulmonary vessels [18], it has been hypothesized that a blood clot formed in situ within the vascular system could play into gas exchange abnormalities and multisystem organ dysfunction [40].
Overall, it is notable that the SARS-CoV-2 elements have been shown to exist in endothelial cells of SARS-CoV-2-infected patients, characterized by the accumulation of inflammatory cells resulting in endothelial cell death [40, 41]. Thus, endothelitis facilitated by SARS-CoV-2 infection and the inflammatory process that accompanies it can induce significant damage across diverse organs resulting in life-threatening complications in COVID-19 patients.
Although it is clear from the preceding sections that the cellular activation causes cytokine storm, which results in the severe inflammatory process and cell death, a view from organoids that are comprised of multiple cell types is needed to explain the underlying reasons for cell death upon infection. Two studies by Suzuki et al. and Han et al. [15, 16] have shown that the cultured organoids showed permissiveness to the SARS-CoV-2 infection, yet they failed to explain the precise mechanism of lung pathology caused by SARS-CoV-2 infection. The approach to creating organoids varied between the two studies discussed herein, and instead of creating them from adult cells, Han’s study [16] used pluripotent stem cells that have the propensity to develop into any cell type in the body. Thus, the organoids cultured in this way include diverse cell types, but Han’s study cautions that the result is less mature and so might not represent adult tissue [16]. Moreover, it is worth emphasizing Monteil’s study [42], which has demonstrated direct infection of human blood vessel organoids, by SARS-CoV-2, differentiated from induced pluripotent stem cells. Further, Varga et al. [41] confirmed the presence of viral elements within the endothelial cells, which concurred with the accumulation of inflammatory cells, reaffirming [40, 41] that SARS-CoV-2 infection induces endothelium at the multiorgan level.
Unless the lung organoids are grown with the immune cells, the immunopathogenesis caused by SARS-CoV-2 in localized environments, such as organs cannot be fully understood. Moreover, it needs to be cautioned that while the organoids can reproduce the tissue-specific pathology of SARS-CoV-2 in organoids, they simply cannot address the systemic symptoms that associate with a generalized viral infection. The question remains why and how the lung cells die during infection? Is it because of damage caused by the virus or self-induced autoimmune destruction?
The total blood lymphocyte count in SARS-CoV-2-infected patients, and that of T cells, is generally a good indicator of infection and discriminating between healthy and infected controls. Lymphopenia is more prominent in symptomatic carriers of SARS-CoV-2 than in their asymptomatic counterparts [43], and it is an important feature of severe SARS-CoV-2 infection, which resolves upon recovery from infection. In the case of infected children, where the mortality rate is very low, lymphopenia is rarely seen. Whereas in the case of older adults, who experience a higher mortality rate, lymphopenia is predominantly observed severe cases [44]. The mechanism by which lymphopenia is caused is not clearly known for SARS-CoV-2. Viruses such as human immunodeficiency virus (HIV), varicella zoster virus (VZV), and polio caused lymphopenia independent of cytokine responses [45], but it remains unclear whether the SARS-CoV-2-induced lymphopenia follows a similar mechanism. One thing is clear that augmented levels of NLR associate with lymphopenia in SARS-CoV-2 disease, its severity, and in defining disease outcomes.
The studies on peripheral blood, single-cell RNA sequencing (scRNA-seq) performed on bronchoalveolar lavage fluid (BALF) and the autopsied tissue has shed some light on the nature of lymphocytic infiltration in the lungs of SARS-CoV-2 patients [46, 47]. From these studies, it is clear that there is a considerable reduction in the numbers of cytotoxic T lymphocytes in the upper respiratory tract, which could be owing to either to hyperactivation of T cells or higher levels of FAS (CD95) expression, and caspase-3, which are the pro-apoptotic molecules [48]. It is to be noted that the virus-specific cytotoxic CD8+ T cells are an effector part of cell-mediated immunity against viruses, including the SARS-CoV-2. What remains to be answered is whether the SARS-CoV-2-induced lymphopenia is an epiphenomenon or is it one of the main driving forces in SARS-CoV-2 pathogenesis?
