 Review
Review

Affiliation:
1Department of Pediatrics, Jackson Memorial Hospital, Miami, FL 33136, USA
ORCID: https://orcid.org/0000-0002-1889-5813
Affiliation:
1Department of Pediatrics, Jackson Memorial Hospital, Miami, FL 33136, USA
ORCID: https://orcid.org/0009-0003-9427-7819
Affiliation:
2Department of Internal Medicine, Jackson Memorial Hospital, Miami, FL 33136, USA
ORCID: https://orcid.org/0009-0005-5785-4044
Affiliation:
1Department of Pediatrics, Jackson Memorial Hospital, Miami, FL 33136, USA
2Department of Internal Medicine, Jackson Memorial Hospital, Miami, FL 33136, USA
ORCID: https://orcid.org/0009-0000-0634-6684
Affiliation:
1Department of Pediatrics, Jackson Memorial Hospital, Miami, FL 33136, USA
ORCID: https://orcid.org/0000-0001-9320-1570
Affiliation:
1Department of Pediatrics, Jackson Memorial Hospital, Miami, FL 33136, USA
3Division of Allergy and Immunology, Department of Pediatrics, University of Miami Miller School of Medicine, Miami, FL 33136, USA
ORCID: https://orcid.org/0000-0003-2066-9476
Affiliation:
1Department of Pediatrics, Jackson Memorial Hospital, Miami, FL 33136, USA
3Division of Allergy and Immunology, Department of Pediatrics, University of Miami Miller School of Medicine, Miami, FL 33136, USA
Email: melissa.gans@med.miami.edu
ORCID: https://orcid.org/0000-0002-3078-0998
Explor Immunol. 2024;4:218–237 DOI: https://doi.org/10.37349/ei.2024.00138
Received: November 21, 2023 Accepted: February 23, 2024 Published: April 23, 2024
Academic Editor: Jochen Mattner, FAU Erlangen-Nürnberg and UKER, Germany
Primary immune regulatory disorders (PIRDs) constitute a subset of inborn errors of immunity and are characterized by lymphoproliferation, autoimmunity, malignancy, and infection. Unlike classical primary immune deficiencies, initial symptoms of PIRDs can manifest as autoimmunity such as cytopenias or enteropathy, which can often prove resistant to conventional treatments and occur years prior to the onset of infectious complications. Raising awareness about PIRDs among specialists and adopting a multidisciplinary approach is crucial for early diagnosis, intervention, and potential prevention of severe organ damage. Significant progress has been made in identifying several PIRDs, which has contributed to a more comprehensive comprehension of their underlying immunological mechanisms. This knowledge has paved the way for targeted therapies focusing on specific molecules, which tend to offer superior disease control compared to traditional immunosuppressants. This review, informed by the latest literature, explores prevalent PIRDs, detailing their clinical manifestations and recent advancements in treatment modalities.
Inborn errors of immunity (IEIs) largely stem from pathogenic germline genetic variants present from birth, though somatic variants can also cause immune dysfunction. Recent strides in high-throughput genetic sequencing have unveiled novel IEI types, previously categorized as primary immunodeficiency disorders [1]. Currently, over 485 IEI variants exist, each displaying diverse phenotypes, with manifestations ranging from infection to malignancy, allergy, autoimmunity, and autoinflammation. These include 55 new monogenic defects since the prior year and 1 autoimmune phenocopy, classified by the International Union of Immunological Societies (IUIS) expert committee [1].
Genetic variations contribute to disease pathology by impacting the gene product, either through the elimination or reduction of protein expression and function (null/hypomorphic), or by modifying the protein to exhibit gain of function (GOF). The mechanisms underlying immune dysregulation in IEI are contingent on the specific variant’s characteristics and the mode of inheritance. Monoallelic variants can instigate disease through haploinsufficiency, negative dominance, or GOF. Conversely, biallelic genetic abnormalities (homozygous, compound heterozygous) lead to autosomal recessive (AR) traits due to loss of expression, loss of function (LOF), GOF, or the acquisition of neomorphic function in the encoded protein. X-linked recessive traits result from LOF or GOF variants on the X chromosome, observed either in hemizygosity in males or in a homozygous state in females [2, 3].
Many studies encompass a broad spectrum of IEIs [1]. However, primary immune regulatory disorders (PIRDs) constitute a specific subset within IEIs, characterized by diverse clinical presentations primarily marked by autoimmunity, lymphoproliferation, autoinflammation, and/or malignancy. These disorders stem from disruptions in immune tolerance pathways that affect multiple levels of the immune system, revealing diverse mechanisms in disease development and the governance of autoimmunity.
A paradigm to explain the underlying pathophysiology of PIRDs is in common variable immunodeficiency (CVID). While CVID is a heterozygous diagnosis, it typically stems from a dysregulation of B cells. If a B cell does not function properly, patients have hypogammaglobulinemia and subsequent recurrent sinopulmonary infections necessitating replacement immunoglobulin (Ig). However, overactivation or abnormal activation of these B cells frequently causes autoimmunity in CVID patients and sometimes lymphoproliferation as well. Defects in T cells, particularly T regulatory cells, neutrophil function, and numerous other pathways within the immune system have similarly been linked to PIRDs [4, 5].
