 Review
Review

Affiliation:
1Laboratory of Molecular Oncology, Institutional Research Training Program, School of Medicine of the National Polytechnic Institute, Mexico City 11340, Mexico
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0009-6902-5680
Affiliation:
2Laboratory of Molecular Oncology, Postgraduate and Research Section, School of Medicine of the National Polytechnic Institute, Mexico City 11340, Mexico
†These authors contributed equally to this work.
Email: nherrera@gmail.com
ORCID: https://orcid.org/0000-0003-0646-6955
Explor Immunol. 2023;3:490–499 DOI: https://doi.org/10.37349/ei.2023.00115
Received: June 01, 2023 Accepted: July 25, 2023 Published: October 16, 2023
Academic Editor: Laura Maggi, University of Florence, Italy
One of the greatest challenges in the study of coronavirus disease 2019 (COVID-19) has been to establish the determining factors in the severity of the disease. Through extensive research efforts, a crucial factor responsible for disease control or exacerbation in COVID-19 has been identified—the regulation of the immune response. The abnormal release of interleukin-1 (IL-1), IL-6, and tumor necrosis factor-alpha (TNF-α) has been extensively studied in the context of the altered immune response observed in severe cases of COVID-19. However, recent attention has turned towards the excessive release of IL-17 and the increased presence of T helper 17 (Th17) cells, the main secretory cells of this cytokine. These factors have garnered interest due to their potential involvement in the cytokine storm observed in severe cases of COVID-19. In this review, it will be delved into the intricate mechanisms by which IL-6 contributes to the differentiation of Th17 cells, resulting in an increase in the population of Th17 cells. Moreover, it will be explored the proportional relationship between the increase of these lymphocytes and the release of IL-17 and other chemokines, which all together play a key role in promoting the chemotaxis and activation of neutrophils. Ultimately, this cascade of events culminates in the generation of tissue damage by neutrophils. Additionally, therapeutic options targeting these lymphocytes and cytokines are explored, providing insights into potential avenues for intervention.
Coronavirus disease 2019 (COVID-19) is a heterogeneous disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus, an enveloped, positive sense, and single-stranded RNA virus. The typical presentation of the disease includes a patient experiencing a fever of 39℃ that has persisted for 5 days, along with coughing and coarse breathing sounds affecting both lungs. However, the symptoms can range from asymptomatic to severe scenarios, often accompanied by respiratory distress, respiratory failure, septic shock, and multiorgan failure [1]. Since the declaration of COVID-19 as a pandemic by the World Health Organization on March 11, 2020, the study of the mechanisms involved in the development of the disease, particularly, those responsible for severe clinical pictures, has been a priority [2]. The heterogeneity of the disease has been one of the main obstacles to establishing these mechanisms.
The duality of the immune response in COVID-19 is represented by the capacity to overcome the disease or the possibility of disease exacerbation mediated by an upregulated immune response [3]. Understanding which factors promote the development of one scenario over the other remains one of the greatest challenges to overcome. Progress in the study of the immunologic dynamics involved in COVID-19 has made it possible to identify that an impaired regulation of the immune response, expressed as a cytokine storm, is largely responsible for the severity of the disease and the development of specific unfavorable clinical pictures such as the acute respiratory distress syndrome (ARDS) [4].
The roles of T helper 17 (Th17) cells and interleukin-17 (IL-17) raise a new focus of study that may broaden the understanding of the development of COVID-19. Th17 cells and IL-17 have been extensively studied in autoimmune diseases. Particularly, in their collaboration with neutrophils, their participation in the generation of oxidative stress, their role in promoting the release of pro-inflammatory cytokines, and their relationship with regulatory T (Treg) cells bound by transforming growth factor-beta (TGF-β); a fact that may clarify the determining factors in the shift toward the regulation of inflammation, or toward the exacerbation of inflammation [5]. These are some of the characteristics that suggest they are part of the immune response dysregulation phenomena identified in severe cases of COVID-19 [6, 7].