Diverse lymphocyte populations in SARS-CoV-2-infected human hosts have been studied, and the CD4+ T, CD8+ T cell subsets, in addition to B cells and NK cells have been shown to play a central role in the immune system functioning. As expected, the healthy controls have higher T cell counts than the infected counterparts [49]. The total amount of T cells, B cells, and NK cells show a significant reduction in the infected group. While it is the T cells that are mostly altered during viral infection, the function of CD4+, CD8+ T cells, and NK cells was observed to be within the normal range, however, a further examination of both helper (CD3+CD4+) and cytotoxic T cells (CD3+CD8+) was found to be below the normal range especially in subjects with SARS-CoV-2 infection, with a normal ratio of T helper/suppressor ratio (Th/Ts). Furthermore, decreased number of regulatory T cells (Treg) (CD3+CD4+CD25+CD127low+) in the SARS-CoV-2 patients, with the effect being more pronounced especially in the critically ill patients with SARS-CoV-2 infection. Following this, a decrease in naive (CD45RA+CD3+CD4+CD25+CD127low+) and induced (CD45RO+CD3+CD4+CD25+CD127low+) Treg cell remained a feature of SARS-CoV-2 patients and again was more evident in critically ill patients. Although no statistical significance was achieved [50], larger studies are needed to reaffirm these observations. Recently, it has been shown that the presence of comorbidities significantly influences the quantity of CD8+ T cells [51]. Nonetheless, these observations in no way undermine the validity, value and, importance of the numeric T-cell counts in diagnosing SARS-CoV-2 infection. While ARDS is a strong predictor of death in the case of older adults (median age 71 years) with SARS-COV-2 infection, the higher number of lymphocytes in circulation is a predictor of better disease outcome [49].
As indicated earlier that the SARS-CoV-2-induced ARDS during severe infection is typified by massive cytokine release and T cell loss. This vigorous host immune response usually is incapacitated in clearing the virus, thereby aggravating respiratory distress, resulting in simultaneous damage to other vital organs followed by death. ARDS now is known to be a major pathogenic trait of SARS-CoV-2 infection potentially resulting in high mortality [17, 24, 25]. This mortality pattern appears to be higher in the older age categories, as opposed to being almost absent in children. It is thus likely that the abnormal immune response to SARS-CoV-2 infection may partly depend on T cell immunological memory, which again is more pronounced in adults when compared against children. Therefore, the immunopathology and massive collateral damage resulting from these host-immune processes contribute to immunopathogenesis in SARS-CoV-2-infected patients [52].
In the context of T cell being the frontline warriors, it is to be cautioned that while the simple lymphocyte enumeration in blood could be a useful marker in identifying patients at serious risk of infection, the consistent and marked absence of ACE2 expression in immune cells implies that the loss of T cells could not be attributed to direct viral infection, although direct infection of lymphocytes by SARS coronaviruses has been sporadically observed [53], may be possible through an alternate receptor [CD147/Basigin/extracellular matrix metalloproteinase inducer (EMMPRIN)] warranting further investigation [33, 54].
Like the antibody-producing B cells, the T cells are the central players in the mounting protective immune response to viral infection [55]. While, infected with SARS-CoV-2 develop severe disease, > 80% of people have only mild to moderate disease and have a full recovery, which is evident from the numbers on global recovery discussed in the preceding sections [56]. Recently, a comprehensive mapping has shown that the functional CD4+ and CD8+ T cells, which target multiple genomic regions of the SARS-CoV-2, are maintained during the mild and severe resolution phase of SARS-CoV-2 infection, and the magnitude of response correlates with the antibody response [57]. Supporting this, a recent study by Peng et al. [58] where they studied T cell memory in 42 patients following recovery from COVID-19 involving 28 individuals with mild disease, 14 with severe disease in addition to 16 unexposed donors. They used IFN-γ-based assays with peptides spanning SARS-CoV-2, except open reading frame 1 (ORF1). Interestingly, although the breadth and magnitude of T cell responses were significantly higher in the severe infection group when compared with mild cases, total and S-specific T cell responses, as seen also in the study by Swadling and Maini [57], correlated with S-specific antibody responses.
