 Review
Review

Affiliation:
1Centre for Excellence in Molecular Biology and Regenerative Medicine laboratory, Department of Biochemistry, Jagadguru Sri Shivarathreeshwara (JSS) Medical College, JSS Academy of Higher Education & Research, Mysore, Karnataka 570015, India
ORCID: https://orcid.org/0000-0003-2236-4588
Affiliation:
1Centre for Excellence in Molecular Biology and Regenerative Medicine laboratory, Department of Biochemistry, Jagadguru Sri Shivarathreeshwara (JSS) Medical College, JSS Academy of Higher Education & Research, Mysore, Karnataka 570015, India
2Division of Infectious Disease and Vaccinology, School of Public Health, University of California, Berkeley, CA 94720, USA
ORCID: https://orcid.org/0000-0001-8470-3114
Affiliation:
3Council of Scientific & Industrial Research – Indian Institute of Chemical Biology (CSIR-IICB), Translational Research Unit of Excellence, Kolkata, West Bengal 700091, India
ORCID: https://orcid.org/0000-0002-2547-1023
Affiliation:
1Centre for Excellence in Molecular Biology and Regenerative Medicine laboratory, Department of Biochemistry, Jagadguru Sri Shivarathreeshwara (JSS) Medical College, JSS Academy of Higher Education & Research, Mysore, Karnataka 570015, India
ORCID: https://orcid.org/0000-0001-9167-9271
Affiliation:
4Department of Respiratory Medicine, JSS Medical College, JSS Academy of Higher Education & Research, Mysore, Karnataka 570015, India
Email: mahesh1971in@yahoo.com; pamahesh@jssuni.edu.in
ORCID: https://orcid.org/0000-0003-1632-5945
Explor Asthma Allergy. 2023;1:174–185 DOI: https://doi.org/10.37349/eaa.2023.00018
Received: December 15, 2022 Accepted: July 28, 2023 Published: November 20, 2023
Academic Editor: Giuseppe Guida, University of Torino, Italy
Steroid-resistant asthma (SRA) is clinically significant, approximately 10–15% of individuals with asthma do not exhibit a positive response to standard treatments. While this subset represents a relatively small proportion of asthma patients, severe refractory asthma places a substantial burden on healthcare resources and contributes significantly to illness and death. Additionally, the quality of life of patients is greatly affected by the adverse effects of excessive steroid consumption, there is a need to identify individuals who do not react well to steroid medication and the ongoing difficulties of these asthma patients in controlling their diseases, which have a large socio-economic impact. The current short article reviews the common molecular mechanisms responsible for steroid resistance in asthma patients.
According to the Global Initiative for Asthma (GINA), asthma is defined as “a heterogeneous disease, usually characterized by chronic airway inflammation with a history of respiratory symptoms such as wheeze, cough, shortness of breath, and chest tightness that vary over time and in intensity, together with variable expiratory airflow limitation” [1]. The prevalence rates for adults wheezing are 8.6%, 4.3% for clinical/treated asthma, and 4.5% for doctor-diagnosed asthma worldwide [2]. In 2019, 262 million people were affected with asthma, and 455,000 deaths were seen [3]. According to the Institute for Health Metrics and Evaluation (IHME), 0.82% of total deaths globally are due to asthma, 1.18% of years lived with disability (YLDs), and 0.85% disability-adjusted life years (DALYs). In India asthma accounts for 2.12% of total deaths, 0.81% of YLDs, and 1.25% of total DALYs [4].