Diverse genes are implicated in these disorders, classified into nine IUIS-defined immune system categories. A study of 2,183 patients with IEIs reported autoimmunity or inflammation in 26% of cases, where autoimmune disease correlated negatively with survival in IEIs particularly marked for autoimmune cytopenia [4]. Delayed PIRD diagnoses are common, driven by prominent autoimmunity overshadowing infection history. Elevating PIRD awareness among specialists and nurturing a multidisciplinary approach is pivotal to enabling early intervention and reducing the risk of end-organ damage. IEI patients frequently encounter severe infectious risks, necessitating immunosuppression. Recent shifts favor precision therapies tailored to specific disrupted immune system-related pathways over broad immunosuppression [3]. These therapies, demonstrated in various PIRDs, rectify abnormal immune responses and mitigate severe autoimmunity, often surpassing conventional immunosuppressants [2, 4].
The long-term ramifications and risks of infection and malignancy necessitate further investigation. For patients with severe phenotypes unresponsive to multiple immunosuppressive regimens, hematopoietic stem cell transplantation (HSCT) or gene therapy offers promising avenues. This journey into precision therapies, understanding immune dysregulation, and prioritizing sustainable solutions highlights medical science’s dynamic nature and unwavering commitment to patient well-being.
This comprehensive review delves into the prevalent clinical features and treatments of recently identified PIRDs. The focus of this review extends to exploring targeted therapies tailored to these specific disorders. Moreover, this discussion predominantly encompasses off-label therapies, emphasizing the innovative approaches being explored in the medical community. The necessity for compliance with regulatory standards is acknowledged. When applicable, Food and Drug Administration (FDA) approved uses for these therapies will be outlined, although many of them remain off-label unless explicitly stated otherwise. This review offers fresh scientific insights and a nuanced view of emerging treatments in the field of PIRDs.
Tumor necrosis factor-α (TNF-α) is a multipotent cytokine involved in macrophage and phagosome activation, monocyte differentiation into macrophage, and neutrophil/macrophage recruitment. Of note, it is also responsible for macrophage and lymphocyte recruitment for granuloma formation and maintenance of this protective system [6, 7]. TNF-α is involved in many of the complications of immune dysregulation seen in PIRDs and thus, anti-TNF-α monoclonal antibodies, such as etanercept, infliximab, and adalimumab have shown potential for use as crucial targeted therapies in the immune dysregulation seen in PIRDs (Figure 1) [8].
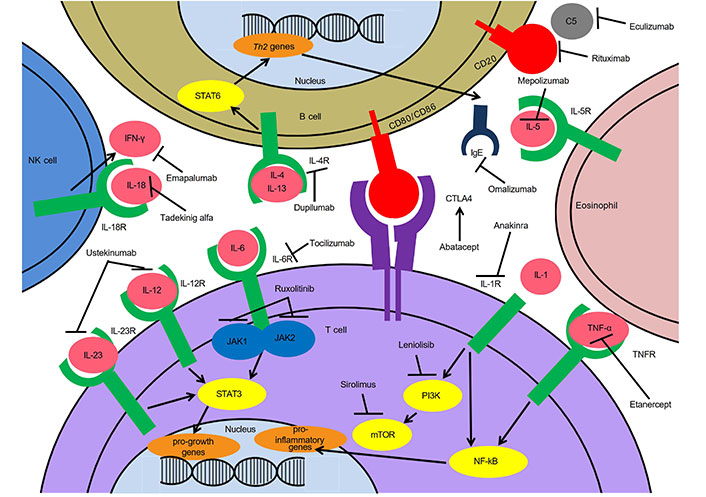
Targeted treatments for immune dysregulation in IEIs. Th2: T helper type 2; STAT6: signal transducer and activator of transcription 6; NK: natural killer; JAK1: Janus kinase 1; PI3K: phosphoinositide 3-kinase; mTOR: mechanistic/mammalian target of rapamycin; NF-kB: nuclear factor kB; IL-5R: interleukin-5 receptor; IL-5: interleukin-5; CTLA4: cytotoxic T-lymphocyte-associated antigen 4; TNFR: TNF receptor; IFN-γ: interferon γ
Granulomatous disease seen in patients with CVID has been associated with increased activation of the TNF-α system resulting in increased granuloma formation, and blockage of excess TNF-α may result in improvement of disease [9]. Etanercept has been shown in case reports to be effective for the treatment of refractory cutaneous granulomas in CVID, likely through downregulating this abnormal immune response [10]. Etanercept has also been shown to cause systemic symptomatic improvement in a patient diagnosed with CVID, juvenile rheumatoid arthritis, and alopecia following one year of treatment [11]. Infliximab has been shown to be an effective treatment of cutaneous and visceral granuloma seen in CVID as well as in ataxia telangiectasia [12–15]. However, all of these are very limited reports.
Treatment with etanercept can be successful in some patients with TNFR-1 associated periodic syndrome (TRAPS) but the response can be varied or absent [16]. In contrast, treatment with anti-TNF-α monoclonal antibodies (adalimumab/infliximab) is not recommended in TRAPS as it is associated with disease flares thought to be due to inherent issues with TNF-α receptor internalization in TRAPS which allows for a hyperinflammatory response on TNF-α antibody binding [17–19].
