 Review
Review

Affiliation:
1Department of Clinical Pharmacy, School of Basic Medicine, China Pharmaceutical University, Nanjing 210009, Jiangsu Province, China
†These authors contributed equally to this work.
Email: hajaraking04@gmail.com
Affiliation:
1Department of Clinical Pharmacy, School of Basic Medicine, China Pharmaceutical University, Nanjing 210009, Jiangsu Province, China
†These authors contributed equally to this work.
Affiliation:
1Department of Clinical Pharmacy, School of Basic Medicine, China Pharmaceutical University, Nanjing 210009, Jiangsu Province, China
2School of Pharmacy, Mongolian National University of Medical Sciences, Ulaanbaatar 14210, Mongolia
Affiliation:
3Department of Pharmaceutical Analysis, China Pharmaceutical University, Nanjing 210009, Jiangsu Province, China
Affiliation:
1Department of Clinical Pharmacy, School of Basic Medicine, China Pharmaceutical University, Nanjing 210009, Jiangsu Province, China
Affiliation:
1Department of Clinical Pharmacy, School of Basic Medicine, China Pharmaceutical University, Nanjing 210009, Jiangsu Province, China
Affiliation:
4Department of Natural Products Development and Formulation, Institute of Traditional Medicine, Muhimbili University of Health and Allied Sciences, Dar es Salaam 65001, Tanzania
Affiliation:
1Department of Clinical Pharmacy, School of Basic Medicine, China Pharmaceutical University, Nanjing 210009, Jiangsu Province, China
Email: mingxu@cpu.edu.cn
Explor Cardiol. 2026;4:1012112 DOI: https://doi.org/10.37349/ec.2026.1012112
Received: April 05, 2026 Accepted: April 30, 2026 Published: June 24, 2026
Academic Editor: Chiara Caselli, Consiglio National Research Council (CNR), Italy
Gasdermin D (GSDMD) has been identified as a potential key effector protein within the inflammatory response and is characterized as a primary executor of pyroptosis through the formation of transmembrane pores. This review evaluates the emerging role of GSDMD-mediated pyroptosis in the pathogenesis of cardiovascular diseases (CVDs), with a specific focus on its potential contributions to atherosclerosis. We examine how the activation of GSDMD by inflammasomes, such as NLRP3 and AIM2, facilitates the release of inflammatory cytokines (IL-1β and IL-18) and damage-associated molecular patterns (DAMPs). Central to this discussion is the proposed role of GSDMD in driving macrophage foam cell death and vascular smooth muscle cell (VSMC) dysfunction. These factors are associated with necrotic core expansion and increased risk of atherosclerotic plaque instability. Furthermore, GSDMD may mediate endothelial dysfunction and disrupt lipid metabolism, and is suggested to participate in systemic signaling via extracellular vesicles. Finally, we highlight the therapeutic potential of targeting GSDMD as a possible strategy to stabilize vulnerable plaques, which may offer new avenues for cardiovascular precision medicine.
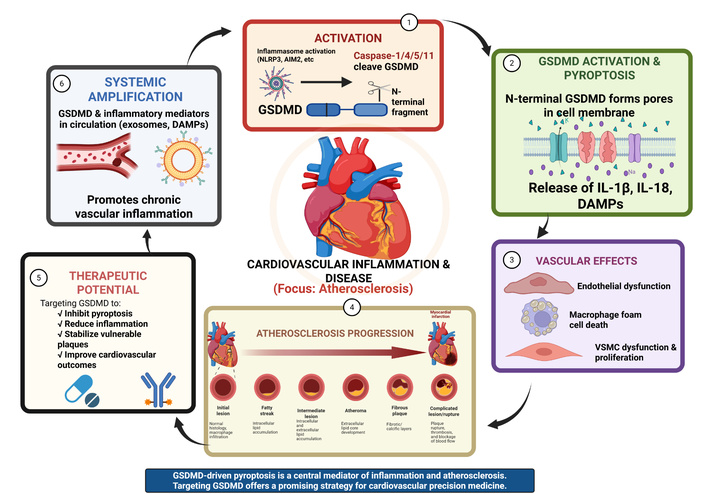
Cardiovascular disease (CVD) remains the leading cause of global mortality and disability. As of 2020, its prevalence has significantly surpassed other health burdens, with a substantial portion of fatalities occurring in low- and middle-income nations. The spectrum of CVD encompasses primary conditions such as coronary artery disease, heart failure (HF), stroke, and peripheral vascular diseases [1–5]. This rising trend is largely driven by Western lifestyle shifts—including poor dietary patterns and sedentary habits—which promote chronic inflammation within the arterial walls. Furthermore, the alarming rise in obesity among children and adolescents is predicted to increase rates of insulin resistance, further exacerbating the global burden of CVD.
This metabolic dysfunction triggers systemic innate immune activation. While traditional risk factors like hypertension, obesity, and smoking are critical, evidence increasingly indicates that inflammation within the arterial walls is a major contributor to atherosclerosis. Pathophysiologically, arteries react to damaging stimuli through adaptive mechanisms, including vascular remodeling and endothelial dysfunction [6, 7]. Recent findings suggest these metabolic stressors converge on Gasdermin D (GSDMD)-mediated pathways, creating a mechanistic link between lifestyle-induced inflammation and vascular damage. Identifying the potential role of these inflammatory pathways provides a new lens for understanding conditions like atherosclerosis and acute myocardial infarction (MI), potentially offering novel therapeutic approaches [8].