The common treatment for inflammatory diseases such as asthma is steroids and the burden of asthma morbidity and mortality is increasing globally due to 5% to 10% of asthma patients, who have greater severity of the disease and insensitivity to high doses of steroids, described as steroid-resistant asthma (SRA) which accounts for 50–80% of all healthcare-associated expenses [5–9]. In 1968, the first instances of glucocorticoid (GC) resistance in asthma were observed and reported in six patients diagnosed with asthma who had poor disease control and did not respond clinically or experience a decrease in blood eosinophilia despite receiving large doses of systemic GCs [10]. SRA is defined as “less than 15% improvement in baseline forced expiratory volume in one second (FEV1) even after two weeks of an adequate dose of prednisolone (40 mg/day)” [11, 12]. The clinical diagnosis of SRA is based on the history, and pulmonary function test with respect to the steroid dose intake to reduce the symptoms. GCs’ primary purposes are to prevent and regulate the emergence of inflammation. Several mechanisms that may vary between patients who can cause resistance to the anti-inflammatory actions of GCs at the molecular level. Host and environmental factors like genetic predisposition, modifications in steroid receptors and/or their ligands, their binding ability, increased expression of transcription factors involved in the inflammatory process, neutrophilia, immunomodulation, respiratory virus and bacterial infections, cigarette smoking, air pollution, allergen exposure, a high-fat diet, and/or obesity can lead to SRA, each factor or interaction of host and environment factors interact with different molecular pathways leading to the severity of the disease (Figure 1) [11, 13, 14].
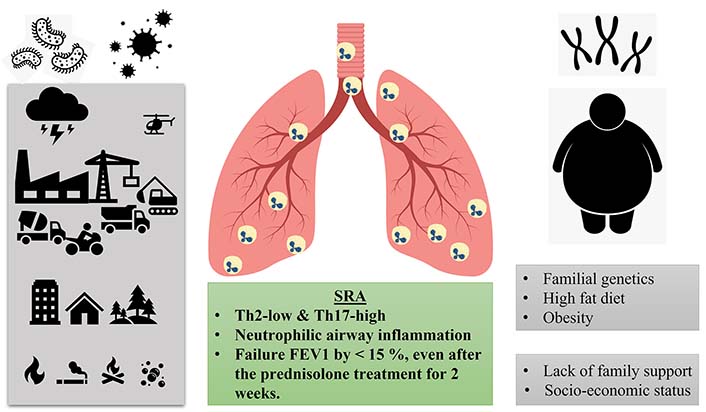
Graphical representation of SRA phenotype. The external factors (bacterial and viral infection, particulate matter generated from the construction, transportation, and vehicles, indoor air pollution like cooking, cooker, and smoking, volcano eruption, occupational like asbestos and silica) and the internal factors responsible the steroid insensitive phenotype. Th2: T helper type 2
Due to its heterogeneity, the term “asthma” should be used to refer to a variety of phenotypes. Two distinct endotypes can be characterized: allergic asthma, atopic which is triggered by Th2 cell responses, and non-allergic, non-atopic (non-Th2) asthma, which is triggered by other immune cells such as neutrophils [15–18]. SRA is characterized by a lack of response to corticosteroid therapy, leading to persistent symptoms and exacerbations. The complex molecular mechanisms underlying steroid resistance play a pivotal role in disease progression and treatment outcomes. In 1968, SRA was first reported in six patients who did not respond to large doses of oral gluco-corticosteroids with poor disease control [11]. Later, detailed investigations revealed that the poor response to steroid treatment arises from the comparatively lower efficacy of steroids in exerting their anti-inflammatory effects compared to their endocrine or metabolic functions showing a low eosinopenic response [19, 20]. SRA is linked to the non-eosinophilic endotypes of asthma, whereas neutrophilic asthma is characterized by the activation of innate immune responses, specifically, this involves the triggering of Toll-like receptor 2 (TLR2) and TLR4 responses, as well as nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein 3 (NLRP3), inflammasome, and interleukin (IL)-1β responses [16, 21]. Anti-inflammatory corticosteroids, like GCs, are the primary treatment for patients with asthma, which can be administered orally or through inhalation [22]. The inhaled corticosteroids (ICSs), which are synthetic and lipophilic, are absorbed into the tissue of the airways upon inhalation, where they bind and