Moreover, recent studies have also shown the evidence of polyfunctional SARS-CoV-2-specific T cells, which is shown to harbour a stem-like memory phenotype, has been observed in the circulation of seronegative individuals who have recovered from an infection, and also the ones who displayed asymptomatic and mild SARS-CoV-2 infection [59]. This implies that in the absence of antibodies, a robust and broad T cell response could be enough to provide immune protection against the virus. Thus, it needs to emphasize that effective cooperation between T and B cell responses seen during the clinical course of SARS-CoV-2 infection might represent not only the key to future vaccine design, but the future efficacy of vaccine and therapeutics for SARS-CoV-2 will largely depend on a profound understanding of viral immunity.
The age, male gender, and pre-existing comorbidities are the main risk factors that correlate with a fatality rate in SARS-CoV-2 infection [6, 7], and this could be attributed to a higher basal proinflammatory status and the progressive inability of the immune system in mounting optimal responses. This could relate to ‘immuno-senescence’- a phenomenon that has been seen predominantly among the aging individuals in Italy, Spain, the UK, Germany, and the USA where the largest number of old people died during the COVID-19 pandemic [60]. This is also evident from the cases of severe interstitial pneumonia in this age globally.
Although the decline in the numbers of both CD4+ and CD8+ T cell numbers might contribute to higher susceptibility to SARS-CoV-2 infection among the older adults [47], weakened adaptive responses coupled with augmented systemic inflammation (Figure 1) could compromise antiviral responses, and in turn, induce mortality [61]. In this context, it is important to iterate that the age-dependent reduction in peripheral blood T cells has been seen in SARS-CoV-2-infected individuals > 60 years of age. Thus, immune-senescence has a strong correlation with mortality with SARS-CoV-2 infection in the older-adults group of individuals. Due to aging, there occurs a complex remodeling of the immune system dependent on age [50, 51], and many cellular changes affecting the T cells in particular have an impact on the development of the CD4+ T follicular helper cells. CD4+ T cell: CD8 T cell ratio, T-cell receptor (TCR) diversity, cytotoxicity of natural killer T cells, memory and effector T cells clonal expansion, immune defence against viral infections, a balance between naive and memory T cells, and the balance in the concentration of soluble cytokines. Usually, the higher inflammation in aged individuals, in addition to an overactive cytokine secretion predispose the old and frail to detrimental effect and poor viral clearance [62]. In this context, it has been hypothesized by Candia et al. [52] that the higher proportion of memory versus naive T cells between adults and children is likely to contribute to collateral tissue damage and cytokine storm that is seen mainly in the severely ill older age category of COVID-19 patients. To confirm these observations, aging has been shown to be characterized by decreased T cell functions along with a parallel augmentation in cytotoxic and monocyte cell functions [63].
