 Original Article
Original Article

Affiliation:
1Department of Medical Sciences, Division of Health Sciences, Leon Campus, University of Guanajuato, Leon, Gto. 37320, Mexico
ORCID: https://orcid.org/0000-0002-6815-7472
Affiliation:
2Department of Genetic Engineering, CINVESTAV Irapuato Unit, Irapuato, Gto. 36824, Mexico
ORCID: https://orcid.org/0000-0002-2933-4720
Affiliation:
3Centro de Investigaciones en Óptica, Asociación Civil, Leon, Gto. 37000, Mexico
ORCID: https://orcid.org/0000-0001-8347-8111
Affiliation:
2Department of Genetic Engineering, CINVESTAV Irapuato Unit, Irapuato, Gto. 36824, Mexico
ORCID: https://orcid.org/0000-0001-6179-3233
Affiliation:
1Department of Medical Sciences, Division of Health Sciences, Leon Campus, University of Guanajuato, Leon, Gto. 37320, Mexico
ORCID: https://orcid.org/0000-0002-5497-1035
Affiliation:
4Department of Medicine, Samuel Oschin Cancer Research Center, Los Angeles, CA 90048, USA
ORCID: https://orcid.org/0009-0001-2978-3438
Affiliation:
5Department of Medicine and Department of Biomedical Sciences, Research Division in Immunology, Cedars-Sinai Cancer Institute, Smidt Heart Institute, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA
ORCID: https://orcid.org/0000-0001-9872-9905
Affiliation:
2Department of Genetic Engineering, CINVESTAV Irapuato Unit, Irapuato, Gto. 36824, Mexico
ORCID: https://orcid.org/0000-0003-2231-5014
Affiliation:
2Department of Genetic Engineering, CINVESTAV Irapuato Unit, Irapuato, Gto. 36824, Mexico
Affiliation:
1Department of Medical Sciences, Division of Health Sciences, Leon Campus, University of Guanajuato, Leon, Gto. 37320, Mexico
ORCID: https://orcid.org/0009-0006-0946-7661
Affiliation:
2Department of Genetic Engineering, CINVESTAV Irapuato Unit, Irapuato, Gto. 36824, Mexico
ORCID: https://orcid.org/0000-0003-3556-8520
Affiliation:
1Department of Medical Sciences, Division of Health Sciences, Leon Campus, University of Guanajuato, Leon, Gto. 37320, Mexico
Email: szaina@ugto.mx
ORCID: https://orcid.org/0000-0001-6589-076X
Explor Cardiol. 2024;2:49–66 DOI: https://doi.org/10.37349/ec.2024.00021
Received: December 28, 2023 Accepted: February 27, 2024 Published: April 10, 2024
Academic Editor: Maria Grazia Andreassi, CNR Institute of Clinical Physiology, Italy
Aim: The DNA of the atheroma is hypermethylated relative to adjacent healthy vascular tissue. A significant portion of hypermethylated loci in the atheroma DNA map to genes related to macrophage function. Reversing macrophage DNA methylation to physiological levels by targeting DNA methyltransferase (DNMT) activity may therefore slow atherogenesis. Here, the anti-inflammatory and anti-atherogenic activity of macrophage-targeted DNMT inhibitor SGI-1027 were tested.
Methods: SGI-1027 was encapsulated into human serum albumin (HSA) nanoparticle (HSANP) functionalized with the PP1 peptide, a macrophage scavenger receptor 1 ligand, fused to a FLAG epitope (S-HSANP-FLAGPP1).
Results: Nanoparticle physico-chemical characteristics predicted good marginalization towards the vascular wall, although SGI-1027 encapsulation efficiency was relatively low (~23%). S-HSANP-FLAGPP1 were rapidly internalized compared to non-functionalized and, surprisingly, functionalized void controls, and induced a shift towards an anti-inflammatory profile of secreted cytokines in human THP-1 macrophages. S-HSANP-FLAGPP1 colonized the atheroma and induced a significant ~44% reduction of atherosclerosis burden in the aortic tree of apolipoprotein E (ApoE)-null mice compared to controls. A reduction in aortic root atherosclerosis was observed, although primarily induced by HSANP irrespective of loading or functionalization. No alteration of body weight, non-vascular tissue gross histology, plasma glucose, triglyceride or cholesterol were observed. HSA whether free or structured in nanoparticles, induced a 3–4-fold increase in high-density lipoprotein (HDL) compared to vehicle.
Conclusions: Unexpectedly, effects that were likely non-epigenetic and induced by HSA per se were observed. HSANP loaded with SGI-1027 were anti-atherogenic but in an anatomical location-dependent fashion. SGI-1027 displayed a novel anti-inflammatory activity in non-proliferating THP-1 cells, implying that those effects are likely unrelated to DNMT inhibition. HSA elevated HDL per se, thus underlining a possible additional advantage of HSA-based nanocarriers.
Atherosclerosis is an inflammatory disorder of the arterial wall and an underlying cause of cardiovascular disease (CVD). One of the pivotal events in atherosclerosis is the infiltration of circulating monocytes into the vascular wall. Once infiltrated, monocytes differentiate into macrophages, which contribute to the onset and maintenance of chronic inflammation [1]. Clinical complications of CVD—myocardial infarction, stroke, peripheral arterial disease—are significant causes of death and disability worldwide [2, 3]. Pharmacological and lifestyle-induced reduction of hyperlipidaemia, a major risk factor for atherosclerosis, have successfully lowered the incidence of clinical complications of CVD [4]. Yet, a sizeable residual CVD risk persists after correction of hyperlipidaemia, thus underscoring the need to test candidate additional therapeutic strategies [5, 6]. Modulation of gene expression by targeting the DNA methylome is among such therapeutic strategies. DNA methylation is a reversible chemical modification of the genome that participates in transcriptional regulation [7, 8]. The most abundant methylated base is cytosine (5-methylcytosine) in a 5’-CG-3’ (CpG dinucleotide) context in mammalian DNA. 5-Methylcytosine is produced by three catalytically active DNA methyltransferases (DNMTs; DNMT1, DNMT3a and DNMT3b), whereas DNA demethylation is operated by a combination of ten-eleven translocation (TET) dioxygenases (TET1, TET2 and TET3)-catalysed 5-methylcytosine oxidation and DNA damage repair in mammals [9, 10]. Complex deviations from physiological DNA methylation profiles have been observed in the atherosclerotic artery of animal models and humans. On the one hand, the DNA of affected artery is hypomethylated when compared with different vascular beds within the same individual or when the same vascular bed is compared between individuals [11–13]. On the other hand, when donor-matched and vascular bed-matched atherosclerotic and histologically normal arteries are compared, DNA hypermethylation is observed of the diseased samples [14–16]. These inconsistencies likely reflect poorly understood dynamic changes in DNA methylation during early atherogenesis [17]. Nevertheless, growing evidence indicates that DNA hypermethylation is a landmark and potential therapeutic target in atherosclerosis. Atherogenic lipoproteins and proinflammatory fatty acids induce DNA hypermethylation in cellular and animal models [18–20]. A comparison of twins discordant for CVD identified DNA hypermethylation of several relevant loci in the affected sibling [21]. Exposure to disturbed blood flow, a landmark of proatherogenic vascular microenvironment, induces DNMT expression in endothelial cells [22]. Furthermore, consistent changes were observed in the DNA demethylase TET2 in atherosclerosis: TET2 expression is decreased during neointimal formation in vivo and by oxidised low-density lipoprotein (LDL) in cellular models [23, 24]. Importantly, a wealth of experimental data assigns a causal role to DNA hypermethylation in atherogenesis. DNMT1 overexpression accelerates atherosclerosis in apolipoprotein E (ApoE)-null mice [25]. Conversely, DNMT inhibition by systemic administration of the nucleotide analogue azacytidine decreases atherosclerosis in mouse models [26–28]. Azacytidine was singled out among >3,000 drugs that promote a contractile, non-atherogenic smooth muscle cell (SMC) phenotype [29]. TET2 overexpression, which is expected to decrease global DNA methylation, is anti-atherogenic [30]. Notably, a set of hypermethylated loci in atheroma DNA map to genes related to macrophage function [14]. Taken together, those findings underline the potential of macrophage-targeted DNMT inhibitors as therapeutics for atherosclerosis. A minimal requirement for the use of DNMT inhibitors in vivo, is effective targeting to the atheroma to avoid widespread DNA hypomethylation and dysregulated gene expression in non-target organs.