GSDMD is characterized as a primary executor of pyroptosis—a pro-inflammatory form of cell death that exacerbates the vascular microenvironment. Within atherosclerotic plaques, GSDMD-activated inflammasomes, such as NOD-like receptor family pyrin domain containing 3 (NLRP3) and absent in melanoma 2 (AIM2), are associated with pathogenic changes [9, 10]. Macrophages are crucial for plaque formation, and GSDMD-driven pyroptosis within these cells is suggested to contribute to plaque development and instability [11]. Specifically, GSDMD impairs reverse cholesterol transport (RCT), leading to cholesterol retention in arterial walls. Furthermore, GSDMD-driven mitochondrial perforation and the subsequent leakage of mitochondrial DNA (mt DNA) trigger the stimulator of interferon genes (STING)-IRF3/NF-κB signaling axis. This process facilitates the formation of GSDMD pores and the release of mature IL-1β, a key proinflammatory cytokine in atherogenesis [12]. Ultimately, GSDMD deficiency has been shown to shift macrophage death from pyroptosis to apoptosis, potentially resulting in smaller, more stable plaques with reduced necrotic cores [8].
Pyroptosis is a highly regulated lytic form of inflammatory programmed cell death. It is primarily characterized by rapid cell swelling, plasma membrane pore formation, and eventual membrane rupture. This process serves as a critical host defense mechanism by eliminating infected or damaged cells [13]. Unlike other forms of cell death, pyroptosis is distinct from apoptosis, a non-lytic “silent” form of cell death involving cell shrinkage and the formation of apoptotic bodies that are cleared without inflammation [14, 15]. While necrosis was traditionally seen as an accidental, uncontrolled cell death, pyroptosis is a genetically programmed process. Furthermore, pyroptosis differs from necroptosis (another programmed necrosis) because necroptosis is caspase-independent and regulated by RIPK1/RIPK3/MLKL pathways [16, 17]. The initiation of pyroptosis is generally understood to occur through canonical pathways involving the assembly of inflammasome—multiprotein complexes that sense danger signals.
The canonical pathway: Involves the activation of caspase-1. Once activated, caspase-1 cleaves both GSDMD and the precursors of IL-1β and IL-18, enabling the maturation and secretion of these cytokines.
The non-canonical pathway: Triggered by intracellular lipopolysaccharide (LPS), which directly activates caspase-4/5 (in humans) or caspase-11 (in mice). These caspases directly cleave GSDMD to induce pyroptosis independently of the traditional inflammasome [18, 19].
Once cleaved, the N-terminal domain of GSDMD forms large transmembrane pores. These pores disrupt osmotic balance, leading to water influx and cell swelling [18]. Recent research has highlighted that the final stage of complete membrane rupture is promoted by the protein Ninjurin-1 (NINJ1), which follows GSDMD pore formation to further amplify the inflammatory response and the release of intracellular contents [20].
The gasdermin family consists of pore-forming proteins that serve as the primary executioners of pyroptosis. This family comprises six members, including GSDMA, GSDMB, GSDMC, GSDMD, GSDME (also known as DFNA5), and DFNB59 (Pejvakin, PJVK) [21, 22]. Gasdermins share a conserved structure composed of an N-terminal cytotoxic domain, a C-terminal repressor domain, and a flexible linker. Under resting conditions, the C-terminal domain inhibits the pore-forming activity of the N-terminal domain. Upon cleavage by specific proteases, the N-terminal fragment is liberated, allowing it to oligomerize and insert into the plasma membrane to form large pores [23, 24] (See Figure 1). GSDMD is the most extensively studied member. When cleaved by inflammasome-activated caspases (such as caspase-1, -4, -5, and -11), GSDMD N-terminal fragments bind to specific acidic membrane lipids, such as phosphoinositides and cardiolipin. These fragments oligomerize to form membrane-embedded pores with an inner diameter of approximately 10–14nm, composed of multiple symmetric protomers [25]. Other gasdermins also contribute to pyroptosis and immune responses through distinct pathways. For instance, GSDME can be cleaved by caspase-3, creating a functional link between apoptosis and pyroptosis [26]. In tumor immunity, GSDME acts as a tumor suppressor that can be activated by chemotherapy to induce an anti-tumor immune response, although it may also cause off-target tissue damage [27, 28]. GSDMB has been shown to promote non- canonical pyroptosis by enhancing caspase-4 activity, which subsequently leads to the cleavage of GSDMD [29]. Beyond cell death, gasdermins play broader biological roles, including the regulation of cell differentiation, coagulation, and the unconventional secretion of leaderless cytokines that bypass the traditional ER- Golgi pathways [22, 24]. Gasdermins have been implicated in various diseases, ranging from cancer to organ-specific pathologies. In the gastrointestinal and respiratory systems, gasdermin regulates mucosal immunity, playing complex roles in inflammatory bowel disease (IBD) and pneumonia [21, 30].