Dynamic shifts occur in the trajectory of immune cells encompassing neonatal age to adulthood, and these shifts predominate changes from a highly diversified set of antigen-specific TCRs to a clonally less diverse composition of the expressed TCR repertoire from a highly diversified set of antigen-specific TCRs, to a less diverse dominated by an oligoclonal collection of TCR molecules, attributable to immune responses arising from past infections [52]. This is consistent with the immune dynamics of the peripheral blood naive CD4+ and CD8+ T cell enumeration, which decline in concomitance with age. Similarly, the circulating memory cells outnumber naive cells around the age of 35, but the frequencies of the same cells decline in the age group 65 and above [64, 65]. This reduced naive T cell repertoire difference in frequency between children and adults appears to be associated with lower T cell activation of both CD4+ and CD8+ T cells, in addition to multifunctional populations of healthy pediatric T-cells upon in vitro stimulation, which is an indicator of less severe toxic shock syndrome (TSS), which associates with morbidity in children as opposed to adults [66], but both TSS and the toxic shock-like syndrome occur in children at a lower incidence than in adults [67]. Moreover, early-life CD4+ T cells (foetus-derived cells), which have the propensity to differentiate into a Foxp3+CD25+ Treg cell phenotype, appear to persist and maintaining an anti-inflammatory profile for extended periods as opposed to adults [68]. Notable is that the thymus-derived Tregs (t-Tregs) and their peripherally induced counterparts (p-Tregs) are characterized by unique Foxp3-dependent and independent transcriptional and epigenetic traits. These traits provide them with the ability and the arsenal to suppress disparate immunological and non-immunological challenges [69]. Thus, it is important to iterate that there may be significant epigenetic shifts in these Treg cells with age that may associate with lower to high susceptibility to SARS-CoV-2 between children and adults. Importantly, since the Treg cells play a vital role in restricting the antiviral response of the host and resulting tissue immunopathology [52], the reduction in their activity may have a significant impact on systemic inflammation as observed in the case of severe COVID disease. Nonetheless, together, these critical immunological and cellular phenotypic differences will contribute to differences in pathogenicity that discriminate SARS-CoV-2 pathogenesis between children and adult.
Multiple organ infection by SARS viruses, SARS-CoV and MERS, that emerged in 2002 and 2012, respectively, has been well documented, and the severity of infection of multiple organs that we have seen globally in case of SARS-CoV-2 is highly reminiscent of its earlier counterparts [53].
Initial studies on the SARS-CoV-2 largely focused on the tropism and pathogenesis of the virus in the lungs. Like its earlier counterparts [SARS-CoV-1 and MERS-coronavirus (MERS-CoV)], it also has a high propensity to infect the pulmonary machinery. Now it is apparent that the SARS-CoV-2 can also infect other organ systems, which occurs either via direct viral infection or indirectly mediated through the immune system where indirect effects of the innate immune system during viral infection play a significant role in the viral spread and subsequent organ destruction. These include the heart, kidneys, intestines, brain, lungs, pharynx, trachea, gut, blood and blood vessels/endothelium, etc. Therefore, the viral load can be seen in body fluids, such as saliva, plasma, urine, faces, semen, cerebrospinal fluid (CSF) and, also breast milk, and is used as one of the markers in detecting the disease load [70, 71]. Interestingly, the organ-specific infection by SARS-CoV-2 correlates with the degree of expression of the ACE2 [72]. How the infection of diverse organs happens remains unknown, but chronic infection in certain individuals, or through the development of transitory reservoirs that virus is able to establish at early stages for viral persistence can result in viral shedding and dispersal in the respiratory tract—a phenomenon already known for Ebola virus and other RNA viruses [73]. It remains unknown as to how the viral burden is established in other organs, and in what manner the destruction of diverse organs, including the brain, occurs? As the knowledge on the mechanism of organ infection is just emerging, it is beyond the scope of this review to discuss each organ, therefore only brain, lungs, and blood are discussed herein.
The very first reports pertaining to neurological complications due to the SARS-CoV-2 emerged from Wuhan, China where the COVID-19 pandemic started. This initial report showed evidence of neurological symptoms in SARS-CoV-2-infected patients. Of the 214 patients screened in the study 36.4% had mild neurological symptoms, whereas 88/214 (41%) with severe infection showed pronounced neurological symptoms, in addition to displaying acute cerebrovascular disease, impaired consciousness, and injury to the skeletal muscle [74]. Followed by this, a Japanese study also showed COVID-19-associated encephalitis where the patient tested negative for SARS-CoV-2 using a nasopharyngeal swab, the viral RNA was detected in the CSF with a concomitant exhibition of abnormalities in the hippocampus and medial temporal lobe [75]. Supporting this, the Beijing Ditan Hospital, in 2020, also reported a similar case with SARS-CoV-2 detected only in the CSF in a patient with encephalopathy. Thus, the neurological aspect of the disease is also becoming increasingly evident, and the detection of the virus in the CSF further emphasizes the neuro-invasiveness of SARS-CoV-2, which is a possible scenario despite the low levels of ACE2 receptor expression in the brain. In this context, it is important to reiterate that sensory and gustatory impairments are seen quite commonly in SARS-CoV-2 patients [76], thereby implying the involvement of the peripheral nervous system, and signifying its potential to cause neurological damage in the central nervous system (CNS).