Anti-TNF-α monoclonal antibodies have been approved for a wider range of diseases than etanercept [20, 21]. Infliximab and adalimumab are both FDA approved for inflammatory bowel disease (IBD) and their use has crossed over to the treatment of PIRD-associated IBD (Table 1). Infliximab has shown promise in the treatment of refractory CVID-associated severe enteropathy resulting in clinical response of weight gain and higher quality of life scores [20, 21]. It was also reported to control IBD-like complications in CTLA4 haploinsufficiency and lipopolysaccharide-responsive beige-like anchor (LRBA) deficiency patients and the IBD-like complications and sacroiliitis in immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome [22–25].
Targeted therapies and clinical use in PIRDs
| Targeted treatment | PIRD |
|---|---|
| Cytotoxic agents | |
| Anti-TNF-α monoclonal antibodies | CVID with ganulomas, IBD, or other autoimmunity |
| TRAPS | |
| PAPA | |
| Blau syndrome [33] | |
| Mevalonate kinase deficiency [26–28] | |
| DIRA [26–28] | |
| Ataxia telangiectasia with granuloma [11–14] | |
| CTLA4 haploinsufficiency [102, 104] | |
| LRBA deficiency [21–24] | |
| Cytokine inhibitors | |
| IL-1 inhibitors | CVID with autoinflammation |
| PAMI | |
| CAPS | |
| HA20 | |
| DIRA | |
| NOMID [36] | |
| HLH | |
| APECED [29] | |
| FCAS [36] | |
| MWS [36] | |
| Majeed syndrome [48] | |
| IL-4R inhibitors | IEIs with significant cutaneous atopy |
| Wiskott Aldrich syndrome [61] | |
| ARPC1B deficiency | |
| DOCK8 deficiency [62–70] | |
| CARD11 deficiency [62–70] | |
| Netherton syndrome [62–70] | |
| RAG1/RAG2 immunodeficiency [62–70] | |
| STAT3 LOF [62–70] | |
| ZNF341 deficiency [62–70] | |
| STAT6 GOF [62–70] | |
| IPEX syndrome with atopic symptoms [62–70] | |
| IL-5 inhibitors | STAT3 LOF with eosinophilic asthma [73, 74] |
| IEI and coexisting atopic asthma | |
| IL-6 inhibitors | Cytokine release syndrome in IEI |
| SAVI [79, 80] | |
| SOCS1 [79, 80] | |
| IL-12/23 inhibitors | CARD14 GOF disease |
| IL-18 inhibitors | Primary HLH in IEI [88] |
| NLRC4 mutation [88] | |
| XIAP deficiency [88] | |
| IFN-γ inhibitors | Primary HLH in IEI |
| Antibodies against specific immune cell molecules | |
| B cell depleting agents | GLILD in CVID [91] |
| IgE depleting agents | STAT3 deficiency |
| Complement depleting agents | CHAPLE disease [99] |
| Small molecule inhibitors | |
| CTLA4 analogs | CTLA4 insufficiency |
| LRBA deficiency | |
| Rheumatoid arthritis [77] | |
| Juvenile idiopathic arthritis [77] | |
| Psoriatic arthritis [105, 106] | |
| JAK inhibitors | STAT1 GOF |
| STAT5b GOF | |
| STAT3 GOF | |
| Kinase inhibitors | |
| mTOR inhibitors | NLRC4 GOF [112] |
| CTLA4 haploinsufficiency | |
| APDS | |
| ALPS [114, 115] | |
| ALPS like syndrome [116] | |
| PI3K inhibitors | APDS [117–119] |
| Immunostimulants | |
| IFN recombinant cytokines (IFN-γ) | CGD [89, 90] |
PAPA: pyogenic arthritis, pyoderma gangrenosum and acne; DIRA: deficiency of IL-1R antagonist; PAMI: PSTPIP1-associated myeloid-related proteinemia inflammatory; CAPS: cryopyrin-associated periodic syndrome; HA20: haploinsufficiency of A20; NOMID: neonatal-onset multisystem inflammatory disorder; HLH: hemophagocytic lymphohistiocytosis; APECED: autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; FCAS: familial cold autoinflammatory syndrome; MWS: Muckle-Wells syndrome; ARPC1B: actin-related protein 2/3 complex subunit 1B; DOCK8: dedicator of cytokinesis 8; CARD11: caspase recruitment domain-containing protein 11; RAG1: recombination activating gene 1; ZNF341: zinc finger protein 341; SAVI: STING-associated vasculopathy with onset in infancy; SOCS1: suppressor of cytokine signaling 1; NLRC4: NOD-like receptor family CARD domain-containing protein 4; XIAP: X-linked inhibitor of apoptosis; GLILD: granulomatous lymphocytic interstitial lung disease; CHAPLE: complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy; APDS: activated PI3K δ syndrome; ALPS: autoimmune lymphoproliferative syndrome; CGD: chronic granulomatous disease
In CGD, however, infliximab is associated with severe infections with typical CGD organisms and death [26]. Adalimumab has been shown to provide a clinical response in patients with mevalonate kinase deficiency through apoptosis of the activated lymphocytes that cause an exaggerated inflammatory response [27]. Adalimumab has also been effective in DIRA, where it is suggested that TNF blocking may have an indirect effect on IL-1 dysregulation [26–29].