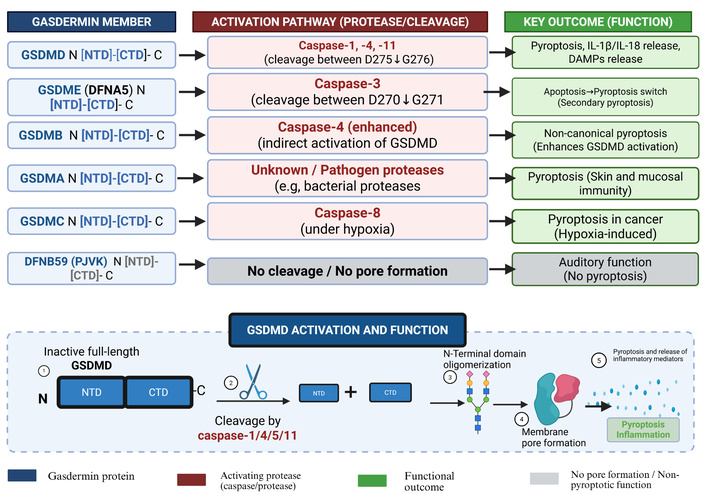
Activation mechanisms and functional outcomes of gasdermin family members: gasdermins share an N-terminal pore-forming domain and a C-terminal repressor domain. Upon cleavage by specific proteases, the N-terminal domain inserts into the plasma membrane to form pores (except DFNB59). GSDMD is cleaved by inflammatory caspases (1/4/5/11), triggering pyroptosis and release of IL-1β, IL-18, and damage-associated molecular patterns (DAMPs). GSDME is cleaved by caspase-3, converting apoptosis to secondary pyroptosis. GSDMB enhances caspase-4 activity to promote non-canonical pyroptosis. GSDMA and GSDMC mediate pyroptosis in epithelial and hypoxic tumor contexts, respectively. DFNB59 lacks pore-forming activity and is involved in auditory function. Created in BioRender. Created in BioRender. Nyame, D. K. (2026) https://www.biorender.com/rzdg8l3
Evidence suggests that tissue damage in many organs is often mediated by GSDMD-dependent pyroptosis. For example, GSDMD-mediated pyroptosis aggravates myocardial hypertrophy and fibrosis, contributing to adverse cardiac remodeling under pressure overload [31]. Furthermore, GSDMD mutations have been found to be protective against renal ischemia - reperfusion injury in acute kidney injury (AKI) [31]. In conditions such as severe acute pancreatitis (SAP), downregulating GSDMD has been proven to reduce pyroptosis-related inflammation [32]. Lastly, GSDMD- driven inflammatory signaling underlies tissue dysfunction associated with Tet2- mutant clonal hematopoiesis [33] (Figure 2).
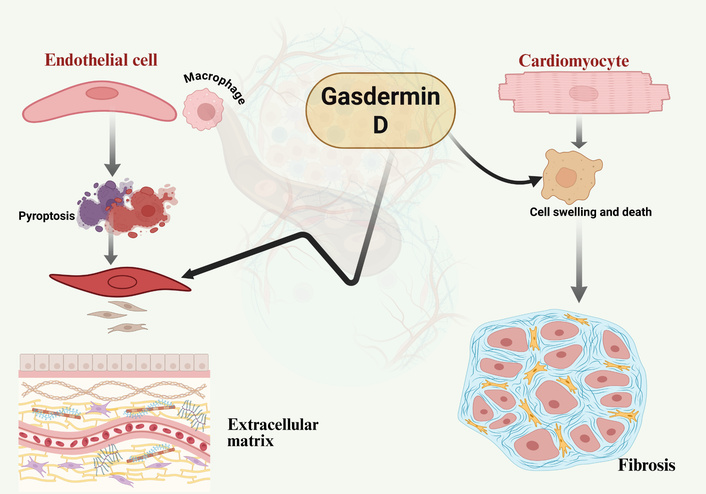
GSDMD contribution to cardiovascular tissue damage: schematic representation of GSDMD activation and its pathological consequences. Upon activation, the N-terminal domain of GSDMD, cleaved by inflammatory caspases, inserts into the plasma membrane to form pores. This process drives pyroptosis in endothelial cells and macrophages, leading to vascular barrier disruption and immune cell infiltration. In vascular smooth muscle cells (VSMCs), GSDMD activation promotes extracellular matrix (ECM) accumulation and fibrosis, while in cardiomyocytes, it triggers cell swelling and death. Collectively, these processes drive progressive vascular and myocardial damage, contributing to atherosclerosis and heart failure. Created in BioRender. Nyame, D. K. (2026) https://biorender.com/q1rzues
GSDMD autoinhibition is maintained by an intramolecular interaction between its C-terminal (C-GSDMD) and N-terminal (N-GSDMD) domains. Crystal structure analysis of the human GSDMD C-terminal domain (resolved at 2.64 Å) reveals that a key loop inserts into the N-terminal domain to stabilize this inhibited state [34]. This interaction masks the positively charged surface of the N-terminal domain, preventing membrane binding. Upon proteolytic cleavage by inflammatory caspases, the N-terminal domain is released and undergoes higher-order oligomerization via charge-charge interactions [35, 36]. These oligomers form dynamic pores (10–14 nm in diameter) across the plasma membrane, disrupting ion homeostasis and facilitating the secretion of pro-inflammatory cytokines, including IL-1β and IL-18. Additionally, internal cysteine residues contribute to the plasticity and modulation of this pore-formation process [37].
The canonical pathway is initiated by the assembly of multiprotein inflammasome complexes (e.g., NLRP3) in response to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [38, 39]. These cytosolic sensors recruit procaspase-1 via the adaptor protein apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD, an adaptor protein) (ASC), triggering its autocatalytic cleavage into active caspase-1 [40, 41]. Active caspase-1 then cleaves GSDMD at a specific aspartate residue (Asp276 in mice) within the linker region, while simultaneously processing pro-IL-1β and pro-IL-18 into their mature forms [42, 43]. Beyond inducing pyroptosis, caspase-1 plays a dual role in neutrophils by modulating both apoptotic and inflammatory signaling to balance host defense [44].
The non-canonical pathway involves the direct sensing of cytosolic Gram-negative bacterial LPS by caspase-4/5 (human) or caspase-11 (mouse) [45, 46]. The binding of LPS to the CARD of these inflammatory caspases triggers oligomerization independently of TLR4 and ASC [47, 48]. These activated caspases directly cleave GSDMD to induce pyroptosis [49]. Crucially, the resulting pore-induced potassium (K+) efflux stimulates secondary activation of the NLRP3 canonical inflammasome, linking the two pathways to amplify the inflammatory response [50, 51].