The immune system is finely regulated by control mechanisms. In COVID-19, loss of this modulation leads to a systemic inflammatory state expressed by an excessive release of pro-inflammatory cytokines: the cytokine storm [4]. This has been identified as one of the determinant factors in the development of severe cases of COVID-19 (Figure 1) [8]. The cytokine storm has been involved in the development of ARDS and multiple organ dysfunction [9]. ARDS is characterized by severe hypoxemia, pulmonary infiltrates, and decreased lung compliance due to alveolar-capillary membrane lesions, resulting from excessive synthesis and release of pro-inflammatory cytokines in the lungs [10]. The aberrant release of pro-inflammatory cytokines increases vascular permeability, causing edema and favoring cellular infiltrate. This cellular infiltrate, especially neutrophils, participates in alveolar-capillary injury by releasing lytic enzymes, and in conjunction with edema, perpetuates ARDS associated with severe COVID-19 [11]. In addition, neutrophils can cause tissue damage by oxidative stress, as neutrophils can form reactive oxygen species (ROS) [12, 13]. This is due to their participation in inflammatory processes; this capacity allows these cells to eliminate infectious agents [14]. However, in the case of a non-regulated immune response, the formation of ROS and the release of lytic enzymes cause tissue damage [15].
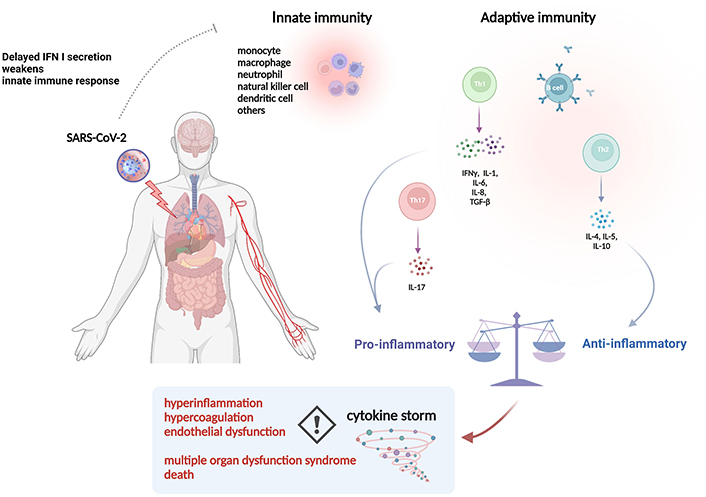
Immune response dysregulation in COVID-19. A dysregulated immune response has been attributed as a determining factor in the aggravation of the disease. Excessive cytokine release perpetuates a systemic inflammatory process and promotes hypercoagulation accompanied by endothelial dysfunction. IFN: interferon
Note. Reprinted from “Effect of extracorporeal hemoadsorpion in critically ill patients with COVID-19: a narrative review,” by Chang K, Li Y, Qin Z, Zhang Z, Wang L, Yang Q, et al. Front Immunol. 2023;14:1074465 (https://www.frontiersin.org/articles/10.3389/fimmu.2023.1074465/full). CC BY.
The cytokine storm orchestrates hemodynamic changes leading to a prothrombotic state [16]. These changes lead to microvascular failure, which ultimately has been recognized as the main culprit of multi-organ failure [17]. While ARDS was initially the prototypical complication of COVID-19, increasing evidence suggests that this illness is a more complex scenario, where, for instance, multiorgan failure syndrome may be of great relevance.
The cytokine storm may favor the prothrombotic state by various mechanisms, including stimulation of hepatic fibrin synthesis and increased release of thromboxane B2 [18]. The major complication of COVID-19, in summary, is the involvement of cytokine storms.
IL-1, IL-6, and tumor necrosis factor-alpha (TNF-α) have been widely described in the cytokine storm observed in COVID-19. These cytokines are the main ones responsible for the inflammatory process at the beginning of the illness [19]. However, there are other cytokines, such as IL-17, that have also been reported as being increased and may have a relevant role in the progression of COVID-19 to the critical stage. A significant factor contributing to the augmentation of Th17 cells is closely tied to the elevation of IL-6 levels, as previously mentioned. This cytokine plays an important role in promoting the differentiation of T CD4+ cells towards Th17, a process that will be further elucidated in subsequent sections of this review [20].