In patients with deficiency of adenosine deaminase 2 (DADA2), TNF inhibition with etanercept or anti-TNF-α monoclonal antibodies is associated with a lower risk of stroke and decreased inflammatory manifestations of the disease [30, 31]. In PAPA syndrome, treatment with adalimumab and infliximab shows efficacy for cutaneous and arthritic manifestations [32, 33]. Adalimumab and infliximab are effective for the ocular and joint symptoms seen in Blau syndrome and may be necessary for preventing irreversible ocular symptoms [34].
Significant side effects of anti-TNF-α monoclonal antibodies are rare but need to be considered in the PIRD population [8]. These include, most commonly, headaches, injection site and infusion reactions, rashes, cytopenias, transaminitis, respiratory tract infections, diarrhea, nausea, and abdominal pain; more serious adverse effects include malignancies (especially lymphoma and skin cancer), congestive heart failure, drug-induced lupus, and demyelinating disorders [8, 35]. Additionally, there are significant immunosuppressing adverse effects from anti-TNF-α monoclonal antibodies with a risk of disseminated bacterial, mycobacterial, fungal, viral, and parasitic infections. Patients actively systemically infected or with a clinically important localized infection should not receive an anti-TNF-α monoclonal antibody. Anti-TNF-α monoclonal antibodies can also reactivate tuberculosis and hepatitis B. However, in patients with PIRD suffering from autoinflammatory, autoimmune, and granulomatous complications of their underlying immune dysregulation without active infection, anti-TNF-α monoclonal antibodies are a potential targeted treatment.
Furthermore, there are multiple non-targeted cytotoxic agents such as methotrexate and calcineurin inhibitors that can be used to treat PIRDs instead of or in combination with anti-TNF-α monoclonal antibodies. For instance, methotrexate which is a folate antimetabolite inhibits the activation and proliferation of lymphocytes. Methotrexate has many potential side effects including but not limited to: gastrointestinal effects, cytopenia, hepatotoxicity, stomatitis, and infection. Methotrexate is sometimes used in combination with anti-TNF-α monoclonal antibodies and calcineurin inhibitors as well. Calcineurin inhibitors inhibit the production of IL-2 which is essential for T cells. Potential side effects of calcineurin inhibitors include but are not limited to: hypertension, dyslipidemia, hyperglycemia, nephrotoxicity, peptic ulcers, and infection. Limited literature exists on the concurrent use of calcineurin inhibitors and anti-TNF-α monoclonal antibodies, indicating a need for further research into combination therapy [36].
Methotrexate primarily inhibits the activation and proliferation of lymphocytes. TNF inhibitors suppress monocytes and myeloid dendritic cells, and tocilizumab has a broader activity and is directed against both the lymphoid as well as the myeloid compartment.
IL-1 is a highly regulated inflammatory mediator involved in many immune dysregulatory disorders and autoinflammatory diseases. IL-1 includes IL-1a and IL-1b cytokines, which signal via the IL-1R. IL-1R antagonist (IL-1RA) is a related cytokine that competes with IL-1a and IL-1b for binding to the IL-1R.
Anakinra is a recombinant IL-1RA that is currently FDA approved for the treatment of immune dysregulation associated with CAPS and DIRA. CAPS include FCAS, MWS, and NOMID. Daily injections of anakinra improve clinical and laboratory manifestations in patients with NOMID [37]. In FCAS, anakinra prevents inflammatory flares, treats daily symptoms, and has been shown to cause resolution of amyloid A amyloidosis-associated nephrotic syndrome [38–40]. Anakinra has also been used for the treatment of HLH in patients with underlying rheumatologic disease and other secondary causes [41–43]. In undifferentiated systemic autoinflammatory disorders, anakinra controls symptoms within 4–6 weeks of starting treatment in most cases.
In CGD, anakinra can be a better alternative to TNF inhibition for the treatment of IBD and granulomas as it can reduce the severity of the disease without intensifying infection [44, 45]. In HA20, a Behcet-like syndrome resulting from dysregulated NF-kB signaling, anakinra is an option for treatment [46, 47]. Anakinra provided symptomatic benefit in a case report of a patient with APECED (Table 1) [1]. IL-10R deficiency leads to early onset IBD refractory to conventional immunosuppressive medications and anakinra treatment has been shown to lead to marked clinical improvement of inflammation [30]. In a patient with NF-kB subunit 1 (NF-kB1) deficiency and CVID phenotype with antibody deficiency, recurrent infections, postoperative fever, and hyperinflammation, anakinra treatment resulted in fever resolution, normalized inflammatory markers, and fewer infections [48]. In Majeed syndrome, an autoinflammatory syndrome comprising chronic osteomyelitis, anemia, and neutrophilic dermatoses, treatment with anakinra can result in clinical improvement including improved bone pain and quality of life [49]. In PAMI syndrome, a rare disease characterized by recurrent fevers, cytopenias, dermatitis, hepatosplenomegaly, arthralgia, failure to thrive, hyperzincemia, hypercalprotectinemia, and elevated inflammatory markers, anakinra can result in improvement in fever, arthralgia, and recurrent infections [48].