Here, a nanoparticle (NP)-based strategy to target a DNMT inhibitor to the atheroma was designed. Human serum albumin (HSA) NP (HSANP) were chosen. HSA is an endogenous protein and building block for the synthesis of highly versatile NP with low immunogenicity that are capable of targeting macrophages [31, 32]. A further reason to use HSA is to simplify the transition to future human studies. Among the available DNMT inhibitors, the non-nucleoside analogue SGI-1027 (N-(4-(2-Amino-6-methylpyrimidin-4-ylamino)phenyl)-4-(quinolin-4-ylamino) benzamide; CAS no. 1020149-73-8) was chosen based on the following rationale: firstly, nucleoside analogues such as the widely used azacytidine and decitabine were avoided, as these act subsequent to incorporation into newly synthesized DNA in replicating cells, thus potentially affecting genome integrity, cell survival and plaque stability [33, 34]; additionally, SGI-1027 is a suitable load for HSANP due to its hydrophobicity [31]. NP were functionalized with the short synthetic peptide PP1, a ligand of human and mouse scavenger receptor 1 (MSR1) for macrophage targeting [35]. The findings are discussed in the context of existing literature and the foreseeable epigenetic therapies for CVD.
HSANP were synthesized by the desolvation method as previously described [36]. Briefly, 100 mg/mL HSA (Sigma-Aldrich, no. A3782) in 10 mmol/L NaCl, pH 9.4 was stirred using an ULTRA-TURRAX® (IKA) system at 200 rpm for 30 min at room temperature. Cross-linking was carried out by adding 3 mL 4% aqueous glutaraldehyde (Merck, no. G7651) at 0.5 ml/min rate under stirring at 150 rpm for 30 min. The resulting NP were purified by 3 cycles of differential centrifugation (10,000 rpm, 7 min). The pellet was dissolved to the original volume in 10 mmol/L NaCl, pH 9.4. SGI-1027-loaded HSA NP (S-HSANP) were obtained by adding 1 mg/mL SGI-1027 in ethanol (1 ml/min rate) with constant stirring at 160 rpm before the cross-linking step. To determine microencapsulation and SGI-1027 loading efficiency, NP were centrifugated at 10,000 rpm for 5 min, the supernatant was discarded and replaced by the same volume of high-pressure liquid chromatography (HPLC)-grade absolute ethanol. Loaded SGI-1027 was extracted from NP by sonication for 15 min, pause for 30 min, further sonication for 10 min and centrifugation at 10,000 rpm for 5 min. The pellet was used to determine HSA microencapsulation efficiency and the supernatant was used to determine SGI-1027 loading efficiency and loading capacity. To measure microencapsulation efficiency, the pellet was resuspended to the original volume in deionized distilled water (ddH2O), transferred to a microtube of known weight, centrifugated at 10,000 rpm for 5 min and the supernatant discarded. The pellet was dried with a MAXI-dry vacuum centrifuge (Heto-Holten) at 1,300 rpm, 30°C for 45 min and weighed to obtain the total amount of HSA incorporated into NP. Microencapsulation yield was calculated as percent HSA in NP of initial amount of HSA used to prepare NP. Free SGI-1027 in the supernatant was determined by HPLC fractionation with a ZORBAX Eclipse Plus C18 column (pore size 130 Å, 1.8 μm, 2.1 mm × 50 mm; Agilent) with a reverse phase of CH3OH, 0.1% trifluoroacetic acid (TFA, HPLC-grade, Sigma, no. 302031)/H2O, 0.1% TFA (v/v) and ultraviolet (UV) detection at 360 nm. Loaded SGI-1027 was quantified against pure standards. Loading efficiency was calculated as percent loaded (i.e., total – free) of total SGI-1027. Loading capacity was the percent loaded SGI-1027 of NP weight.
Following synthesis and loading with SGI-1027, NP were functionalized with the FLAGPP1 fusion peptide, containing in N-terminus to C-terminus order: a 3-glycine spacer, a FLAG epitope (italics) and the PP1 peptide (underlined): GGGDYKDDDDKLSLERFLRCWSDAPA (> 95% purity; Biomatik) [35]. Functionalization was carried out as previously described with minimal modifications [37]. Briefly, 800 μL FLAGPP1 (5 mg/mL in ddH2O) were incubated with 200 μL N-(3-dimethylaminopropyl)-N-ethylcarbodiimide in the dark for 15 min at 24°C under constant shaking at 700 rpm on a benchtop mixer. One mL NP was added, and shaking was continued for 1 h. The reaction was stopped by centrifugation at 300 rpm for 5 min. NP were purified by 3 cycles of centrifugation (10,000 rpm, 7 min), resuspended to the original volume in ddH2O. Functionalization was verified by immunogold labelling with InnovaCoat GOLD (Expedeon, 40 nm). The conjugation solution was prepared according to the manufacturer’s instructions with the anti-FLAG monoclonal antibody (FG4R; ThermoFisher Scientific, no. MA1-91878) at 1:3,000 dilution. One mL functionalized NP was centrifugated at 10,000 rpm for 5 min. The pellet was resuspended in 1 mL conjugation solution, incubated for 30 min at 4°C under constant shaking and washed three times in distilled water by centrifugation at 10,500 rpm for 5 min. Antibody binding was revealed by transmission electron microscopy (TEM; Morgagni M-268, Philips/FEI). Samples were spotted on 200 mesh formvar/carbon coated copper grids (Ted Pella) and incubated for 10 min. Grids were dried at room temperature for 5 min. No positive contrast was used. Operating conditions were: 80 kV high voltage (EHT) in high magnification (magnification: 1,000–140,000×), working pressure 5 × 10–3 Pa (5 × 10–5 Torr). Micrographs were captured in TIFF format, 1,376 × 1,032 pixels size, grayscale. In this format, 0 was assigned to black and 255 to white. Image analysis was performed in ImageJ (https://imagej.net/software/imagej/, MacOS version) with shape descriptors plugin. FLAGPP1-functionalized NP loaded with SGI-1027 or void are henceforth referred to as S-HSANP-FLAGPP1 or HSANP-FLAGPP1, respectively. In all cases, NP were kept in refrigeration until used.