GSDMD activity is finely tuned by various PTMs. For instance, AMPK phosphorylates the N-terminal domain at Ser46 to inhibit oligomerization, whereas TRAF1-mediated ubiquitination facilitates its membrane translocation [38, 39]. Similarly, the neddylation of the caspase-1 CARD domain by Nedd8 is essential for efficient self-cleavage [52, 53]. Negative regulators, including the E3 ubiquitin ligase Nedd4, Irgm2, and GATE-16, act as checkpoints to prevent excessive inflammation and sepsis [54, 55]. Furthermore, metabolic signals like cyclic AMP (cAMP) and proteases such as Cathepsin B modulate caspase-11 activation, while inhibitory proteins like SERPINB1 prevent spontaneous caspase oligomerization [56–58].
Dysregulated GSDMD activation contributes to several pathologies, including myocardial hypertrophy and fibrosis [59]. Beyond acute inflammation, caspase-4 activation by cytoplasmic LPS is linked to cellular senescence and the senescence-associated secretory phenotype (SASP) via the GSDMD and p53 pathways [60]. Pharmacological targeting of these pathways—using agents such as Korean Red Ginseng saponins—offers potential therapeutic avenues for managing chronic inflammation and lethal sepsis [61].
The NLRP3 inflammasome is a central component of the innate immune system, serving as an elaborate sensor for numerous environmental and endogenous danger signals. The NLRP3 protein consists of an N-terminal pyrin domain (PYD), a central nucleotide-binding and oligomerization domain (NOD, also known as NACHT domain), and a C-terminal leucine-rich repeat (LRR) domain, classifying it as a member of the NLR family [62, 63]. The NACHT domain is required for ATP-dependent oligomerization, while the LRR domain acts as a regulatory scaffold that maintains the protein in an autoinhibited conformation during resting states [64] (Figure 3). Activation follows a stringent two-step mechanism:
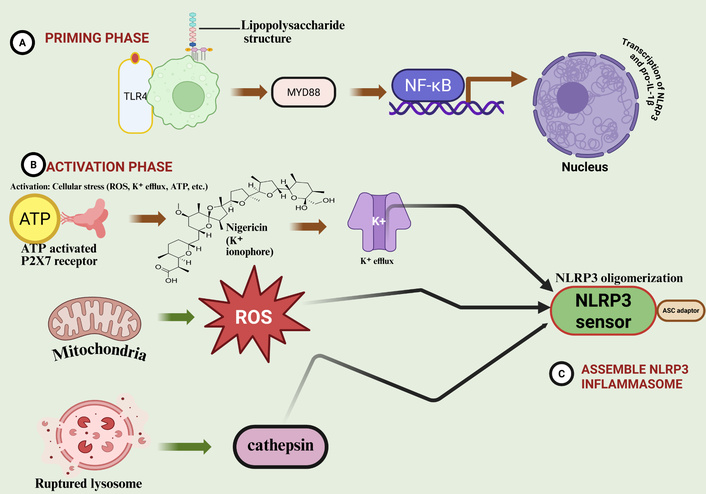
Activation and assembly of the NLRP3 inflammasome. The NLRP3 inflammasome activation is a tightly regulated two-step process. (A) Priming phase: recognition of pathogen-associated molecular patterns (PAMPs) by TLR4 triggers the NF-κB signaling pathway, inducing the transcription of NLRP3 and pro-inflammatory cytokines like pro-IL-1β. (B) Activation phase: diverse cellular stressors, including potassium (K+) efflux via the P2X7 receptor, mitochondrial reactive oxygen species (ROS), and cathepsin release from ruptured lysosomes, serve as secondary signals. (C) Assembly: These signals converge to promote NLRP3 oligomerization and the recruitment of the ASC adaptor and pro-caspase-1, forming the active inflammasome complex, which subsequently activates GSDMD. Created in BioRender. Nyame, D. K. (2026) https://biorender.com/s87d74a
Priming (Signal 1): Triggered by TLR, NLR, or cytokine receptors (e.g., TNFR), leading to NF-κB-dependent transcriptional upregulation of NLRP3 and pro-IL-1β [65–67]. PTMs, such as deubiquitination by BRCC3, are critical during this phase to license NLRP3 for assembly [68, 69].
Activation (Signal 2): Triggered by diverse stimuli, including extracellular ATP (via the P2X7 receptor), crystalline substances (e.g., uric acid or cholesterol crystals), and lysosomal rupture [70–72]. A central unifying event is K+ efflux, which induces a conformational shift in NLRP3, allowing it to recruit the adaptor protein ASC [73, 74].
Unlike the versatile sensing of NLRP3, the AIM2 inflammasome specifically detects double-stranded DNA (dsDNA) in the cytosol. AIM2 is composed of a C-terminal HIN-200 domain that binds dsDNA and an N-terminal PYD domain that initiates downstream signaling [75–77].
Upon detecting dsDNA—whether from microbial pathogens (e.g., Francisella, Listeria) or host mitochondrial damage—AIM2 undergoes oligomerization [78, 79]. This process is regulated by type I interferons and guanylate-binding proteins (GBPs), which facilitate the liberation of DNA from intracellular vesicles [80]. Additionally, the stability of AIM2 is managed by the deubiquitinase USP21, which prevents premature proteasomal degradation [81].
Modern immunology recognizes that inflammasomes do not operate in isolation but interact through complex signaling networks.