Various neurological manifestations and subsequent damages have been observed in patients as a result of infection with viruses like enteroviruses, influenza viruses (H1N1), measles virus, SARS-CoV-1, MERS-CoV, and, recently, SARS-CoV-2, which have also been proven in the cellular and animal model systems. The neurological damage may range from cognitive problems to motor dysfunction, in addition to a range of CNS-related anomalies, such as acute necrotizing haemorrhagic encephalopathy (ANHE), encephalitis, meningitis, Guillain-Barré syndrome, audio-visual disabilities, and anxiety. It has been hypothesized that these virus-associated neurological anomalies are triggered because of massive inflammation that is caused because of peripheral viral infection [77] (Figure 1).
Previous reports on severe fatal cases have already demonstrated the neuro-invasiveness of the SARS-CoV-1 through autopsy studies, further attesting to its neurotropic behaviour. Consistent with these findings, both SARS-CoV-1 and -2 enter the host cell through the ACE2 receptor, which, coincidentally, is expressed at low levels in the brain and the ACE2 receptors present on the capillary endothelium could facilitate entry into the CNS. Although the exact route of SARS-CoV-2 into the CNS is debatable, a report by Hoffman [78] has shown that apart from the ACE2 receptor, the SARS-CoV-2 can use transmembrane protease serine 2 (TMPRSS2) as well as endosomal cysteine proteases that help S-protein binding. It is believed that the olfactory aspiration assists neurotropic transgression and given the anatomy of the nasal cavity and its proximity to the forebrain it appears more plausible that viral entry follows this route. Mechanistically, upon breaching the blood-brain barrier, the viral dissemination into the brain tissue is either mediated by the recruitment of pathways with specific neurotransmitters across the cerebrum, or via a neuronal retrograde mechanism where the virus can invade the peripheral neurons. This allows the ultimate entry into the brain and CNS and viral spread via the connected network of neuronal transport machinery [79]. There is also a possibility raised for neuro-invasion via the enteric pathway, suggesting that SARS-CoV-2 can prime the enteric nervous system (microbiome) or may reach the CNS via intestinal vagal afferents [80]. Thus, the role of host-microbiome or microbial metabolites may have some role, considering the neural-gut communication with the vagus nerve.
Further studies are needed to attest to the full-scale of neuro-invasiveness by SARS-CoV-2. As noted in earlier sections, viremia has been observed in the case of SARS-CoV-2-infected patients, in addition to viremia in the CSF in the case of individuals displaying neurological abnormalities. Thus, it is possible that the virus may be able to breach the blood-brain barrier through the hematogenous route as a result of cytokine storm that is caused in severe cases of SARS-CoV-2 infection [81].
It is well known that viral genetic diversity plays a vital role in viral entry, host-pathogen interaction, and pathogenesis linked to host immune evasion, drug resistance, and evading neutralizing and vaccine responses. But how, in the case of SARS-CoV-2, genetic diversity is associated with diverse disease outcomes, manifestations and comorbidities remain unclear.