Morphometric analysis was performed by TEM. Seven μL resuspended NP were contrasted by adding 2.5% uranyl acetate (electron microscopy science) and incubated for 15 min. Sixty images of each NP version were analysed. Diameter, area, and perimeter were recorded.
ζ-Potential, polydispersity index (PI) and hydrodynamic ratio were obtained for technical triplicate and experimental duplicate, by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Panalytical), at room temperature and a scattering angle of 13° after a 1:10 dilution in distilled water, pH 7.4.
Typically, 1 × 106 human THP-1 monocytes (ATCC no. TIB-202™) were cultured in 60 mm × 15 mm cell culture dishes in RPMI medium (ThermoFisher, no. 11875093) supplemented with 10% foetal bovine serum (ThermoFisher, no. 16140071), 1% penicillin/streptomycin (ThermoFisher, no. 15140130), 2 mmol/L glutamine at 37°C, 5% CO2. THP-1 monocytes were differentiated to macrophages by supplementing the medium with 50 ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma, no. P8139) for 4 h, followed by fresh 25 ng/mL PMA until complete differentiation (~4 days). Cells were maintained in serum-free RPMI medium for 30 min before the uptake assay. NP were labelled with eosin Y, a high-affinity HSA binding dye HSA [38]. NP were incubated with 1 μl/mL eosin Y (Sigma, no. 230251) at 4°C for 15 min with gentle shaking, followed by three washes and centrifugation at 10,000 rpm for 5 min with distilled water. NP were sonicated for 5 min immediately before the assay. Following incubation with labelled NP, cells were washed three times with PBS, fixed with 4% paraformaldehyde (Sigma, no. 16005) for 20 min, and washed three times with PBS. Cell viability was determined by trypan blue (Sigma, no. T6146) exclusion. Cellular uptake of eosin Y-labelled NP was determined by multiphoton microscopy and flow cytometry. Prior to microscopy, cell membranes were stained with 5% SynaptoRed C2 (Sigma-Aldrich no. S6689) in ddH2O for 30 min. Several washes at room temperature were performed until no colour in solution was observed, followed by three washes in ice-cold PBS. Nuclei were stained with 1% 4’-6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich no. D9542) for 10 min. In all cases, staining was performed using the same protocol and reagent concentrations. All sample staining and washes were performed under gentle agitation. Multiphoton microscopy was performed immediately following staining. Detection of eosin Y-labelled NP emission spectrum was performed using the LSM 880-NLO multiphoton microscopy system (Zeiss) equipped with Ti:Sapphire laser (Chameleon Vision II, Coherent) capable of tuning in the 690–1,060 nm range. Images were acquired by separating the emission into three channels, blue or UV (371–440 nm), green/yellow (450–550 nm), and red (560–730 nm). The “lambda mode” scanning for detection of eosin Y-labelled NP spectral emission was performed by excitation at 810 nm. For SynaptoRed C2, an Argon laser was operated at 0.18% of power and 53 µm open pinhole with excitation at 488 nm and emission detection at 683 nm. Operating conditions for DAPI detection were Chameleon laser operated at 1.0% power with excitation at 710 nm and emission detection at 355 nm. Observation was carried out with immersion objective 60×/1.3, NA ∞–0.17, Plan-Neofluar (Zeiss).
For flow cytometry, NP-stimulated cells were harvested with 1 mL PBS, 5 mmol/L ethylenediaminetetraacetic acid (EDTA) at 4°C for 10 min with shaking. Cells were transferred to an Eppendorf tube and fixed with 2% paraformaldehyde for 15 min at room temperature. After three washes with PBS by centrifugation at 3,500 rpm for 5 min, cells were resuspended in PBS and kept in the dark until analysed. Labelled cells were detected using a 450–580 nm excitation-emission wave in a FACSCanto™ II (BD Bioscience) flow cytometer. The FlowJo software (v10.4.1) was used for analysis. Data were represented as the percentage of cells positive for eosin Y fluorescence per 106 cells.
Interleukin-1β (IL-1β), IL-6, IL-8, IL-10 and interferon α (IFNα) polypeptides were determined by immunocapture-based quantitative PCR (qPCR; PreciseDX Lab, Chihuahua, Chi., Mexico), a multiplex assay. Briefly, 200 μL medium were used for the analyte capture, using a 1:10 dilution of monoclonal antibodies (Santa Cruz Biotechnology): anti-IL-1β (H-153; sc-7884), anti-IL-6 (E-4; sc-28343), anti-IL-8 (C-11; sc-376750), anti-IL-10 (E-10; sc-8438), anti-IFNα (FL-189; sc-20106). After a 15 min incubation at room temperature, 400 μL PBS were added. Samples were centrifuged at 10,000 rpm for 5 min, the supernatant was discarded, and a proprietary secondary antibody coupled to a synthetic DNA fragment was added following manufacturer’s instructions (Thunder-Link Plus Oligo Conjugation System, Expedeon). After 2 washing cycles, qPCR was performed, using Phire Hot Start II DNA Polymerase Master Mix Green (F126L, Thermo Scientific) following a 3-step reaction cycle: initial denaturation at 98°C, 30 s; 30 cycles: 98°C for 5 s, 64°C for 5 s, 72°C for 5 s; final extension at 72°C for 60 s. Samples were quantified in triplicate.
In preliminary experiments aimed to detect subcutaneously injected HSANP in peripheral blood, whole tail blood and Tris-buffered saline (TBS) with 2% Tween (TBS-T; 2.5 μL each) were mixed and carefully spotted on a nitrocellulose membrane. After drying at room temperature, equal loading was estimated by Ponceau red (Merck, no. 09276) staining. Membranes were blocked with TBS-T, 5% fat-free milk for 1.5 h. After incubation with a 1:2,500 dilution of an anti-HSA, HRP-conjugated monoclonal antibody (ab24458, Abcam) for 30 min at room temperature, the membrane was washed three times with TBS-T, 0.1% fat-free milk for 30 min at room temperature. Antibody binding was revealed with ECL™ Prime Western Blotting Detection (Amersham). HSA was determined by comparison with a standard curve using free HSA and the average value at time 0 was subtracted from all other time point data.