Non-canonical to canonical bridge: Caspase-11/4/5 activation by LPS triggers GSDMD pores, which subsequently induce K+ efflux, thereby serving as a secondary signal for NLRP3 activation [82].
cGAS-STING synergy: Mitochondrial dysfunction releases DNA that can simultaneously trigger the cGAS-STING pathway and the AIM2 inflammasome, creating a robust pro-inflammatory feedback loop [83].
PAN-optosis: Master regulators such as ZBP1 and TAK1 coordinate a unified death signaling complex. This integration allows the cell to switch between pyroptosis (NLRP3), apoptosis (caspase-8), and necroptosis depending on the physiological context, ensuring optimal pathogen clearance [84, 85].
To prevent chronic inflammation and autoimmune pathologies, several regulatory “brakes” are employed:
Autophagy and mitophagy: These processes act as critical negative regulators by clearing assembled inflammasome complexes and damaged mitochondria, reducing ROS-mediated activation [69].
MicroRNA (miRNAs) interference: miRNAs, such as miR-223-3p, target the 3’ UTR of NLRP3 and caspase-1 to prevent excessive priming and activation [86].
Redox control: The Nrf2 transcription factor suppresses NLRP3 activation by inducing antioxidant genes that mitigate oxidative stress [87].
Immune checkpoints: Proteins like SERPINB1 prevent spontaneous assembly of the ASC speck, ensuring the inflammatory response is only mounted upon reaching a definitive activation threshold [58].
Dysregulation of NLRP3 and AIM2 is implicated in a spectrum of diseases, ranging from neurodegenerative disorders (Alzheimer’s, Multiple sclerosis) to metabolic syndromes (type 2 diabetes, gout) [81, 88, 89]. In oncology, inflammasome activity influences the tumor microenvironment by modulating cytokine milieus (IL-1β/IL-18) and adaptive immune recruitment [90]. Targeted inhibitors, like MCC950 and glyburide, are promising therapeutic approaches to reduce sterile inflammation in disorders like ischemia/reperfusion (I/R) injury and autoimmune myopathies [91].
SSc and atherosclerosis are increasingly recognized as related pathologies within the CVD spectrum. They share a common mechanistic substrate characterized by chronic inflammation, immune dysregulation, and progressive vascular injury [92]. The NLRP3-caspase-1-GSDMD axis is central to atherosclerosis; cleaved GSDMD assembles into membrane pores, inducing pyroptosis and releasing pro-inflammatory cytoplasmic contents, which exacerbate vascular inflammation [93]. Similarly, SSc is characterized by widespread endothelial dysfunction and vasculopathy, typically initiated by anti-endothelial cell (EC) antibodies and oxidative stress [94]. This injury impairs vascular relaxation, enhances leukocyte adhesion, and increases the risk of both organ fibrosis and accelerated atherosclerosis in patients with SSc [95, 96] (Figure 4).
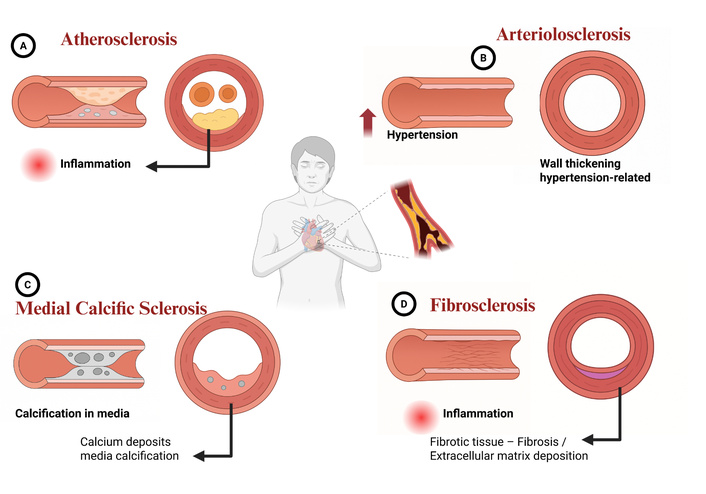
Types of sclerosis in cardiovascular diseases. (A) Atherosclerosis: Characterized by the buildup of fatty plaques (atheromas) within the inner lining (intima) of the artery, where inflammation is a key driver. The cross-section reveals significant narrowing of the arterial lumen, obstructing blood flow. (B) Arteriolosclerosis: Involves the thickening of the walls of small arteries and arterioles, typically resulting from chronic hypertension. The thickened wall significantly reduces the lumen diameter. (C) Medial calcific sclerosis (Mönckeberg’s sclerosis): Marked by calcium deposits within the middle layer (tunica media) of the artery. These calcifications cause vessel stiffening but typically do not obstruct the lumen. (D) Fibrosclerosis: Thickening of the arterial wall due to excessive formation of fibrotic tissue and deposition of extracellular matrix (ECM), driven by chronic inflammation. The cross-section illustrates the fibrotic layer narrowing the vessel opening. Created in BioRender. Nyame, D. K. (2026) https://biorender.com/hpn09xz
A key link between these conditions is the role of endothelial GSDMD in enhancing vascular damage. Recent studies have shown that GSDMD pores allow the cytosolic release of mtDNA, which triggers an immune signaling cascade via the cGAS-STING pathway [97]. This feed-forward loop leads to increased production of mitochondrial reactive oxygen species (ROS) and STING-dependent inflammatory signaling, which directly promotes atherogenesis [97]. Although this mechanism has not been specifically studied in SSc yet, it is highly probable, as clinical reports have documented augmented arterial stiffness and increased carotid intima-media thickness in SSc patients—both of which are hallmarks of accelerated vascular aging and subclinical atherosclerosis [98].