The study conducted by Ghazavi et al. [21] involving 63 patients with COVID-19, divided into severe and mild cases and 33 healthy subjects (controls), showed an association between high levels of TGF-β and IL-17 and the severity of COVID-19. They showed that observed that both groups presented elevated concentrations of TGF-β and IL-17 compared to the control group. However, while the increase of TGF-β was higher in the severe group compared to the mild group, the increase of IL-17 was higher in the mild group compared to the severe group. This may suggest a dual role for TGF-β; as an inducer of the Th17 response in mild cases, but also as a regulator of inflammation in severe cases. Thus, a higher peak of IL-17 was observed in mild patients, followed by a progressive decrease, but still elevated concentrations of the cytokine in relation to the control group. The bridge between TGF-β and IL-17 may be Th17 cells, since TGF- β is an important cytokine for differentiation of Th17 cells, and in turn, Th17 cells are majorly responsible for IL-17 synthesis [22]. In addition, the study conducted by Sadeghi et al. [23] involving 40 patients with critical COVID-19 admitted to the intensive care unit (ICU) and 40 healthy subjects, was able to identify an increase in both the number of Th17 cells and IL-17 levels in the group of patients with COVID-19, compared to the control group. Moreover, they identified an increase in the expression of genes associated with Th17 cell differentiation, such as related orphan receptor gamma t (RORγt).
Chávez-Ocaña et al. [24] found in their exploratory observational study, elevated levels of IL-17 in severe and critical patients with an average of 312 ng/L and 297 ng/L respectively. When comparing values of IL-17 from healthy versus critical patients, the significant difference was P < 0.0001 [24]. Additionally, they analyzed which parameters were associated with mortality: demographic characteristics (age, sex, and comorbidities), laboratory data (albumin, leukocytes, lymphocytes, platelets, and ferritin), days of hospital stay, ILs (IL-2, IL-6, IL-7, IL-10, and IL-17), and P-selectin in 40 Mexican patients admitted to medical emergencies with a confirmed diagnosis of COVID-19, a complete clinical record, and who signed the informed consent. Twenty severe (they required intermediate care with non-invasive ventilation) and twenty critically ill patients (they required mechanical ventilation) were classified, and these were subsequently compared with healthy and recovered subjects. A significant difference was found between the hospitalized groups in the parameters of age, ferritin, days of hospital stay, and death with P values = 0.0145, P = 0.0441, P = 0.0001, and P = 0.0001, respectively. In the determination of cytokines and P-selectin, a significant difference was found between the following groups: recovered patients and healthy volunteers compared with hospitalized patients in severe and critical condition. Importantly, IL-7 remained elevated one year later in recovered patients. Taken together, these values determined at the time of hospital admission could be useful to monitor patients closely and evaluate in-hospital progress, hospital discharge, and out-of-hospital progress.
The differentiation of T CD4+ lymphocytes is cytokine-dependent, specifically for Th17 cells, in which case IL-6, IL-23, TGF-β, and IL-1β are mainly responsible [25]. TGF-β participates actively in two cell differentiation pathways: towards Th17 and Treg [26]. By considering the collaboration with other cytokines, it may be understood the dual role of TGF-β in the differentiation of T lymphocytes (Figure 2) [27]. Thus, the collaboration of TGF-β with IL-6 biases differentiation towards Th17 [28–30]. IL-6 activates signal transducer and activator of transcription 3 (STAT3), a regulator of RORγt expression, which is the main transcription factor responsible for Th17 differentiation. This feature allows IL-6 to induce differentiation towards Th17 independently, without the collaboration of other cytokines [20]. However, the TGF-β-mediated differentiation mechanism towards Th17 is not very clear. An indirect action by suppressing the expression of T-box expressed in T cells (T-bet) and GATA-binding protein 3 (GATA 3) has been proposed, which would lead to the suppression of Th1 and Th2 differentiation [31, 32]. Thus, if differentiation towards Th1 and Th2 is suppressed, the action of IL-6 in inducing differentiation towards Th17 would be enhanced [26, 31–33]. In addition, the concentration of TGF-β is another factor that has been studied for its importance in the differentiation towards Th17 or towards Treg [30].