High fat diet-fed nullizygous ApoE-null mice in a C57BL/6 background, a model of hyperlipidaemia-induced atherosclerosis, were used [39]. Mice were housed in groups of 4–5 in a controlled environment with 12 h day-light cycle and ad libitum access to food and water. Twenty-week-old female and male mice were fed for 8 weeks with a high fat diet (30% fat) that was elaborated by us. The rationale for not using a commercial diet was to avoid oxidation or microorganism contamination during the lengthy importation process and storage. We deemed those advantages to be reasonable trade offs, compared to the small batch-to-batch variation of the in-house diet, based on industrial, high-volume production lard. The high fat diet was elaborated by mixing pulverized standard chow (Rodent 5001, LabDiet) with pork lard (0.16:1 lard:chow ratio, w:w). The resulting paste was pelleted and stored at 4°C for no longer than 5 days before use. Mice were treated with HSANP (n = 20), HSANP-FLAGPP1 (n = 10), S-HSANP-FLAGPP1 (n = 20), vehicle (n = 20), or an unstructured mix of SGI-1027 and native HSA (SGI-HSA; n = 10) by weekly injections during the 8-week high-fat diet feeding. That sample size exceeded the one used in comparable studies where a 40% reduction of the size of atheroma—deemed here as the lowest biologically acceptable effect—was observed [27]. NP were sonicated for 5 min prior to injection. Overnight-fasting mice were sacrificed 1 week after the last injection. Glucose was measured in a tail blood drop (One Touch Ultra 2, Johnson & Johnson) immediately prior to sacrifice. Sacrifice was performed by inducing anaesthesia in isoflurane vapor in a glass container. As the mouse reached unconsciousness, decapitation was carried out. Total blood was collected at decapitation in EDTA (Vacutainer, BD) for plasma total cholesterol, triglycerides, and high-density lipoprotein (HDL) determination by the Clinical Analysis Laboratory, Department of Medical Sciences, University of Guanajuato, following standard colorimetric methods. The aortic root, the remaining aorta (arch, thoracic, abdominal to the caudal bifurcation), liver, kidney, adipose tissue, ventricular (lower) heart portion, were dissected and processed for downstream analysis.
Liver, kidney, adipose tissue, and ventricular heart portion were transferred to 300 μL RNAStill (RNA stabilization agent, RBC) at dissection, stored at –20°C and transferred to 1% formaldehyde. The aortic root was dissected, washed in PBS, and fixed in 1% formaldehyde. Fixation was for at least 24 h prior to paraffin embedding. Serial sections (6 µm) of the aortic root were stained with haematoxylin and eosin (HE), Masson trichrome, or periodic acid-Schiff-staining (PAS) according to standard protocols. Three sections were used to determine cellularity, basal lamina, and collagen using a U-DO3 microscope (Olympus Optical CO, LTD); images were digitalized and examined with ImageJ software (https://imagej.net/software/imagej/). For this purpose, all images were taken at 20×, and the regions of interest were traced where loss of continuity of media smooth muscle was observed. Original images were converted to 8 bits, and a threshold was obtained. This shadow was overlaid on the original image, to visually separate the cells. Cellularity and foam cell counting were limited for those with complete nuclei and identifiable cytoplasm. PAS-stained sections were used for the determination of the lesion area, using the ImageJ software version 1.52 (NIH, USA). Smooth muscle was stained with an anti-smooth muscle actin (SMA) antibody (rabbit-anti-mouse, Abcam, no. 5694). Briefly, samples were dewaxed by heating at 65°C for 1 h, rehydrated in a xylene-ethanol train [three changes of xylene during 15 min each, one change of xylene-ethanol (1:1), two changes of 100% ethanol, one change of 95% ethanol and two changes of 70% ethanol during 5 min each], and washed with ddH2O. Antigen retrieving was performed by heating the samples in a 1× citrate solution (Electron Microscopy Sciences, no. 64142-08) at 94°C for 45 min. After cooling, samples were washed twice with PBS for 15 min, and endogenous peroxidase was blocked by incubation in 3% H2O2 (Merck, no. 516813). Samples were washed in PBS and then in PBS, 0.05% Triton X-100 (PBST) for 10 min. Samples were blocked with a 5% BSA solution at 4°C for 45 min. After blocking, samples were incubated with the anti-SMA primary antibody (1:200) diluted in PBST, 1% BSA, at 4°C overnight. Samples were rinsed twice with PBS for 15 min and with PBST for 5 min, followed by incubation with an anti-rabbit-HRP secondary antibody (GeneTex-GTX 83399 OneStep Polymer HRP) according to manufacturer’s instructions. Staining was performed with a DAB substrate system (Abcam, no. ab64238) according to manufacturer’s instructions, followed by washing in ddH2O for 5 min. Counterstaining was performed using haematoxylin (Sigma-Aldrich, no. HHS32) and a final incubation in 0.25% ammonium water, followed by dehydration (70% ethanol for 5 min, three times; 100% ethanol for 5 min, two times; and three changes of xylene for 5 min). Samples were mounted using Permount mounting medium solution (Fisher Chemical, SP15). All images were acquired with a Nikon Eclipse 80i microscope or EVOS Auto FL2. Liver, kidney, and heart tissue sections (7 µm) were stained with HE and examined by a trained pathologist. For the assessment of the aortic lesion area, whole mount oil red O (ORO) staining was performed essentially as described [40]. Images were digitalized and the lesion area was calculated using the ImageJ software. The lesion area was expressed as a percentage of the area of lesions (dark red), relative to the total surface area of the aortic lumen. For NP detection in the aorta, 6 µm paraffin sections of the aortic root of mice treated with vehicle or S-HSANP-FLAGPP1 were deparaffinized and prepared as stated for immunohistochemistry (IHC). Immunogold was used for the detection of the FLAG epitope in S-HSANP-FLAGPP1, using an anti-FLAG antibody coupled to 40 nm gold NP. Immunogold was prepared as described in the previous section and incubated with the tissue overnight at 4°C. After three 10-min washes with PBS, samples were stained with DAPI (1:1,000, 20 min), followed by two washes with PBS. Samples were mounted using Permount mounting media. Images were acquired in a Leica Stellaris 8-STED Confocal Microscope (60× oil-objective) by separating the emission in two channels, blue for DAPI (400–450 nm), and red for gold (590–650 nm). Gold-labelled NP were excited at 530 nm. At the sites with signal detection, zoom-in (3.0) was performed, and 14 consecutive images were acquired along the Z-axis. A three-dimensional image of the signal detected was extracted from one of these Z-stacks. Images were collected and further analysed with the FIJI software (https://imagej.net/software/imagej/, Windows version). Contrast and colour adjustment were performed using the same parameters across all images.
The data were tested for normal distribution using the Kolmogorov-Smirnov with Lilliefors correction test or Shapiro-Wilk test accordingly. The Kruskal Wallis test with Bonferroni for multiple comparisons post hoc or Mann-Whitney U test were used for non-normally distributed data. Paired Student’s t test and ANOVA with Bonferroni post hoc were used in the case of normal distribution. Significance threshold was set at P = 0.05 (two-tailed). Tests were performed with SPSS v20 (IBM).