The NF-κB pathway largely governs inflammatory signaling in both SSc and atherosclerosis, driving the recruitment of macrophages and T cells to the vascular wall. This process is finely tuned by miRNAs and other noncoding RNAs that modulate cytokine production (TNF-α, IL-1β, IL-6) and vascular smooth muscle cell (VSMC) proliferation [99]. Given the central role of the NLRP3-GSDMD axis in executing pyroptosis, targeting this pathway represents a promising therapeutic avenue. Anti-inflammatory biologics, particularly those inhibiting TNF-α and IL-1β, effectively reduce vascular inflammation and plaque progression. These treatments potentially attenuate both the fibroproliferative changes in SSc and the associated accelerated cardiovascular risks [100].
Macrophage pyroptosis is a decisive factor in the progression and eventual rupture of atherosclerotic plaques. Within the lipid-rich microenvironment of a plaque, macrophages—the predominant immune cells—undergo GSDMD-mediated lysis, transforming stable lesions into vulnerable, pro-inflammatory sites [101].
The mitochondria-STING axis: Beyond simple cell lysis, recent evidence reveals that macrophage-derived GSDMD mediates mitochondrial membrane damage. This results in the release of mtDNA into the cytosol, further activating the cGAS-STING-IRF3/NF-κB signaling axis [102]. This creates a robust feed-forward loop that amplifies pro-inflammatory cytokine production and recruits additional immune cells.
Endoplasmic reticulum (ER) stress and metabolic triggers: External metabolic stressors, such as homocysteine, contribute to this process by inducing ER stress and impairing calcium homeostasis. Such cellular disturbances generate excess mitochondrial ROS, a crucial secondary signal for NLRP3-GSDMD activation [103].
Therapeutic stabilization: The GSDMD pathway provides significant therapeutic opportunities; genetic deletion in vivo (Gsdmd-/-models) or pharmacological targeting of this pathway reduces necrotic core size and stabilizes plaques [101]. Novel approaches, such as lysosomal zinc nanomodulation, prevent macrophage pyroptosis by suppressing ROS production and remodeling the immune microenvironment within the vascular walls [104].
The vascular endothelium consists of a monolayer of ECs that play a vital role in maintaining vascular tone, barrier integrity, and blood fluidity. Endothelial dysfunction involves the loss of these homeostatic functions and is characterized by impaired vasodilation—often due to decreased nitric oxide (NO) bioavailability—increased permeability, and pro-inflammatory activation [105–107].
EC injury is primarily driven by pyroptosis. The overproduction of ROS causes oxidative damage to mitochondria and cellular DNA, which triggers the assembly of the NLRP3 inflammasome. This assembly activates caspase-1, which cleaves GSDMD to form membrane pores, resulting in lytic cell death and the release of pro-inflammatory cytokines [106, 108].
In vitro and in vivo observations, this process disrupts the physical barrier of the vessel and recruits immune cells, thereby accelerating atherosclerotic plaque deposition and arterial stiffening [101]. Furthermore, chronic conditions such as diabetes and hypertension exacerbate oxidative stress, driving phenotypic shifts like the endothelial-to-mesenchymal transition (EndMT), which further destabilizes plaques.
Recent research highlights protective signaling networks that can mitigate this damage:
The melatonin/RORα/miR-223 axis: Melatonin signaling promotes miR-223 transcription, which suppresses pyroptotic proteins and adhesion molecules like ICAM-1, effectively restoring NO levels and vascular health [109].
Pharmacological interventions: Agents such as Trimetazidine and various antioxidant therapies show promise in improving endothelial nitric oxide synthase (eNOS) activity and reducing GSDMD-mediated damage, offering a potential strategy to reduce the overall CVD burden [110].
VSMCs possess remarkable plasticity, allowing them to switch from a quiescent, contractile phenotype to a highly proliferative, synthetic state in response to environmental stimuli. While this modulation is necessary for normal vascular repair, it is significantly dysregulated in pathologies such as atherosclerosis, hypertension, and medial thickening [111]. The abnormal migration and hyperproliferation of dysfunctional VSMCs are tightly regulated by several molecular pathways, including miRNAs and long non-coding RNAs (lncRNAs) that control the cell cycle and inflammatory signaling [112].
Pathological fibrosis results from excessive ECM deposition by synthetic VSMCs, causing increased vascular stiffness and decreased compliance. A key mediator is the ECM glycoprotein biglycan, which promotes VSMC proliferation and migration by modulating cell cycle regulators such as cdk2 and p27 [113]. Overexpression of these matrix components increases arterial susceptibility to injury and contributes to long-term fibrotic changes.
EGFR signaling: The EGFR in VSMCs is a key mediator of arterial wall stiffening. Chronic Angiotensin II stimulation activates EGFR, leading to medial thickening and fibrosis. Notably, the genetic deletion of EGFR in VSMC models protects against hypertension-induced vascular remodeling, suggesting its critical role in maintaining vascular architecture [114].
Metabolic triggers: In patients with chronic kidney disease (CKD), uremic toxins promote VSMC dysfunction through phenotypic switching, senescence, and pathological calcification. These alterations are further exacerbated by oxidative stress and chronic inflammation, creating a pro-fibrotic environment [115].
The molecular interplay between VSMCs and macrophages significantly influences plaque stability. This crosstalk, mediated via soluble factors and extracellular vehicles (EVs), facilitates ECM remodeling and sustains the local inflammatory response [116]. In the context of pyroptosis, VSMC-derived signals can activate macrophage inflammasomes, creating a feedback loop that drives necrotic core expansion and vascular aging.