According to recent reports the SARS-CoV-2 has shown to be mutated into at least 30 different genetic variants, and of these 19 are novel with a set of 6 different mutations spreading differently in different parts of the globe. It is believed that the mutations virus has acquired have already shown its capability in substantially changing its pathogenicity in different geographical locales across the globe [82, 83]. For example, one mutated and lethal strain of SARS-CoV-2 circulated across Europe, and the USA, and other parts of the world [84]. At least three clades are known, characterized by geographic and genomic specificity. For example, the clade G prevalent in Europe, carrying a D614G mutation in the S protein–the initial point of contact for virus–host interaction. In the context of the current aggressive spread in the Indian sub-continent, another variant, the B.1.617 first emerged in Maharashtra towards the end of 2020 and has spread throughout India and to at least 40 different countries worldwide. Apparently, this variant is highly transmissible, and termed as a variant of concern by the WHO as it is moderately resistant to antibodies but has low severity among those who are already vaccinated. The sub-lineage of this variant, which also has three strains—L452R, E484Q, and P681R, and the last one these (P681R), has been reported to cause increased tissue damage and is more pathogenic [85].
It is essential to mention that mutation in the S protein D614G has been recognized to be of utmost importance because it has been the dominant mutation since the spread of SARS-CoV-2 began in Europe in early February [86]. Moreover, following its introduction into new geographical regions, it continued to remain dominant. They show evidence in favour of multiple strain infection possibly occurring through the process of recombination between locally circulating strains, which may have implications in SARS-CoV-2 transmission, dissemination, pathogenesis, immune interventions (such as vaccine design), and drug treatments (emergence of drug resistance), etc. S mutation pipeline does show a possible relationship with the emergence of a more transmissible form of SARS-CoV-2 [86]. Thus, the priority should be to delineate dynamically changing patterns of mutation in concert with viral evolution, which can help map positive selection for S variants crucial for future vaccine development.
A new variant referred to as SARS-CoV-2 variant under investigation, year 2020, month 12, variant 01 (VUI 202012/01) has been identified in the UK, followed by Denmark, Australia, Iceland, Italy, and the Netherlands [87]. Recent analysis indicates that although the variant may have high transmissibility, it remains to be established if this variant may associate with the severity of COVID-19 symptoms, response to antibody, or vaccine efficacy. This variant is also characterized by the presence of around 14 different non-synonymous mutations and three deletions. It is, therefore, posited that these mutational changes may alter the transmissibility of the virus, but the immunopathological implications of these mutations remain unclear.
One of the mutations-N501Y has an altered amino acid within the six key residues in the receptor-binding domain (RBD), and it originated independently in the UK and South Africa. Another mutation of biological relevance, the P681H, has also been found in the RBD. The deletion at position 69/70 has been shown to affect the detection ability of some diagnostic PCR assays that use the S gene as a target. Although most PCR assays worldwide use multiple targets, the impact of the variant on diagnostics is not significant. Noteworthy is that the newly identified variant VUI-2020/12/01 circulating in several countries appears to display different outcomes.
So far, the genetic diversity among the SARS-CoV-2 strains is low. Still the underlying reasons for its efficient and rapid spread across the globe pin-point virus’s ability to undergo natural selection, thereby acquiring favourable mutations through recombination causing antigenic drift. All these attributes could play a definitive role in viral evasion of the host selection pressure, host immunity, drug and vaccine pressure, viral adaptation into the host, and eventually its transmissibility and spread. Over and above, as the virus adapts to different geographical regions of the globe, the antigenic drift has the potential to exacerbate the accumulation of mutations that are immunologically advantageous for the virus, which may not only be lethal pathogenically but also play into affecting vaccine strategies. Thus, a clear understanding of viral diversity and evolutionary transitions, which virus makes, is vital to understanding viral immuno-pathogenesis.
Overall, it still remains to be established whether these mutations confer any distinct selective advantages in the context of host adaptation, infectious potential, geographic spread, association with co-morbidities, positive selection, or viral transmission is not clear. Nonetheless, the blueprint of these mutations is very valuable, and as more viral sequences become available the presently opaque view of viral genetic diversity in infection, transmission, and pathogenesis will become more evident.