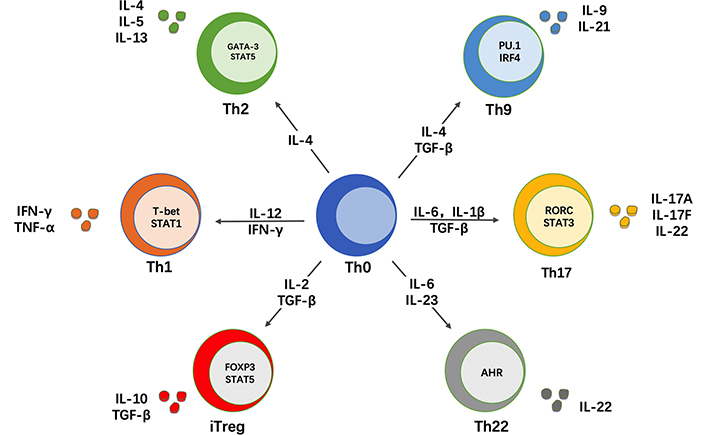
Cytokines involved in the differentiation of CD4+ T lymphocytes. The cytokine environment, among other factors, determines CD4+ T cell differentiation. The participation of TGF-β in two differentiation pathways is dependent on the collaboration with other cytokines: in the case of Th17 cells, with IL-6. PU.1: Spi-1 proto-oncogene; IRF4: IFN regulatory factor 4; RORC: retinoic acid-related orphan receptor C; FOXP3: forkhead box P3; iTreg: inducible Treg; AHR: aryl hydrocarbon receptor
Note. Reprinted from “The multifaceted role of Th1, Th9, and Th17 cells in immune checkpoint inhibition therapy,” by Lee J, Lozano-Ruiz B, Yang FM, Fan DD, Shen L, González-Navajas JM. Front Immunol. 2021;12:625667 (https://www.frontiersin.org/articles/10.3389/fimmu.2021.625667/full). CC BY.
The cytokine profiles analyzed in patients with severe COVID-19 may guide towards factors leading to differentiation towards Th17 (such as TGF-β and IL-6) and the development of severe disease. High concentrations of TGF- β, and IL-6 were identified in severe patients of COVID-19 recently admitted to the ICU, found by Agrati et al. [34]. In addition, the study conducted by Ghazavi et al. [21] identified a relationship between IL-17 and TGF-β.
Dysregulation of neutrophil activity in COVID-19 has been identified as part of the pathophysiology of the disease [35]. In severe disease, the elevated number of neutrophils and their exacerbated activity is part of the complex phenomenon of dysregulation of the immune response leading to multisystem inflammatory disorder [36]. The interaction between neutrophils and Th17 cells may guide us in understanding the mechanisms involved in neutrophil dysregulation.
The interaction between Th17 and neutrophils, particularly in chemotaxis, facilitates mutual cell migration. Th17 cells can participate in neutrophil chemotaxis via C-X-C motif chemokine ligand 8 (CXCL8) [5]. In addition, chemokine secretion by epithelial and endothelial cells mediated by IL-17A and IL-17F derived from Th17 cells also stimulates neutrophil chemotaxis [7]. In turn, neutrophils participate in Th17 chemotaxis by releasing C-C motif chemokine ligand 2 (CCL2) and CCL20, showing a mutual collaboration [37].
Aberrant cytokine release by neutrophils may exacerbate the inflammatory process (Figure 3) in COVID-19 [38]. In addition, excessive production of neutrophil extracellular traps (NETs) may cause tissue damage [37, 39]. This process is accompanied by excessive neutrophil infiltration into the alveoli, leading to tissue damage by enzymatic release and production of ROS [40].