Vascular calcification is an actively regulated, cell-mediated process characterized by the deposition of calcium phosphate minerals within arterial walls. It manifests in two distinct layers:
Intimal calcification: Primarily associated with atherosclerosis, lipid accumulation, and chronic inflammation within the tunica intima [117, 118].
Medial calcification: Linked to Mönckeberg’s arteriosclerosis and elastin degradation within the tunica media. This form significantly reduces vascular compliance and serves as a hallmark of aging, diabetes, and CKD [119].
In both in vivo and in vitro models, arterial stiffness arises from structural remodeling, including VSMC phenotypic switching, collagen deposition, and fibrosis. Clinically, this stiffness is quantified by pulse wave velocity (PWV) [120]. Pro-calcification factors, such as oxidative stress and inflammatory cytokines, stimulate VSMCs to undergo osteogenic differentiation. Recent studies suggest that GSDMD-mediated pyroptosis may exacerbate this transition by releasing intracellular signals (DAMPs) that accelerate mineral deposition and vascular senescence [121].
In CKD patients, hyperphosphatemia and the reduction of calcification inhibitors such as fetuin-A and matrix Gla protein (MGP) drive rapid medial calcification [122, 123]. This leads to increased systolic blood pressure and pulse pressure, which damage the microvasculature of the heart, brain, and kidneys, ultimately contributing to HF and cognitive decline [124].
MI is characterized by irreversible myocardial cell death resulting from prolonged, severe ischemia. While timely reperfusion is essential to salvage the ischemic myocardium, the restoration of blood flow paradoxically induces additional damage, known as myocardial I/R injury. This secondary damage can expand infarct size and severely worsen cardiac function through oxidative stress, intracellular calcium overload, and systemic inflammation [125, 126].
Mitochondrial dysfunction plays a central role in I/R injury. The mitochondrial quality control system—encompassing fusion, fission, and mitophagy—is critical for cardiomyocyte survival; its disruption leads to excessive ROS production and triggers cell death pathways [127]. Recent research emphasizes that GSDMD-mediated pyroptosis is a major contributor to I/R-induced cardiomyocyte loss. During reperfusion, the sudden influx of ROS and calcium triggers the NLRP3 inflammasome, which activates caspase-1. This process causes GSDMD to be cleaved by caspase-1, leading to membrane pore formation, cell lysis, and the massive release of pro-inflammatory cytokines (IL-1β, IL-18) and DAMPs. This “pyroptotic storm” exacerbates local tissue damage and promotes adverse cardiac remodeling [128].
Molecular players: Thioredoxin-interacting protein (TXNIP) and various lncRNAs have emerged as key modulators of oxidative stress and inflammatory signaling during I/R [129].
Cardioprotective strategies: Novel interventions aiming to inhibit these pathways show promise. Mild therapeutic hypothermia targeting mitochondrial apoptotic and pyroptotic pathways have been shown to reduce infarct size. Additionally, hydrogen sulfide (H2S), an endogenous gasotransmitter, serves as a potent cytoprotective agent by modulating oxidative stress and suppressing GSDMD activation [129].
DCM is a distinct clinical entity characterized by structural and functional myocardial abnormalities in diabetic patients. It occurs independently of coronary artery disease, hypertension, or valvular heart disease. DCM initially manifests as diastolic dysfunction due to cardiac stiffness and progresses to clinical HF with either preserved (HFpEF) or reduced ejection fraction (HFrf) [130, 131].
The pathogenesis of DCM involves a complex interplay of metabolic and cellular abnormalities driven by hyperglycemia and insulin resistance.
Oxidative stress and advanced glycation end-products (AGEs): Hyperglycemia promotes the formation of AGEs and excessive ROS production, leading to mitochondrial damage and interstitial collagen accumulation [132, 133].
GSDMD-mediated pyroptosis: Recent in vivo and in vitro research has identified GSDMD-mediated pyroptosis as a critical driver of cardiomyocyte loss. High glucose levels activate the NLRP3 inflammasome, triggering caspase-1 to cleave GSDMD. The resulting N-terminal fragments form membrane pores, leading to cell lysis and the release of pro-inflammatory cytokines like IL-1β and IL-18, which accelerate myocardial fibrosis and remodeling [134].
Lipid metabolism: Increased free fatty acid (FFA) metabolism leads to lipid accumulation in cardiomyocytes (lipotoxicity) and energy inefficiency [135].
Clinically, DCM presents with left ventricular hypertrophy, cardiomyocyte apoptosis, and coronary microvascular dysfunction [135]. While standard HF therapies remain the baseline, emerging options show significant promise:
SGLT2 inhibitors & GLP-1 agonists: These agents provide substantial cardiovascular protection beyond glucose lowering [134]. SGLT2 inhibitors, in particular, have shown remarkable efficacy in reducing HF hospitalizations.
Experimental approaches: Investigational therapies targeting mitochondrial preservation, oxidative stress reduction, and the inhibition of pyroptotic pathways (such as NLRP3 inhibitors) are currently being evaluated in rodent models to mitigate DCM progression [134].
Circulating GSDMD-dependent EVs function as pivotal mediators of inflammasome-driven signaling across various tissues. These vesicles carry the activated p30 N-terminal fragment of GSDMD, which acts as a molecular “trigger” for inflammatory cell death in recipient cells. By encapsulating these potent fragments, EVs protect them from degradation and facilitate the propagation of pyroptosis far beyond the initial site of injury [136].
The presence of GSDMD within circulating EVs enables systemic activation of downstream inflammatory pathways, amplifying injury through organ crosstalk:
Lung-to-brain injury: In neonatal models of hyperoxia, EVs enriched with activated GSDMD and caspase-1 are released from damaged lung tissue. Upon uptake by neural cells, these EVs induce neuroinflammation and pyroptotic death, linking pulmonary insult to remote brain injury (lung-to-brain axis) [136].