Although the host immune responses play a vital role in limiting SARS-CoV-2 infection, the transcriptomic basis of this control remains incompletely characterized and poorly understood. A recent study by Su et al. [88] on immune cells, and plasma clinical measurements, and multi-omics of 139 COVID-19 patients at different stages of disease severity. Their data showed a major shift between mild and moderate disease, in addition to elevated levels of inflammatory signaling accompanied by the loss of specific classes of metabolites and related processes. In the stressed plasma environment, they identified multiple rare immune cell phenotypes which display enhancement as the disease becomes severe. This study also showed how the different classes of immune cells can coordinate immune responses to SARS-CoV-2. This immune response when visualized on one axis, as the study did by condensing 120,000 immune features on a single axis, the coordinates independently aligned with the major plasma composition changes, with clinical metrics of blood clotting, and with the sharp transition segregating mild and moderate COVID-19 disease [88].
Further, these single-cell transcriptomic data also demonstrated that the COVID-19 severity was associated with changes in polyfunctionality in CD8+ T cells, in particular the monotonic changes which reduce in patients with severe disease and is consistent with the highest percentages of effector phenotypes in moderate disease. Polyfunctional T cells produce a diverse panoply of cytokines and can release a relatively higher amount of cytokines as opposed to other T cells, thereby dominating an immune response [89, 90]. Apart from polyfunctional CD8+ T cells defining the severity of SARS-CoV-2 disease, there are two distinct CD4+ T cell sub-populations, which are associated with COVID-19 disease severity, as evident from the CD4+ T cell-derived transcriptomic data [88]. For instance, the naive-like cells were elevated [transcription factor 7 (TCF7), chemokine receptor 7 (CCR7)/CD197 surface protein], which showed a reduction in COVID-19 patients thereby implying augmented activation of CD4+ T cells in SARS-CoV-2 infection, as reported by Mathew et al. [48]. More studies are needed on transcriptome from SARS-CoV-2 patients to define the global interpretation of Su’s observations [88], which correlate largely with independently published immunopathologic observations from SARS-CoV-2-infected subjects.
Although several vaccines (mRNA and non-mRNA-based) have shown promise in vaccines, there is a complete absence of effective therapeutics for SARS-CoV-2. The nanotechnology-based tools have shown considerable promise in preclinical studies, and the field is wide open to its use in material science to medicine. A variety of pathogens against which the nanotechnology-based tools have been trialed include the respiratory syncytial viruses (RSV), HIV type 1 (HIV-1), herpes simplex virus (HSV), human papillomavirus (HPV), and others. Nanoparticles, usually smaller than a micrometer, are of particular interest given their unique features such as low cost, pliable to modification, size, and facile preparation.
There are several nanotechnology-based strategies that may prove to be effective in anti-SARS-CoV-2 diagnostics and therapeutics. Although the polymeric, inorganic self-assembling materials, peptide-based nanoparticles have shown some promise against SARS-CoV-2 with respect to rapid diagnosis, the nanotechnology-based strategies for COVID-19 using polymeric-based nanostructured materials are thought to be a powerful tool that can be deployed against the SARS-CoV-2 owing to its safety profile, protective encapsulation of drugs, bio-compatibility and bio-degradability, and flexibility of modification to size, shape, and properties, which include drug administration (FDA) approved poly (lactic-co-glycolic acid) (PLGA) and polyethylene glycol (PEG) are FDA approved polymers [91, 92]. Nanotechnology can also improve vaccine development, and the most promising outcome using nano-liposomes has been seen in the mRNA (S protein of SARS-CoV-2) vaccine which is encapsulated in nanoliposomes with specific physicochemical properties [93].