Canakinumab is an IgG1 monoclonal antibody against IL-1b that inhibits the binding of IL-1 to the IL-1R complex. It is long-acting and given by subcutaneous injection every one to two months. It is approved by the FDA for CAPS (FCAS and MWS), TRAPS, hyperimmunoglobulin D syndrome (HIDS), and familial Mediterranean fever. Rilonacept is a recombinant IL-1R that binds to and inhibits IL-1a, IL-1b, and IL-1RA. Rilonacept is approved by the FDA as weekly injections for the treatment of patients with CAPS (FCAS and MWS) and DIRA. In CAPS, treatment improves symptoms of disease and normalizes levels of serum amyloid A, an important risk factor for amyloidosis [50]. Case reports demonstrate benefits in treating colchicine-resistant familial Mediterranean fever with anakinra and rilonacept [51]. In periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome, IL-1 blockade is experimental with use in refractory cases with anakinra and canakinumab shown to improve symptoms and decrease inflammatory markers during flares [52, 53]. Likewise in the syndrome of undifferentiated recurrent fever (SURF), IL-1 blockade is the most effective biologic therapy [54]. In PAPA syndrome, anakinra and canakinumab are both effective in controlling flares [55].
Inhibition of IL-1 is well tolerated with injection site reactions being the most common adverse events. Anakinra has a good record of safety, including in the setting of bacterial sepsis [56–58]. Opportunistic infections are rare with IL-1 blockade as compared with other immunosuppressants, especially anti-TNF-α monoclonal antibodies, including in populations at risk of Mycobacterium tuberculosis reactivation [56, 59]. Discontinuation of IL-1 inhibitors during infection is not mandatory due to relative safety and risk of flares on discontinuation.
Dupilumab is a monoclonal antibody that binds to IL-4Rα and inhibits signaling of both IL-4 and IL-13, thereby blocking signaling and achieving regulation of immune function. It decreases the expression of more than 800 genes affected in atopic dermatitis with potent inhibition in the Th2-associated chemokines and does not have a significant decrease in Th1-associated genes (Figure 1) [60, 61]. Dupliumab is FDA approved for multiple atopic conditions including atopic dermatitis, eosinophilic esophagitis, asthma, and sinusitis with nasal polyposis. Dupilumab for PIRDs with significant cutaneous atopy has shown some promise and success [62]. There are case reports that show dupilumab use in Wiskott Aldrich syndrome, ARPC1B deficiency, DOCK8 deficiency, CARD11 deficiency, Netherton syndrome, RAG1/RAG2 immunodeficiency, STAT3 LOF, ZNF341 deficiency, STAT6 GOF, and IPEX syndrome with improvement in atopic symptoms [63–71]. Patients with PIRDs have a higher risk for eczematous dermatitis that presents earlier in life and is resistant to treatment [72]. As dupilumab avoids broad immunosuppression and has a favorable side effect profile, it is a good option for these patients.
Anti-IL-5 monoclonal antibodies are also emerging in the treatment of monogenetic causes of severe atopy associated with PIRDs. PIRDs can show both eosinophilia and variable severity of atopy [73]. Reslizumab, mepolizumab, and benralizumab are currently FDA approved for severe eosinophilic asthma. Mepolizumab is also approved for eosinophilic granulomatosis and polyangiitis and recently, in 2020, gained an indication for FIP1L1-PDGFRA negative hypereosinophilic syndrome [74, 75]. Benralizumab (IL-5Rα) has also gained a recent orphan drug designation for eosinophilic esophagitis. There is a case report showing the successful treatment of eosinophilic asthma with benralizumab in a patient with STAT3 LOF [76]. As anti-IL-5 agents also have minimal immunosuppression with a favorable side effect profile, these are also good options for patients with PIRDs and coexisting atopic asthma. There are increasing monogenetic etiologies for severe atopy such as STAT6 GOF disease and IL-4R, IL-5, and IgE inhibitors might be useful for these diseases [77]. However, currently, there are very limited cases where using anti-IL-5 monoclonal antibodies has successfully resolved symptoms for patients with PIRD.
Tocilizumab, an anti-IL-6R monoclonal antibody, is currently used in multiple inflammatory autoimmune diseases. It has FDA approval for rheumatoid arthritis, systemic juvenile idiopathic arthritis, giant cell arteritis, and cytokine release syndrome and has emergency approval for coronavirus disease 2019 (COVID-19). IL-6 acts on multiple cell types and has numerous functions including hematopoiesis, immune inflammatory response, nervous system response, hepatocyte stimulation, induction of acute phase reactants, and antiviral antibody response (Figure 1) [78]. Blockade of the IL-6 pathway using tocilizumab in a patient with a STAT3 GOF disease resulted in clinical improvement and decreased Th17 cells [79]. There are also reports for the use of tocilizumab in a 3-year-old patient with SAVI and a 42-year-old patient with SOCS1 [48, 80]. Moreover, some side effects associated with tocilizumab are increased plasma cholesterol, increased plasma alternative lengthening of telomeres (ALT), and less common hypertension and hypothyroidism [81]. Anti-IL-6 agents do have immunosuppressive effects, more so than anti-IL-1 agents. The common adverse events of IL-6 inhibitors include nasopharyngitis, headache, upper respiratory tract infection, gastritis, rash, arthralgia, extremity pain, fatigue, and nausea. However, the more prevalent serious adverse events of IL-6 inhibitors include infection and gastrointestinal perforation. Laboratory abnormalities include neutropenia, thrombocytopenia, dyslipidemia, and elevated liver enzymes. Notably, there is no significant increase in rates of malignancy, autoimmunity, tuberculosis reactivation, or hepatitis reported in the literature [82]. The risk of tuberculosis reactivation and primary infection appears to be minimal, although certain trials may have excluded patients with latent tuberculosis [83]. Other IL-6 inhibitors currently in development include siltuximab (FDA approved for Castleman disease) and sarilumab (FDA approved for polymyalgia rheumatica).