TEM and DLS analysis showed the following characteristics for HSANP: ~300 nm or ~100 nm diameter, as determined by TEM or DLS analysis, respectively; acceptable size homogeneity; markedly negative surface charge (Table 1). Loading with SGI-1027 did not significantly affect any of those parameters (see Figure S1 for representative chromatograms of SGI-1027 determination by HPLC). Functionalization with FLAGPP1 significantly increased NP hydrodynamic diameter by ~3-fold when HSANP and S-HSANP-FLAPP1 were compared (P = 0.022) and promoted heterogeneity in NP size and ζ-potential (P = 0.016). Gross parameters dependent on the structure of the NP per se were not measured in unfunctionalized NP. Representative TEM images of functionalized and control NP are shown in Figure S2. HPLC-assessed efficiency of SGI-1027 loading onto S-HSANP-FLAGPP1 was ~23%. As for S-HSANP-FLAGPP1 composition, the SGI-1027:HSA ratio (µg:g) was ~0.9.
NP physicochemical characteristics
| Variable | HSANP | S-HSANP | P |
|---|---|---|---|
| Diameter (nm)a | 308.3 ± 106.8 | 311.7 ± 109.7 | n.s. |
| Roundnessa | 0.8 ± 0.2 | 0.8 ± 0.1 | n.s. |
| Shape factora | 0.5 ± 0.1 | 0.4 ± 0.1 | n.s. |
| Hydrodynamic diameter (nm)b | |||
| Not functionalized | 104.2 ± 14.4 | 141.8 ± 4.6 | n.s. |
| Functionalized with FLAGPP1 | 296.4 ± 72.6 | 447.1 ± 131.2 | 0.028 |
| Polydispersity indexb | |||
| Not functionalized | 0.5 ± 0.1 | 0.6 ± 0.1 | n.s. |
| Functionalized with FLAGPP1 | 0.1 ± 0.1 | 0.9 ± 0.1 | 0.013 |
| ζ-Potentialb | |||
| Not functionalized | –43.7 ± 0.5 | –42.2 ± 0.3 | n.s. |
| Functionalized with FLAGPP1 | –39.5 ± 1.3 | –44.6 ± 0.8 | n.s. |
| Microencapsulation efficiency (%)c | 55.5 | 56.0 | n.s. |
| Loading efficiency (%)c | Not applicable | 22.6 | n.s. |
Values are mean ± SD. HSANP and S-HSANP, void and SGI-1027-loaded NP, respectively. a n = 60 individual NP per group; b means of technical triplicate measure; c mean from a technical duplicate measure. Kruskal-Wallis test with Bonferroni correction and multiple comparisons post hoc. n.s.: statistically not significant
No significant effect of NP on cell viability was observed (Figure S3). This observation supports our initial hypothesis that the chosen dose of SGI-1027 is within the non-toxic range. Flow cytometry and fluorescence microscopy analysis of NP-challenged THP-1 macrophages revealed a marked, incubation time-dependent uptake of FLAGPP1-functionalized NP in the order S-HSANP-FLAGPP1 > HSANP-FLAGPP1, with readily detectable uptake of S-HSANP-FLAGPP1 at the 15 min timepoint. By contrast, HSANP uptake was comparatively slow (Figure 1A and B; Figure S4). Internalized NP were mostly in cytoplasmic droplet-like structures, although optical section data were consistent with nuclear localization of a subset of S-HSANP-FLAGPP1 (Figure 1C).
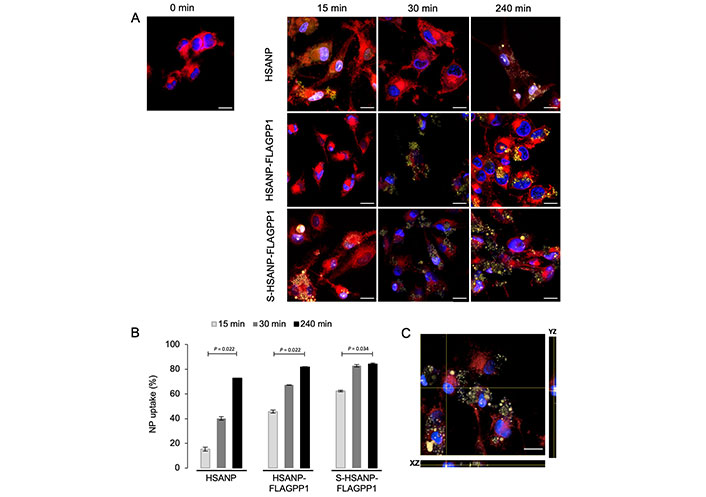
NP uptake by THP-1 macrophages. A. Representative images of fluorescence microscopy analysis of cells challenged for the indicated time lengths. Blue: DAPI (nuclei); red: SynaptoRed C2 (cell membranes); yellow: eosin Y-labelled NP. Bar = 16 µm; B. flow cytometry-based analysis of NP uptake by THP-1 macrophages treated as in A. Notice the comparatively faster S-HSANP-FLAGPP1 uptake, n = 3. Kruskal-Wallis test with pairedcomparisons test post hoc; C. confocal microscopy-generated single-cell orthogonal view of eosin Y fluorescence. Notice the presence of eosin Y fluorescence within the nucleus. Bar = 16 µm. HSANP: void NP; HSANP-FLAGPP1: functionalized void NP; S-HSANP-FLAGPP1: SGI-1027-loaded and functionalized NP
NP elicited distinct effects on the secreted cytokine profile. HSANP-FLAGPP1 and HSANP induced weak changes, whereas S-HSANP-FLAGPP1 modestly increased IL-1β (~50%), but markedly (2–4-fold) augmented IL-10, IL-8 and IFNα in a time-dependent fashion (Figure 2). S-HSANP-FLAGPP1 induced a 2.7-fold higher IL-10/IL-1β ratio compared to HSANP-FLAGPP1 or HSANP, suggestive of a net anti-inflammatory effect (Figure 2). Selected cytokines (IL-10, IFNα) show inconsistent values at the 30 min timepoint, yet given the relatively small variation and the fact that all cytokines were measured in parallel, the observed imperfect time-dependent response is likely due to unidentified biological underlying causes. Indeed, cases of non-linear time-dependent cytokine expression and secretion have been observed in several models (see for example: [41–44]).