activate GC receptors (GRs). The GRs bind to GC response elements (GREs) in turn which can regulate gene expression, thereby exerting an anti-inflammatory effect by interacting with transcription factors like nuclear factor kappa B (NF-κB), activating protein-1 (AP1), and leucine zipper. NF-κB is a crucial regulator of immune genes and pro-inflammatory cytokines, and evidence suggests that GR can reduce inflammation by inhibiting NF-κB activity (Figure 2) [23–25]. GR inhibits the phosphorylation of mitogen-activated protein kinase (MAPK) proteins, which is essential for their activation [26]. GR indirectly inhibits protein synthesis by decreasing RNA stability, which encodes cyclooxygenases [26]. GR is a homo-dimer that belongs to the nuclear receptor type 1 family with a common structure that consists of a highly conserved DNA-binding domain (DBD), activation function-1 (AF-1) located in the N-terminal region of the receptor, ligand binding domain (LBD) situated at the C-terminal region of the receptor, and a ligand-dependent AF-2, which is regulated by binding of GC hormones and co-regulatory proteins [27, 28]. The human GR gene consisting of nine exons is located on chromosome 5q31.3 with the protein-coding regions between exons 2 and 9 [29]. The human GR gene has three different promoters: 1A, 1B, and 1C, and the GR gene contains multiple GC boxes but does not contain TATA or CCAAT boxes [30]. Four distinct isoforms, GRα, GRβ, GRγ, and GRδ, are produced by alternative splicing of the primary transcript [pre-messenger RNA (mRNA)] of GR among these GRα and GRβ are dominant forms. The only isoform that is physiologically active is GRα and translocated to the cytoplasm, it is widely expressed in tissues and facilitates the genomic action of GCs. GRβ is ineffective in binding to GC due to the non-functional LBD, which instead remains inside the nucleus where it acts as a dominant negative inhibitor [31–34]. Studies have shown that GRβ expression is increased by tumor necrosis factor alpha (TNF-α) and IL-1, which may interfere with the binding of GR to DNA that influences the GC sensitivity and formation of the GRβ/GRα heterodimer weakens the functionality of GRα. GRβ has been shown to have a potential role in GC resistance in various diseases [32]. The GR protein structure is similar to the nuclear receptor family and consists of three primary domains: 1) a variable N-terminal domain (consisting of 421 amino acids), 2) a central DBD (consisting of 65 amino acids), and 3) a C-terminal domain (consisting of 250 amino acids) and both the DBD and the LBD contain the nuclear localization motif [27]. The GCs have three primary mechanisms, which include: 1) the binding of heterodimer receptors to the GRE for the activation of transcription, 2) GR heterodimer binding to the negative GRE which inhibits the target gene expression, and 3) transactivation or trans-repression by physical interaction with the other transcription factors [35]. The GR translocates into the nucleus when it binds with the ligands. GRα stays inside the cytoplasm through its interaction with various proteins like immunophilin p23, p59, heat shock protein 90 (Hsp90), and Hsp70. When the ligand binds to the receptor the protein complex separates, exposing the signals of nuclear localization and changing the shape of the receptors. As a result, the GR is translocated through nuclear pores into the nucleus [36, 37]. The GR protein undergoes post-translational modifications which modulate the receptor activity. GRα has a basal level of phosphorylation and increases in response to the GC binding and has six known phosphorylation sites (serine 113, 141, 203, 211, 226, 404) located on the N-terminal binding domain of the receptor [38]. Phosphorylation at different locations has different effects on the response of gene activity. The phosphorylation plays another post-translational modification