Ustekinumab is a monoclonal antibody that targets both IL-12 and IL-23 and is FDA approved for psoriasis, psoriatic arthritis, and IBD. IL-12 is associated with a Th1 immune response and a strong cytokine induction of IFN-γ and TNF-α. IL-23 is associated with Th17 differentiation [84]. There are multiple case reports of this medication being used successfully to treat patients with CARD14 GOF disease (Table 1) [85]. There was noted improvement in skin manifestation in a patient with Netherton syndrome [86]. Currently, multiple anti-p19 subunit of IL-23 (IL-23p19) antibodies, risankizumab, brazikumab, mirikizumab, tildrakizumab, and guselkumab, and anti-IL-17 antibodies are in stages of development [87]. These may be future therapeutic options for PIRD patients.
Tadekinig alfa is a new, non-FDA approved recombinant human IL-18 binding protein (r-hIL-18BP). It is currently going through a phase three trial in HLH (Figure 1). Specifically, this may be helpful in HLH patients with elevated IL-18, specifically those with NLRC4 mutation, XIAP deficiency, and macrophage activation syndrome [88].
Emapalumab, a human IgG1 monoclonal antibody targeting IFN-γ, is FDA approved and has been shown to be efficacious in treating primary HLH (Figure 1) [89]. Emapalumab is currently FDA approved for those patients with refractory, recurrent, or progressive disease or intolerance with conventional HLH therapy. However, it is increasingly being used off-label as first line therapy. Emapalumab can be immunosuppressing and there is a concern about unmasking a hidden mycobacterial or leishmania infection. Other HLH treatments can be used in conjunction with emapalumab for HLH. Beyond emapalumab, alemtuzumab, an anti-CD52 monoclonal antibody, also can be used as second line therapy for refractory HLH [90, 91].
Advances in immunology and molecular biology have paved the way for the use of monoclonal antibodies targeting specific cell markers in treating immune dysregulation. B cell depletion therapies, via mechanisms like signaling disruption, complement-dependent cytotoxicity, and antibody-dependent cellular cytotoxicity (ADCC), target CD20 which is expressed by B cells across various developmental stages. Rituximab, a first generation chimeric IgG1 monoclonal antibody against CD20, is FDA approved for certain types of lymphoma, leukemia, rheumatoid arthritis, and granulomatosis with polyangiitis. However, it is used off-label in many autoimmune diseases and is considered off-label first line treatment of GLILD in CVID [92]. Rituximab is often combined with an antimetabolite such as azathioprine or mycophenolate mofetil to treat CVID GLILD [93]. Second generation anti-CD20 agents ocrelizumab and ofatumumab as well as third generation obinutuzumab have been approved for the treatment of chronic lymphocytic leukemia (CLL) and follicular lymphoma (FL) by FDA [31]. In laboratory studies, it has been demonstrated that obinutuzumab exhibits greater potency in inducing direct cell death (DCD) and more effectively triggers ADCC against target cells when compared to rituximab [94].
Omalizumab, a recombinant humanized IgG1 monoclonal antibody that binds to IgE, effectively inhibits mast cell cross-linking and subsequent degranulation (Figure 1). Omalizumab is FDA approved for asthma and chronic urticaria. This therapeutic is also commonly used in atopic conditions in IEI such as CVID [95] but its off-label application in conditions involving pathogenic IgE has shown significant promise [96]. Notably, two case reports have documented encouraging treatment responses to omalizumab in patients with STAT3 deficient hyper IgE syndrome [97, 98].
Eculizumab, a humanized anti-C5 monoclonal antibody, comprises an IgG-κ with a fragment crystallizable (Fc) component combining IgG2 and IgG4 elements. Eculizumab is FDA approved for paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. This engineered antibody halts aberrant complement activation on host cells, a hallmark of the life threatening CHAPLE disease [99]. CHAPLE disease, also known as CD55 deficiency, is characterized by CHAPLE. Presently, an open-label clinical trial is underway to assess pozelimab, a fully humanized monoclonal anti-C5 antibody targeting a different epitope, for CHAPLE disease [100]. These treatments hold promise for CHAPLE disease, a condition once considered fatal. Eculizumab, by reducing the production of C5b and inhibiting membrane attack complexes (MACs) formation, is associated with an elevated susceptibility to bacterial infections, particularly opportunistic infections and meningococcus. Considering individuals with genetic deficiencies in terminal complement proteins (such as MAC) face a higher risk of infections at baseline, particularly from Neisseria meningitidis, patients undergoing eculizumab treatment undergo meningococcal vaccination as a precautionary measure before initiating the therapy [101, 102].