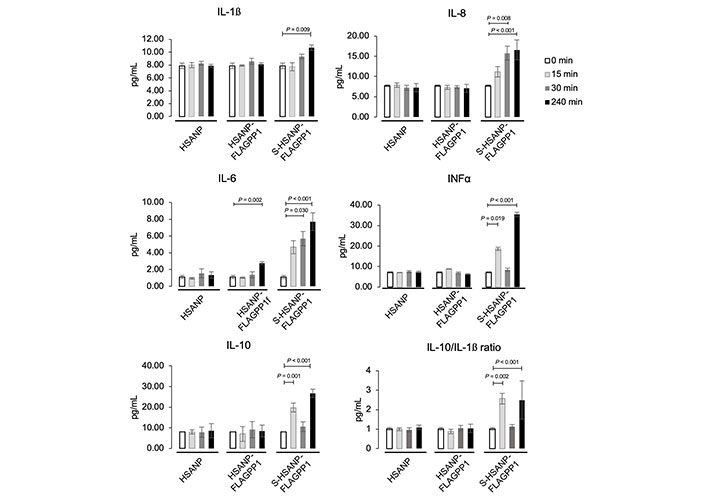
NP-induced modulation of secreted cytokine profile in THP-1 macrophages. Data are mean ± SD. HSANP, void NP; HSANP-FLAGPP1, functionalized void NP; S-HSANP-FLAGPP1, SGI-1027-loaded and functionalized NP (n = 3). Kruskal-Wallis test, paired-comparisons-Bonferroni adjusted post hoc
Since repeated injection into the tail vein poses ethical and practical issues in dark-skinned mice, alternative administration routes were first assessed. Intraperitoneal administration was discarded based on concerns that peritoneal macrophages would sequester FLAGPP1-functionalized NP. Therefore, the feasibility of the subcutaneous route was tested, based on the successful reduction of intimal hyperplasia by subcutaneously injected polymeric NP [45]. Six-week-old female C57BL/6 mice were injected below the dorsal neck skin with 150 μL HSANP (1 mg protein/mL in PBS) or PBS (n = 3 each). Although ApoE-null mice for subsequent NP testing (see below) were used, the subcutaneous route was evaluated in WT mice based on animal availability. To our knowledge, it has not been determined whether capillary permeability is affected in ApoE-null mice. If anything, hyperlipidaemia-associated endothelial dysfunction may facilitate NP perfusion through the capillary endothelium of subcutaneous tissue [46]. Fifty μL tail tip peripheral blood were obtained immediately before the injection (time 0) and at four time points up to 48 h post-injection. A significant increase in HSA with post-injection time was detected in peripheral blood (P = 2.3 × 10–4, ANOVA and Bonferroni post hoc test) (Figure S5A). Detected HSA at 48 h corresponded to ~20% of the total injected NP.
A weekly, single-dose protocol for in vivo administration of NP to ApoE-null mice was designed. The protocol aimed to achieve a 7 µmol/L SGI-1027 concentration in the total peripheral blood volume calculated based on recipient mouse body weight (BW). That SGI-1027 concentration is within the reported 6–13 µmol/L IC50 range and below the 20 µmol/L toxic dose observed in rat hepatocarcinoma cells [47, 48]. A schematic view of the protocol of ApoE-null mice treatment with NP is shown in Figure 3A.
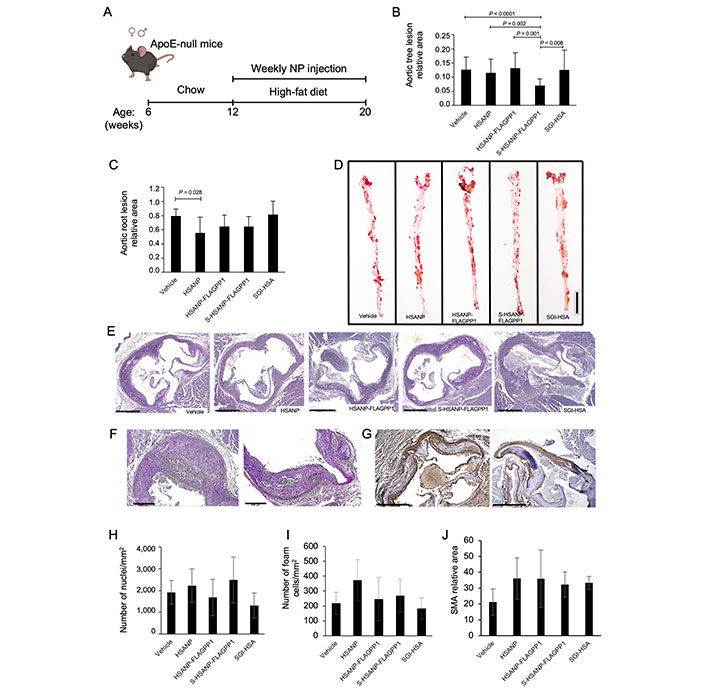
Effects of NP administration in vivo. A. Schematic view of the experimental design of ApoE-null mice treatment with NP (mouse drawing: BioRender); (B, C). quantitative analysis of atheroma size in whole mount aortas (n = 20 per group) and aortic roots (n = 10 per group), respectively; D. representative images of ORO-stained atherosclerotic lesions in whole mount aortas; (E–G). representative images of PAS atherosclerotic lesions in aortic roots. Representative images used for nuclei, foam cell and SMA quantification (PAS staining, and SMA-IHC); (H–J). cellularity (n = 10 per group), foam cell (n = 10 per group) and SMA (n = 5 per group) abundance. Graph data are mean ± SD. Kruskal-Wallis test, Bonferroni post hoc or one-way ANOVA test with Bonferroni post hoc. HSANP: void NP; HSANP-FLAGPP1: functionalized void NP; S-HSANP-FLAGPP1: SGI-1027-loaded and functionalized NP; SGI-HSA: unstructured mixture of SGI-1027 and native HSA. Bars in D, E, F and G are 5 mm, 500 μm, 200 μm and 500 μm, respectively
The effects of NP (HSANP, HSANP-FLAGPP1, S-HSANP-FLAGPP1), and SGI-1027 mixed with free HSA (SGI-HSA) on general well-being, metabolic parameters, and non-vascular tissue gross morphology were assessed first. No obvious alteration of mobility or fur appearance was detected. No significant change in BW was observed at treatment endpoint, compared to first day of treatment (Table 2). In one case, a tangible lump was detected at the injection site. The treatment did not elicit any statistically significant change in plasma glucose, total cholesterol, or triglycerides. Conversely, all treatments induced a ~3–4-fold increase in plasma HDL relative to vehicle, although differences reached statistical significance only for HSANP-FLAGPP1 and SGI-HSA.