of GR, which is the ubiquitination and proteasomal degradation, to regulate the turnover of GR. The GR, activated hormone-bound receptor enters the nucleus where it dimerises and the DBD with the zinc finger motif binds to the GRE which brings the conformational changes in the GR and facilitates the GR to interact with the other coactivators like p300, cyclic adenosine monophosphate response element binding protein, Switch/Sucrose Nonfermentable (SWI/SNF), steroid receptor coactivator-1, and nuclear receptor coactivator-1 (NCoA-1) activates transcription of the genes by chromatin unwinding [34, 35]. The anti-inflammatory response is generated by corticosteroids generated by APs like secretory leuko-protease inhibitors, annexin-1, inhibitor of NF-κB alpha (IκBα), and IL-10. The endogenously produced GC is involved in the various physiological processes in the cells like epithelial cells, immune cells, neurons, cardiomyocytes, and hepatocytes. GC regulates multiple pathways like inflammation, amino acid metabolism, carbohydrate metabolism, and programmed cell death [35, 38]. Mechanisms associated with steroid resistance comprise both genetic as well as acquired factors from the environment of the individual that affect steroid sensitivity [39]. Observational study shows that hospitalizations increase the time of greater air pollution, which is in accordance with the fact that many asthma patients occasionally experience exacerbations [40]. Air pollution increases the oxidative stress in the lungs which leads to inflammation and enhances asthma symptoms [41]. Evidence from epidemiological research typically supports a link between increased usage of bronchodilators like short-acting beta agonists (SABAs) and maintenance drugs like ICS and higher exposure to air pollution [42]. Steroid resistance in asthma patients is inherited from one generation to another, which indicates that hereditary factors determine corticoid sensitivity [43]. Several pathways and mechanisms have been investigated in ten years to elucidate the molecular causes of steroid resistance; however, the pathways contributing to SRA were discussed here. The study suggests that 13-hydroxy octadecadienoic acid, a metabolite of omega-6 fatty acids causes symptoms of severe asthma in mice and is unresponsive to treatment with steroids [42, 44]. This finding is especially important since it has demonstrated that asthma patients’ serum has higher levels of 13-hydroxy octadecadienoic acid than controls. This suggests that 13-hydroxy octadecadienoic acid is an internal factor that leads to steroid resistance in patients with asthma. Therefore, nutritional supplementation may potentially play a significant impact in the development of steroid-resistant traits in asthma patients. The common molecular and cellular mechanisms responsible for steroid resistance in asthma were discussed here [45].
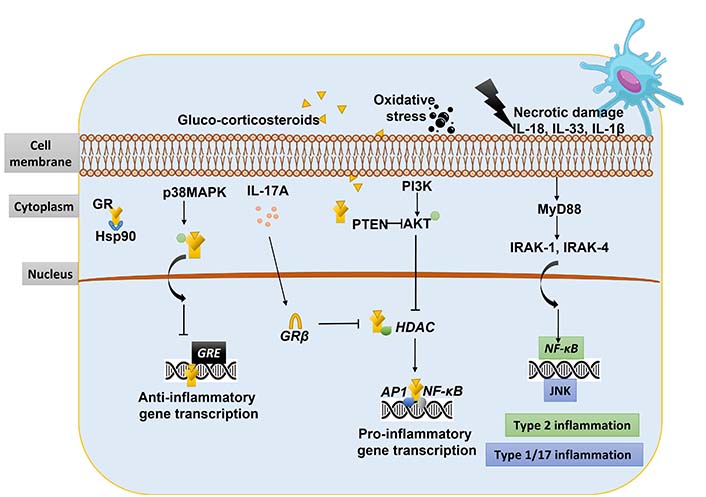
Molecular mechanism of steroid resistance in asthma. PI3K: phosphoinositide 3-kinase; PTEN: phosphatase and tensin homologue; AKT: protein kinase B; MyD88: myeloid differentiation primary response 88; IRAK-1: IL-1 receptor-associated kinase 1; HDAC: histone deacetylase; JNK: c-Jun N-terminal kinase
Studies have shown that IL-2, IL-4, and IL-13 are upregulated in the airways of SRA patients, physiologically this cytokine combination reduces the GR binding affinity and nuclear translocation in the inflammatory cells and T cells. Serine 226 on the GR is phosphorylated by IL-2, IL-4, IL-5, and IL-13, while p38MAPK also induces the phosphorylation of GR which inhibits nuclear translocations [10, 43]. Studies have shown that the p38MAPK inhibitor enhances the response to steroids in peripheral blood mononuclear cells (PBMCs) and alveolar macrophages of patients with severe asthma [46]. Additionally, the phosphorylation of GR at serine 226 by TNF-α-induced JNK inhibits