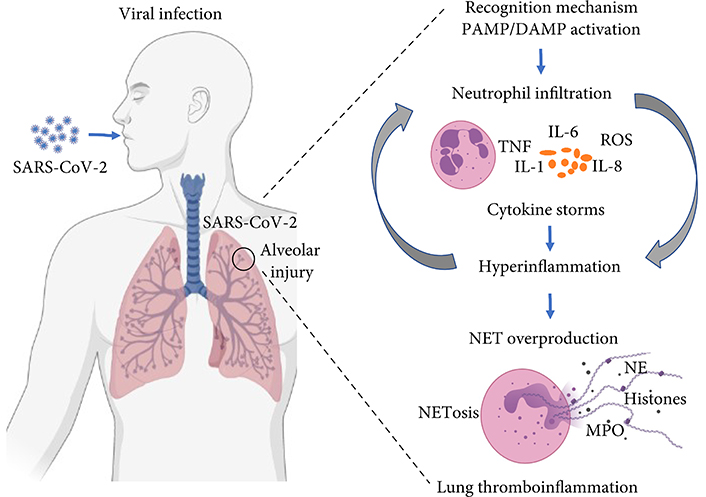
Dysregulation of neutrophil activity in COVID-19. In severe cases of the disease, neutrophils release excessive cytokines, and ROS, as well as produce extracellular neutrophil traps, leading to an exacerbated inflammatory response and tissue damage. NE: neutrophil elastase; PAMP: pathogen-associated molecular pattern; DAMP: danger-associated molecular pattern; MPO: myeloperoxidase
Note. Reprinted from “COVID-19 and neutrophils: the relationship between hyperinflammation and neutrophil extracellular traps,” by Borges L, Pithon-Curi TC, Curi R, Hatanaka E. Mediators Inflamm. 2020;2020:8829674 (https://www.hindawi.com/journals/mi/2020/8829674/). CC BY.
One of the leading actions to address the COVID-19 pandemic is the development of therapeutic options. Many research efforts into the behavior of SARS-CoV-2 and the characteristics of COVID-19 have led to the development and subsequent approval of several therapeutic options. Currently, the treatment for COVID-19 relies on two main approaches. The first one involves targeting the specific agent responsible for the disease, SARS-CoV-2, through the use of antivirals like Remdesivir. The second approach focuses on mitigating the harmful effects of immune response dysregulation. This can be achieved through therapies involving corticosteroids (e.g., dexamethasone) or biologics (e.g., baricitinib) [41].
Biological therapy is founded upon our understanding of immune response dysregulation. For instance, extensive research has been conducted on the use of monoclonal antibodies targeting key pro-inflammatory cytokines such as IL-6 and TNF-α [42–44]. Nevertheless, the heterogeneity of the disease highlights the importance of continuing research focused on therapeutics. Therefore, understanding the involvement of Th17 cells and IL-17 in the disease has led to the proposal of potential therapeutic options [45].
One of the proposed therapies targets the Janus kinase (JAK)/STAT pathway, which is key in Th17 cell differentiation and M2 macrophage polarization [46, 47]. The meta-analysis performed by Wijaya et al. [48] showed that, in five studies, with a total of 1,190 patients, the use of JAK inhibitors was associated with a reduction in mortality (OR: 0.51, 95% CI: 0.28–0.93, P = 0.02; I2: 7.8%, P = 0.354). The use of monoclonal antibodies against IL-17, such as netakimab, has also been proposed as a therapeutic option [49]. Finally, IL-6 blockade has been one of the most extensively studied therapeutic options [50]. The fact that this cytokine is part of the exacerbated inflammatory response in COVID-19 has been the main key to the study of IL-6 blockers such as tocilizumab and sarilumab [51]. However, the direct involvement of IL-6 in hyperinflammation is not the only consideration for the use of blockers. This is because IL-6’s involvement in the differentiation of Th17 lymphocytes indicates that it might also mitigate abnormalities arising from an impaired Th17 response [52].
It has been well established that Th17 cells are a subgroup of Th cells that are relevant for host defense against fungal and bacterial infections by means of IL-17, which is a very relevant cytokine for clearing extracellular pathogens. However, inappropriate signaling of this cytokine has been linked to ARDS, a major inducer of mortality in the setting of many infections. This may be due to increased alveolar endothelial and epithelial damage with resultant hypoxemia. Increased Th17 activity and IL-17 signaling, or unregulated Th17 responses may play a role in the chronic inflammation of COVID-19. Given the high morbidity and mortality seen in critically ill patients with COVID-19, new candidates to target are needed. Th17/IL-17 is a duet relevant that should be targeted in this illness.
ARDS: acute respiratory distress syndrome
COVID-19: coronavirus disease 2019
IL-1: interleukin-1
ROS: reactive oxygen species
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
STAT3: signal transducer and activator of transcription 3
TGF-β: transforming growth factor-beta
Th17: T helper 17
TNF-α: tumor necrosis factor-alpha
Treg: regulatory T
MAPV and NEHG equally contributed to: Conceptualization, Writing—original draft, Visualization. Both of the authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