BW, serum glucose and lipid profile of NP-treated ApoE-null mice
| Treatment | Sex (n) | Initial BW (g) | Final BW (g) | Glucose (mg/dL) | Cholesterol (mg/dL) | Triglycerides (mg/dL) | HDL (mg/dL) |
|---|---|---|---|---|---|---|---|
| Vehicle | F (10) | 29.1 ± 3.4 | 31.0 ± 3.3 | 143.2 ± 18.0 | 434.4 ± 65.4 | 152.8 ± 72.8 | 2.7 ± 2.6 |
| M (10) | 31.5 ± 2.3 | 31.5 ± 4.6 | 149.2 ± 41.9 | 448.9 ± 95.2 | 182.0 ± 78.5 | 8.4 ± 3.3 | |
| Total (20) | 30.3 ± 2.9 | 31.3 ± 3.9 | 146.2 ± 29.9 | 441.6 ± 80.3 | 167.4 ± 75.7 | 5.6 ± 4.1 | |
| HSANP | F (10) | 28.4 ± 1.9 | 31.2 ± 3.6 | 143.9 ± 22.3 | 506.3 ± 139.3 | 169.1 ± 45.6 | 15.5 ± 9.2 |
| M (10) | 33.3 ± 1.6 | 34.5 ± 2.0 | 137.9 ± 9.5 | 446.8 ± 96.1 | 163.1 ± 41.2 | 15.4 ± 1.2 | |
| Total (20) | 30.8 ± 1.8 | 32.9 ± 2.8 | 140.9 ± 15.9 | 476.5 ± 11.7 | 166.1 ± 43.4 | 15.4 ± 12.3 | |
| HSANP-FLAGPP1 | F (5) | 31.1 ± 1.7 | 36.3 ± 30.7 | 175.0 ± 37.4 | 539.3 ± 224.2 | 250.1 ± 216.5 | 19.7 ± 6.3 |
| M (5) | 29.5 ± 1.6 | 30.7 ± 2.4 | 127.0 ± 19.4 | 394.8 ± 107.2 | 125.6 ± 56.2 | 14.2 ± 8.1 | |
| Total (10) | 30.3 ± 1.6 | 33.5 ± 2.9 | 151.0 ± 28.3 | 467.1 ± 165.7 | 187.8 ± 136.3 | 16.9 ± 11.6 (P = 0.028) | |
| S-HSANP-FLAGPP1 | F (10) | 27.0 ± 3.9 | 29.0 ± 3.7 | 148.7 ± 20.8 | 464.1 ± 108.3 | 147. ± 61.2 | 15.4 ± 11.8 |
| M (10) | 32.3 ± 3.5 | 31.7 ± 4.2 | 144.3 ± 28.1 | 489.1 ± 215.9 | 161.6 ± 46.1 | 16.3 ± 2.1 | |
| Total (20) | 29.6 ± 3.7 | 30.3 ± 4.0 | 146.5 ± 24.5 | 154.7 ± 53.6 | 476.6 ± 162.1 | 15.9 ± 13.8 | |
| SGI-HSA | F (5) | 29.6 ± 2.9 | 33.8 ± 2.2 | 177.0 ± 38.4 | 555.4 ± 300.9 | 261.2 ± 197.2 | 22.7 ± 13.5 |
| M (5) | 28.2 ± 2.1 | 31.6 ± 2.5 | 131.8 ± 23.0 | 311.7 ± 239.3 | 111.8 ± 91.6 | 12.0 ± 8.6 | |
| Total (10) | 28.9 ± 2.5 | 32.7 ± 2.3 | 154.4 ± 30.7 | 433.6 ± 270.1 | 186.5 ± 144.4 | 17.3 ± 15.4 (P = 0.020) |
F: female; M: male. Data are mean ± SD. Significance refers to comparison with vehicle control. Only significant P values are shown. ANOVA, Bonferroni post hoc test
The ability of NP to accumulate in the atheroma and off-target, i.e., in non-vascular bed tissue was also estimated. FLAGPP1-functionalized, but not unfunctionalized, NP could be detected in the aortic atheroma, particularly at the edge of the lesion (Figure S5B–D). As for off-target effects, tissue gross morphology, as assessed by HE staining was examined. Liver, kidney, or heart morphology was not detectably altered (Figure S6).
Atherosclerosis was quantified in whole mount aortic arch and descending aorta, and in aortic root sections. S-HSANP-FLAGPP1 elicited a marked reduction of lesion occupancy of the aortic arch and descending aorta compared to vehicle (~44%, P < 0.001) and to all other treatments (Figure 3B and C). In the aortic root, S-HSANP-FLAGPP1 reduced lesion size by ~40% (P = 0.028), in comparison with control mice (Figure 3D and E). HSANP and HSANP-FLAGPP1 elicited similar effects (~44% and ~35%, respectively). Aortic root plaque cellularity, number of foam cells or SMA abundance were not significantly different from vehicle controls in any of the treatments (Figure 3F–J).
This study reports for the first time the synthesis and functional evaluation of HSA NP loaded with a non-nucleoside analogue DNMT inhibitor and functionalized with a MSR1 peptidic ligand. The primary objective of the study was to test the hypothesis that targeting the DNMT inhibitor SGI-1027 to the macrophage, decreases atherosclerosis in ApoE-null mice and mitigates the proinflammatory phenotype of cultured THP-1 macrophages, as result of genomic DNA hypomethylation. The results showed a more complex outcome: SGI-1027-loaded HSANP decreased atherosclerosis but in an anatomical location-dependent fashion; SGI-1027 induced a rapid anti-inflammatory response in non-proliferating cultured macrophages, thus likely independent of DNMT inhibition; furthermore, HSA per se—whether structured into NP or free—induced a marked increase in circulating HDL.
From a structural viewpoint, the physico-chemical properties of HSANP predict good in vivo stability and marginalization—i.e., the ability to adhere to the vascular wall: non-neutral charge, µm-region size, and imperfectly spherical shape [49, 50]. Functionally, our findings provide several novel insights. First, the general concept that DNMT inhibitors are anti-atherogenic was confirmed, but with a few caveats [27, 28]. DNA demethylation may contribute to the effects of SGI-1027-loaded HSA NP in vivo, but the relatively rapid anti-inflammatory response observed in non-replicating cultured THP-1 macrophages likely reflects a non-epigenetic activity of SGI-1027, as DNMT inhibition results in DNA hypomethylation in daughters of proliferating cells when DNA replication outpaces the DNA methylation maintenance machinery [51]. Notably, the nucleoside analogue DNMT inhibitor azacytidine can elicit responses that are independent of DNMT inhibition [52]. At any rate, this is the first report of an anti-inflammatory activity of SGI-1027, namely an increase in secretion of the potent anti-inflammatory cytokine IL-10, the pleiotropic IL-6, and the ratio between IL-10 and the inflammatory cytokine IL-1β. These responses are in principle relevant in the context of atherogenesis. IL-10 decreases the proinflammatory response in vascular lesions, preventing intimal hyperplasia by inhibiting the activity of the nuclear factor NF-kappaB (NF-κB) transcription factor, the chemokine monocyte chemoattractant protein 1 (MCP1), and the growth factor basic fibroblast growth factor (bFGF) [53–55]; IL-10 also reduces the expression of matrix metalloproteinases, modulates lipid metabolism in macrophages and limits monocyte recruitment to the plaque [56–58]. Taken together, our data suggest that SGI-1027 may represent a previously unappreciated double-edged tool that targets both the DNA methylome and the macrophage inflammatory status, perhaps by rapid transcriptional regulation or biochemical modulation of cytokine RNA translation and secretion. Further studies are warranted to clarify the molecular mechanisms underlying that response. Also, the ability of the PP1 peptide to be internalized by human macrophages and to colonize the murine atheroma was confirmed [35]. Interestingly, our data indicate that loaded SGI-1027 increases PP1-mediated uptake, possibly by conferring physico-chemical properties to NP that facilitate PP1 binding to MSR1 or promote receptor-independent uptake.