its binding with GRE in PBMC that are isolated from patients with severe asthma [47, 48]. MAPK phosphatase-1 (MKP-1) expression was shown to be significantly downregulated in the alveolar macrophages of individuals who had a poor response to steroids and to be adversely correlated with p38MAPK activity. Corticosteroids and long-acting beta agonists (LABAs), such as formoterol, can stimulate the MKP-1 and protein phosphatase 2A (PP2A), endogenous inhibitors of the JNK and p38MAPK pathways, which counteracts the inefficiency of GR caused by phosphorylation [10, 49]. Elevated levels of inducible nitric oxide synthase (iNOS) have been observed in patients with severe asthma and smoker’s nitric oxide has been shown to alter the ligand binding site of GR by nitrosylation of the Hsp90 interaction site, which prevents GR from moving into the nucleus [50, 51]. It needs to be investigated whether or not this applies to people who are steroid-resistant. Microbial superantigens like staphylococcal enterotoxins, which also boost GRβ expression, may be responsible for some patients’ steroid resistance when they have severe non-allergic asthma [10]. In patients with GC resistance, the expression of GRβ, a dominant negative isoform of GR, is increased in PBMC, lymphocytes, and neutrophils [31].
In patients with steroid resistance and severe asthma, there has been literature that suggests that HDAC2 expression is reduced in alveolar macrophages, airways, and peripheral lungs [52, 53]. The formation of peroxy-nitrite due to nitrative and oxidative stress can result in the nitration of tyrosine residues on HDAC2, leading to its ubiquitination, degradation, and subsequent inactivation [54, 55]. Additionally, oxidative stress can trigger the phosphorylation of PI3K-δ, which can further phosphorylate and inactivate HDAC2 (Figure 2) [56]. The clinical relevance was confirmed in a small group of subjects the steroid anti-inflammatory benefits were found to be reduced, and their adverse effects were also diminished when HDAC2 was inactivated [39]. This was linked to GR’s failure to acetylate lysine residue as a result that prevents the transactivation of genes, which is necessary for both the anti-inflammatory effects and the adverse effects of GCs [39]. HDAC2 is called upon to deacetylate chromatin and alter its structural properties, hence suppressing gene expression. The isoform of PI3K-δ is a critical determinant of steroid resistance in asthma patients, PI3K-δ phosphorylates the AKT, and this phosphorylated AKT induces the HDAC2 gene to undergo phosphorylation and inactivation. At the peripheral lung region and the alveolar macrophages of patients with severe asthma and SRA, HDAC2 is also expressed to a lesser degree. PI3K plays a significant function in this. As a result, HDAC2 can be selectively upregulated and activated by inhibiting PI3K. The cell’s endogenous PI3K inhibitor, known as PTEN, is a tumor suppressor. PTEN overexpression results in decreased PI3K activity and increased nuclear HDAC2 expression (Figure 2) [57].
Cells produce extracellular vesicles called exosomes that contain metabolites, proteins, lipids, and nucleic acids. They influence different facets of cell biology act as a mediator of adjacent and distant intercellular communication and affect cell biology. Exosomes produced by the airway epithelium in response to ovalbumin (OVA) are powerful mediators that increase aryl hydrocarbon receptor (AHR) and cause macrophage, neutrophil, and eosinophil infiltration or activation in the airways [58]. Recent research has shown that neutrophil-derived exosomes can increase the migration and proliferation of airway smooth muscle cells in response to lipopolysaccharide (LPS) stimulation, which may contribute to airway remodeling in asthma [59]. According to this research, exosomes may play a significant role in the development of neutrophilic asthma [59].