The CTLA4 molecule, present on all T cells, serves as a key immune checkpoint of T cell activation responsible for regulating T cell proliferation and effector functions [103, 104]. LRBA plays a pivotal role in the translocation of CTLA4 to the cell surface and protects it from degradation (Figure 1) [103]. The deficiency of LRBA leads to a notable reduction in CTLA4 expression, consequently resulting in overlapping features seen in both LRBA deficiency and CTLA4 insufficiency, attributed to compromised function of the cellular CTLA4 pool. Abatacept (also recognized as CTLA4-Ig) and belatacept, soluble fusion proteins comprised of the extracellular domain of the human CTLA4 linked to the modified Fc (hinge, CH2 and CH3 domains) portion of IgG1, have exhibited significant efficacy in managing immune dysregulatory phenotypes associated with LRBA deficiency and CTLA4 insufficiency off-label (Figure 1) [103, 105]. Abatacept is currently FDA approved for arthritis and graft versus host disease. This agent disrupts the interaction between CD28 on T cells and CD80/86 on antigen-presenting cells, thus reducing unwarranted T cell activation. Patients with CTLA4 insufficiency and LRBA deficiency can have severe life threatening cytopenias in addition to a CVID-like phenotype. Abatacept is employed as an off-label targeted therapy for managing LRBA deficiency and CTLA4 deficiency and can help prevent a life threatening cytopenia while awaiting stem cell transplant. Its primary utilization lies in the treatment of arthritis, with FDA approval extending to adult rheumatoid arthritis, juvenile idiopathic arthritis, and adult active psoriatic arthritis [106, 107]. Abatacept is essential for these patients to prolong survival and can be used as a bridge to HSCT.
JAK inhibitors disrupt the JAK-STAT signaling pathway which is a response for systemic inflammation in many autoimmune disorders and immune dysregulation in PIRDs. For disorders with upregulation of the JAK-STAT pathway, these are ideal targeted agents (Figure 1). STAT1 GOF disease classically characterized by chronic mucocutaneous candidiasis, autoimmune cytopenias, and endocrinopathies can be treated with JAK inhibitors [108]. STAT3 GOF also characterized by a combined immunodeficiency with cytopenias can also be treated with JAK inhibitors [109]. STAT5b, which can present with early onset severe atopy and eosinophilia, is additionally managed well with JAK inhibitors [110]. JAK1 GOF has a similar phenotype to STAT5b and can also be successfully managed with JAK inhibitors [111]. JAK inhibitors are predominantly linked to infectious adverse events, encompassing viral (such as herpes and influenza), fungal, and mycobacterial infections, with respiratory and urinary tract infections being the primary organ locations affected. Additionally, adverse events like embolism and thrombosis, potentially related to polycythemia vera and myelofibrosis, neoplasms, and gastrointestinal perforation events are notable. In addition, a rise in adverse event reporting is observed concerning musculoskeletal and connective tissue disorders [112]. Furthermore, topical and systemic JAK inhibitors are FDA approved for several autoimmune disorders and severe atopic dermatitis and there will likely be more broadened applicability of these small molecule inhibitors in the upcoming years.
mTOR inhibitors include sirolimus, everolimus, and temsirolimus, which work by blocking the response of T and B cell activation by cytokines (Figure 1). These are FDA approved and commonly used as an immunosuppressant in patients after transplant. In diseases of immune dysregulation, mainly sirolimus has been shown to have a large effect [113]. Sirolimus reduces the secretion of IL-1b and IL-18 by phagocytic cells in NLRC4 GOF disease presenting with neonatal macrophage activation syndrome and results in clinical improvement [113]. In a cohort of patients with CTLA4 haploinsufficiency, sirolimus improved cytopenias, splenomegaly, and lymphadenopathy in 13 patients [114]. Sirolimus mainly ameliorates benign lymphoproliferative disease in patients with APDS as well as in ALPS [115, 116]. In ALPS, treatment response also extends to autoimmune manifestations such as cytopenias, arthritis, and colitis [116]. mTOR inhibitors however are not as specific as other targeted therapies but are useful for more broad immune effects, particularly in autoinflammation. Patients with ALPS-like disease are recommended to undergo management similar to those with a confirmed diagnosis. Treatment outcomes have been observed in individuals with ALPS-like conditions characterized by elevated double-negative T cells (DNT) associated with CTLA4 haploinsufficiency, LRBA, tripeptidyl peptidase II (TPP2), adenosine deaminase 2 (ADA2), IL-2RB deficiencies, RAS-associated autoimmune leukoproliferative disease (RALD) syndrome, or CARD11 GOF, who were treated with sirolimus and exhibited positive responses [117].
For patients with APDS either secondary to genetic defect in phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta (PIK3CD) or phosphatidylinositol-3-kinase regulatory subunit 1 (PIK3R1), leniolisib is a small molecule selective PI3K inhibitor that essentially downregulates the pathway that is upregulated in APDS. APDS is another CVID mimicker that also has impaired T cell function, invasive viral infections, cytopenias, arthritis, enteropathy, and lymphoproliferation including often early onset lymphoma. Leniolisib has been shown to be effective in decreasing lymph node size and spleen size, and improving key immune cell subsets and is generally well tolerated with minimal adverse events [118–120]. This molecule also serves as a notable illustration of a targeted disease-modifying medication designed for addressing a specific PIRD. It has recently gained FDA approval as the inaugural treatment for APDS in individuals aged 12 years and older, marking a significant development in the field.