Vascular propagation: This EV-mediated transfer connects local innate immune activation to widespread vascular and tissue damage.
Consequently, GSDMD-dependent EVs are recognized as key players in inflammatory disease progression and represent promising targets for systemic intervention [136].
GSDMD inhibitors have emerged as promising agents to block pyroptosis by targeting various stages of GSDMD activation and pore formation:
GI-Y1: This molecule selectively binds to the Arg7 residue of the GSDMD-N domain. It prevents mitochondrial binding and inhibits pore formation on the plasma membrane. GI-Y1 has demonstrated significant cardioprotective effects in MI/R models [137].
NU6300: This inhibitor covalently reacts with Cys-191, blocking both cleavage and palmitoylation. This action prevents the membrane localization and oligomerization of the pore-forming N-terminus, showing efficacy in sepsis and colitis models [138].
Caspase inhibitors prevent the proteolytic cleavage of full-length GSDMD into its active, pore-forming fragments.
VX-765: A potent caspase-1 inhibitor currently in clinical testing. It effectively reduces GSDMD cleavage in neurotoxicity and IBD models [139, 140].
Caspase-8 inhibitors: These are particularly useful in conditions where TNF-induced lethality or specific infections trigger GSDMD cleavage via caspase-8 [141, 142].
Palmitoylation inhibitors: Targeting GSDMD palmitoylation at conserved cysteines (using agents like 2-bromopalmitate) prevents membrane localization and cytokine release [143].
Regulatory modulators: Proteins such as TRIM21(an E3 ubiquitin ligase and the m6A writer METTL3 regulate GSDMD stability and mRNA silencing, offering novel epigenetic avenues for cancer and inflammatory therapies [144].
The cargo of EVs reflects the physiological state of their origin cells, making them ideal candidates for biomarker development. In cardiovascular and inflammatory diseases, the detection of circulating EVs containing GSDMD-related fragments (such as the p30 N-terminal fragment) and inflammasome components provide non-invasive biomarkers for disease monitoring and personalized medicine [145].
EVs possess low immunogenicity and natural targeting capabilities, providing versatile platforms for the delivery of GSDMD inhibitors directly to fibrotic or inflamed vascular tissues.
Synergistic therapy: The co-delivery of classic anti-inflammatory agents (e.g., colchicine or IL-1β blockers) with GSDMD inhibitors can potentiate therapeutic efficacy [146].
Bioengineering: Surface modification of EVs can further enhance their specificity for damaged ECs or atherosclerotic plaques.
Translating EVs-based therapies into clinical practice requires addressing several critical hurdles:
Standardization: There is a lack of standard protocols for EV isolation and characterization [147, 148].
Scaling: Difficulties persist in large-scale production and in ensuring immune compatibility.
Advanced models: Utilizing organoid-derived EVs and integrating multi-omics data (proteomics, transcriptomics, and metabolomics) are essential for validating therapeutic targets and improving the relevance of preclinical data to human physiology [149].
In summary, GSDMD is a fundamental mediator of chronic inflammation and tissue injury in CVDs, playing a central role in atherosclerosis and myocardial remodeling. By executing pyroptosis—a potent pro-inflammatory form of lytic cell death—GSDMD facilitates the release of critical cytokines (such as IL-1β and IL-18) and DAMPs8, which exacerbate vascular remodeling, endothelial dysfunction, and plaque instability.
GSDMD activation, triggered via both canonical (caspase-1) and non-canonical (caspase-4/5/11 and caspase-8) pathways, directly contributes to cardiomyocyte loss, pathological fibrosis, and vascular barrier disruption. Furthermore, GSDMD-mediated VSMC phenotypic switching and dysfunction are significant factors in plaque rupture mechanisms. Ultimately, targeting the GSDMD-mediated pyroptotic pathway and its associated signaling cascades offers a transformative therapeutic strategy to mitigate the inflammatory drivers central to cardiovascular pathology.
AGEs: advanced glycation end-products
AIM2: absent in melanoma 2
ASC: apoptosis-associated speck-like protein containing a CARD (an adaptor protein)
CKD: chronic kidney disease
CVD: cardiovascular disease
DAMPs: damage-associated molecular patterns
DCM: diabetic cardiomyopathy
dsDNA: detects double-stranded DNA
EC: endothelial cell
ECM: extracellular matrix
EGFR: epidermal growth factor receptor
eNOS: endothelial nitric oxide synthase
ER: endoplasmic reticulum
EVs: extracellular vehicles
GSDMD: Gasdermin D
HF: heart failure
I/R: ischemia/reperfusion
IBD: inflammatory bowel disease
K+: potassium
lncRNAs: long non-coding RNAs
LPS: lipopolysaccharide
LRR: leucine-rich repeat
MI: myocardial infarction
miRNAs: microRNAs
mtDNA: mitochondrial DNA
NLRP3: NOD-like receptor family pyrin domain containing 3
NO: nitric oxide
PAMPs: pathogen-associated molecular patterns
PTMs: post-translational modifications
PYD: pyrin domain
ROS: reactive oxygen species
SSc: systemic sclerosis
STING: stimulator of interferon genes
VSMC: vascular smooth muscle cell
HBM: Writing—original draft. MTN: Writing—original draft. TB: Writing—review & editing. DKN: Visualization. KZ: Project administration. ZH: Project administration. BPM: Conceptualization. MX: Conceptualization, Supervision. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 685
Download: 22
Times Cited: 0