Although previously Lin et al. [94] have shown the possibility of safe delivery of a nano-vaccine, based on synthetic biodegradable PLGA and dielaidoylphosphatidylethanolamine-polyethylene glycol (DEPE-PEG) polymers against MERS, the impediment is that the polymeric nanoparticles are rapidly taken up by the reticuloendothelial system which can be ameliorated via surface modification using PEG. While delivery issues remain challenging, the nanosized feature of the nanomaterials can cause severe problems in lung and respiratory systems through an accumulation of therapeutic agents at the unwanted sites. Nonetheless, the potential of the use of nanomaterials in drug, vaccine, diagnostic, and precision medicine development presents endless possibilities in future medicine, including in treating viral infections. Also, it is important to mention that the role of nanoparticles in understanding immunopathology remains a viable alternative that can be achieved by targeting the intracellular and cellular sites expressing receptors that SARS-CoV-2 uses, and which are involved in the host immuno-and pathophysiology.
Immunology is a vast field and, therefore in this review, we have attempted to bring the most known aspects of immunopathogenesis described in the past year and a half of the COVID-19 pandemic. Since the virus is new, it is still early days for a profound understanding of immune pathogenesis the virus causes in its host, as the knowledge is constantly evolving. It is posited that a detailed understanding of immune responses in SARS-CoV-2-infected patients is central to defining effective treatments, predictive prognostics, and diagnostics, and in developing a new generation of biomarkers for predicting disease progression/severity, in addition to understanding disease heterogeneity and severities.
Nonetheless, remarkable progress has been made in a spate of the last 14 months, with at least 6 vaccines now competing for the global vaccination strategy. We may not have understood the immunology of SARS-CoV-2 fully, which is a critical part of any vaccine or therapeutic strategy and framework but have successfully defined the efficacy of diverse vaccines using new designs. Time would tell if these vaccines based on the S protein of the virus will prove to be efficacious in the long-term, and what side effects they incur in vaccinees.
We still need to understand the actual role of T cells in SARS-CoV-2 infection, and the role it plays in effective viral clearance from the host, T cell immunodominance, and immune-prevalence of SARS-CoV-2 epitopes in infected patients in terms of disease severity characterized by ARDS. Because the same immunity is dramatically impaired in severe cases and what triggers it, and how we can overcome this will be a great addition to designing effective vaccines and therapeutics. Therefore, defining a comprehensive set of epitope specificities is vital because it will determine if different SARS-CoV-2 antigens are immunodominant-a critical information in vaccine design. In concomitance, a clear understanding of B cell immunodominance will also be vital during SARS-CoV-2 infection to unveil antibody evolution to non-neutralizing viral targets that will be useful for vaccine design and in inducing neutralizing-S-specific memory B cells.
Further, a profound understanding of the dynamic shifts in T cell immunity that occur with age and the differences between children and adults will provide deeper insights that can translate into treating SARS-CoV-2 infection effectively. Lastly, artificial intelligence can play a critical role in understanding the pathogenesis caused by SARS-CoV-2, but coordinated global collaboration across all aspects of SARS-CoV-2 is needed. This knowledge will prepare us not only for the next pandemic, but will also define the roadmap to future strategies for effective treatments.
Lastly, it needs to be looked at whether the SARS-CoV-2 is an endothelial disease, as a number of studies based on SARS-CoV-2 patients have shown the evidence of inflammation-guided endothelial destruction and spread to diverse organs in critically ill SARS-CoV-2-infected patients.
ACB: alveolar-capillary barrier
ACE: angiotensin-converting enzyme
ARDS: acute respiratory distress syndrome
CNS: central nervous system
COVID-19: coronavirus disease 2019
CSF: cerebrospinal fluid
IL: interleukin
MERS: middle east respiratory syndrome
PEG: polyethylene glycol
S: spike
SARS: severe acute respiratory syndrome
SARS-CoV: severe acute respiratory syndrome coronavirus
TCR: T-cell receptor
Treg: regulatory T cells
NS created the skeleton and the first draft and wrote all major sections. NS, SRB, and MMS contributed equally at all stages of the review, with SRB conceptually designing and drawing the Figure 1. All authors contributed to manuscript revision and approved the final version. The manuscript has been read and reviewed by all three authors on the manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