A further caveat is that HSA NP induce different effects on atheromas depending on the location along the aorta, with stronger effects in the aortic tree compared to the aortic root. Additionally, the responses observed in the latter showed little specificity for NP composition. The observed extent of decrease of the aortic tree lesion burden is comparable to results of other studies using DNMT inhibitors, such as azacytidine [27, 28] although the treatment period was shorter in our study (8 weeks compared to 30 weeks and 12 weeks, respectively). Other studies using chemotherapeutic agents such as paclitaxel or methotrexate showed a comparable decrease of lesion area [59]. Overall, the data point to heterogeneity of ApoE-null atheromas across different locations along the aorta. Atherosclerosis tends to appear at the aortic root earlier than in other aortic location in ApoE-null mice, implying that our NP design may be little effective against fast proliferating atheromas, whether due to intrinsic limitations of SGI-1027 or constrains imposed by HSA [39]. These considerations will guide future efforts to improve the efficacy of HSA NP-mediated drug delivery. At any rate, the observation that HSA NP irrespective of payload or functionalization, but not unstructured HSA, tend to decrease the size of aortic root atheromas, suggests an unexpected anti-atherogenic activity of HSA NP per se. Based on the ability of serum albumin to avidly bind fatty acids, the anti-atherogenic activity of HSANP may be due to increased lipid sponging by HSA aggregates compared to individual HSA polypeptides, resulting in favourable changes in cellular free lipid pool within the atheroma. Our data show that the anti-atherogenic activity of HSA NP is the result of proportionate deceleration of the expansion of the atheroma, rather than targeted reduction of any of its individual components—cellularity, foam cells, smooth muscle. That outcome is reassuring from the viewpoint of prospective translational applications, as it suggests that a reduction of the atheroma burden is achieved without compromising the stability of the lesion.
An additional layer of the anti-atherogenic effect of HSANP is plasma HDL increase. That effect is elicited by HSA per se, irrespective of whether structured in NP or free. The data echo evidence that hypoalbuminaemia is a proposed predictor of CVD [60], and serum albumin is directly associated with HDL in humans [61, 62]. That association is partially under genetic control and to our knowledge, the effect of the diet has not been tested. Our data suggest that the antiatherogenic effect of HSA in the aortic root is mechanistically uncoupled from HDL increase, as unstructured HSA promotes the latter, but not the former. Our model of serum albumin perfusion may help to understand the functional relationship of hypoalbuminaemia with lipoprotein metabolism, diet, and CVD risk.
As for off-target effects, the absence of any change in BW, homeostasis of plasma glucose, cholesterol or triglycerides, or any gross histological morphology of non-vascular bed tissues, suggests that HSANP can deliver their cargo specifically to target organs without inducing any systemic gross metabolic alterations. Although it cannot be excluded that SGI-1027 induces apoptosis in a subset of cells, THP-1 cell viability and in vivo data suggest that SGI-1027 is well tolerated in the conditions applied here.
Among limitations of our work, is the use of glutaraldehyde as cross-linker, which limits future translational applications. Furthermore, the efficiency of SGI-1027 loading needs improving. Also, no detailed information on the effects of NP on the vascular DNA methylome and gene expression, or circulating inflammatory markers, could be obtained. Critically, longer duration of treatment is necessary to fully appreciate tolerance, toxicity, and any undesired secondary effect. Future studies will aim to correcting those limitations. Particularly, NP with amenable physico-chemical characteristics for human studies should be tested, exploiting the wide choice of available nanomaterials and candidate bioactive loads [63].
In conclusion, our study provides encouraging proof of concept that treatment with NP-loaded DNMT inhibitors may be a valuable strategy to prevent or treat atherosclerosis. Also, a novel anti-inflammatory activity of SGI-1027 was demonstrated, that is likely independent of DNMT inhibition.
ApoE: apolipoprotein E
BW: body weight
CVD: cardiovascular disease
DAPI: 4’-6-diamidino-2-phenylindole
ddH2O: deionized distilled water
DLS: dynamic light scattering
DNMT: DNA methyltransferase
HDL: high-density lipoprotein
HE: haematoxylin and eosin
HPLC: high-pressure liquid chromatography
HSA: human serum albumin
HSANP: human serum albumin nanoparticle
IFNα: interferon α
IL-1β: interleukin-1β
MSR1: mouse scavenger receptor 1
NP: nanoparticle
PAS: periodic acid-Schiff-staining
SMA: smooth muscle actin
TBS-T: Tris-buffered saline with 2% Tween
TEM: transmission electron microscopy
TET: ten-eleven translocation
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/101221_sup_1.pdf.
We thank the personnel of Cedars-Sinai Medical Center Microscopy Core, Los Angeles, CA, USA; Dr. Jiani Zhu, Postdoctoral Scientist, Medicine Department, Cedars Sinai Medical Center, Los Angeles, CA, USA; and the personnel of the Flow Cytometry Core, XXI Century National Medical Center, Mexico City, Mexico, for their generous assistance. This article is in memory of Dr. Jorge A. Martínez García, excellent colleague and pathologist, generous collaborator who has given assistance and advice during the early phase of this study.
ACMS: Investigation, Writing—original draft. AMG, DCC, and GdCRM: Investigation. RCJ and EKK: Supervision, Validation. LSS: Investigation, Supervision, Validation. DK and DRR: Investigation, Formal analysis. AMA: Validation, Supervision. GL: Supervision, Validation, Writing—review & editing. SZ: Conceptualization, Supervision, Writing—review & editing.
SZ filed a patent application regarding the SGI-1027-loaded, PP1-functionalized HSA NP described here. The patent (patent number: MX/a/2019/015004) is of no financial interest to the subject matter and material of the manuscript. All other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The laboratory animal protocol was approved by the committee for ethics in research of the University of Guanajuato (approval no. CIBIUG-P10-2017) and performed in agreement with the official Mexican guideline for the use of laboratory animals NOM-062-ZOO-1999.
Not applicable.
Not applicable.
The data analyzed in this study was obtained from our own experimental work. Requests for access to these datasets should be directed to Silvio Zaina, szaina@ugto.mx.
Work was supported by the Mexican Council for Humanities, Science and Technology (CONAHCyT) Proyectos de desarrollo científico para atender problemas nacionales grant no. [584] to SZ; and by the University of Guanajuato Convocatoria Institucional de Investigación Científica grant no. [045/2019] to SZ. CONAHCyT fellowships supported DCC (Postdoctoral), ACMS (PhD), AMG (MSc) and GdCRM (“Ayudante a SNI3/Emérito”). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