IFNs, a group of soluble low-molecular-weight cytokines, are recognized for their multifaceted roles in immune regulation, antiviral defense, and modulation of hematopoiesis [121]. Of particular interest is IFN-γ, which exerts a crucial influence on immune responses against both viral and bacterial infections [122]. The modulatory effects of IFN-γ on immune cell function and its ability to enhance microbial clearance are leveraged for therapeutic interventions in treating PIRDs.
The emergence of IFN-γ as a therapeutic candidate for PIRDs is exemplified by its successful application in CGD, a PIRD characterized by defective phagocyte superoxide production [123]. A clinical trial employing recombinant IFN-γ has demonstrated its efficacy in reducing serious infections and improving patient outcomes in individuals with CGD and prompted its FDA approval for CGD [124]. Using IFN-γ has been considered by many to be the standard of care treatment in CGD despite this trial not being repeated in over 30 years. IFN-γ is not always well tolerated by patients with significant fever and bone pain with administration. Despite these advancements, the therapeutic potential of other IFN subtypes in addressing IEIs warrants further exploration.
In cases involving patients with autoimmunity, particularly those presenting with refractory cytopenias and lymphoproliferation, considering the possibility of PIRDs is crucial for achieving an early diagnosis. Utilizing a multidisciplinary team approach with targeted therapies becomes essential to ensure optimal disease control and favorable outcomes by maximizing the potential for immune system restoration.
It’s important to note, however, that comprehensive, long-term data on the efficacy of targeted therapies is still needed, and many medications are being used off-label for these rare and understudied diseases. This data is crucial for a thorough understanding of these drugs’ ability to manage disease symptoms in comparison to definitive treatments like HSCT or gene therapy. There are no guidelines and poor data in the literature on when to do a targeted treatment for a PIRD and when to move to HSCT. Some patients are only mildly affected and as this article discusses in depth, there are significant adverse effects from most of these targeted therapies. Immunologists need to discern for their patients when the risks of the disease process outweigh the risks of the targeted therapy. For those who clearly need treatment, there is the decision if the targeted treatment is sufficient or the patient should progress to HSCT. Multi-institutional registries tracking responses to therapies and outcomes from transplants are underway and need to provide better guidance to immunologists.
The field of targeted treatments for immune dysregulation in PIRDs continues to expand. PIRD patients with immune dysregulation should undergo a thorough examination, and genetic diagnostics should be conducted when necessary and feasible. Early and accurate diagnosis is crucial for PIRD patients, especially those with immune dysregulation. However, the treatment often presents challenges, requiring a delicate balance between increased susceptibility to infection and the additional suppression of the immune system [124]. Not long ago, treatment options for PIRD patients remained limited.
ALPS: autoimmune lymphoproliferative syndrome
APDS: activated phosphoinositide 3-kinase δ syndrome
CAPS: cryopyrin-associated periodic syndrome
CARD11: caspase recruitment domain-containing protein 11
CGD: chronic granulomatous disease
CHAPLE: complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy
CTLA4: cytotoxic T-lymphocyte-associated antigen 4
CVID: common variable immunodeficiency
DIRA: deficiency of interleukin-1 receptor antagonist
FCAS: familial cold autoinflammatory syndrome
FDA: Food and Drug Administration
GLILD: granulomatous lymphocytic interstitial lung disease
GOF: gain of function
HLH: hemophagocytic lymphohistiocytosis
HSCT: hematopoietic stem cell transplantation
IBD: inflammatory bowel disease
IEIs: inborn errors of immunity
IFN-γ: interferon γ
Ig: immunoglobulin
IL-1RA: interleukin-1 receptor antagonist
IL-5: interleukin-5
IL-5R: interleukin-5 receptor
IPEX: immunodysregulation, polyendocrinopathy, enteropathy, X-linked
JAK1: Janus kinase 1
LOF: loss of function
LRBA: lipopolysaccharide-responsive beige-like anchor
mTOR: mechanistic/mammalian target of rapamycin
MWS: Muckle-Wells syndrome
NF-kB: nuclear factor kB
NLRC4: NOD-like receptor family caspase recruitment domain-containing protein 4
NOMID: neonatal-onset multisystem inflammatory disorder
PAPA: pyogenic arthritis, pyoderma gangrenosum and acne
PI3K: phosphoinositide 3-kinase
PIRDs: primary immune regulatory disorders
RAG1: recombination activating gene 1
STAT6: signal transducer and activator of transcription 6
Th2: T helper type 2
TNF-α: tumor necrosis factor-α
TRAPS: tumor necrosis factor receptor-1 associated periodic syndrome
NM: Conceptualization, Methodology, Resources, Writing—original draft, Writing—review & editing. MG: Conceptualization, Methodology, Resources, Validation, Writing—review & editing, Supervision. VP, MW, JG, and TS: Investigation, Writing—original draft. GIK: Conceptualization, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