The NLRP3-inflammasome plays a vital role in the innate immune system’s response to microbial infection and cellular injury by promoting activating caspase-1 and the release of pro-inflammatory cytokines IL-1 and IL-18. In neutrophilic asthma, the expression of NLPR3 and caspase activity increases, and higher levels of IL-1β protein in their sputum are observed. The neutrophil chemo-attractants IL-8 and IL-1β were found to be correlated with SRA which leads to the IL-1β-dependent inflammatory response [60]. Recent studies have shown that asthma patients with the clinical indicators of severe SRA exhibit elevated levels of IL-1β and NLPR3 mRNA expression in their sputum samples [61]. A correlation was seen between airway obstruction and neutrophil occurrence with the IL-1β and NLPR3 expression. Another group of researchers has shown the obesity-driven SRA and NLRP3-inflammasome relation, which shows the upregulation in the gene expression of the NLPR3 and nucleotide oligomerization domain 1 (NOD1) [62]. Individuals with asthma who were not obese were fed saturated fatty acids-rich food. Following 4 h of eating, there was an observed elevation in neutrophils, TLR4, and NLRP3 expression. Consequently, the NLRP3-inflammasome plays a vital role in regulating the IL-1β pathway in patients with SRA [63].
Wenzel [16] classified asthma patients with neutrophilic inflammation and obesity-induced asthma as exhibiting non-Th2/type 2 asthma (Figure 2). The chemokine C-X-C motif chemokine ligand 8 (CXCL8, IL-8), a strong chemoattractant involved in dragging the neutrophils, was also reported to be present in higher concentrations in asthma patients’ sputum [64]. Asthma severity was associated with an increase in the production of IL-8 and IL-17. The studies have shown that the IL-17A mRNA expression produced from Th7 was elevated in the sputum of asthma patients [64]. IL-17, the pro-inflammatory cytokine, guides the neutrophil-predominant phenotype which leads to steroid-refractory asthma [65]. The preclinical model studies show that the adoptive transfer of Th17 cells demonstrates severe airway neutrophilia and airway hyper-responsiveness compared to Th2 cells and is much more resistant to corticosteroid therapy [66]. In response to infections, tissue, or organ damage, the innate immune system activates neutrophils which migrate to the site of injury, where they aggregate to form neutrophil extracellular traps (NETs). According to Brinkmann et al. [67], the concept of unique neutrophil cell death, or “NETosis”, first emerged in 2004. According to reports, NET formation and autophagy are elevated in the peripheral blood cells, broncho alveolar lavage (BAL) fluids, and sputum granulocytes of allergic asthma patients [66–68]. NETosis has been shown to promote airway blockage, harm the alveolar-capillary network, and disturb the host cellular matrix, all of which contribute to lung disease [64]. Recently, it has been suggested that the development of NETs in asthma results in steroid resistance situations via an IL-17 [69]. The neutrophilic cytoplasts cause the Th17 cell-mediated inflammation in the mouse model, as shown by Krishnamoorthy et al. [69]. Patients with severe asthma demonstrated elevated neutrophilic cytoplast and NET levels, which were positively correlated with the lungs’ IL-17 levels. Therefore, it is crucial to determine whether neutrophilic cytoplasts and NETs are responsible for the steroid-refractory asthma phenotype [70]. Leukotriene-B4 (LTB4) and respiratory infections induce airway neutrophilia. LTB4, a pro-inflammatory lipid mediator, is produced through the metabolism of arachidonic acid which has an anti-apoptotic effect on the neutrophils [71]. GCs increase the longevity of the neutrophils as well as the LTB4, reports have shown that increased LTB4 levels in nasal, serum, and BAL fluid samples when medicated with corticosteroids in asthma patients [71, 72]. Respiratory infections like Chlamydia pneumoniae have a serious impact on asthma having frequent exacerbations along with neutrophilia which makes the patient steroid-resistant [73]. Clinical studies have shown a common association between the presence of Haemophilus influenzae and the development of IL-17-driven neutrophilia in the airways of asthma patients [74]. Infections induced by viruses increase the severity of the disease which leads to an elevation in the infiltration of mixed immune cells and a significant reduction in the FEV1, which leads to the patient being incompetent to respond to the corticosteroids (Figure 1) [75, 76]. Group 2 innate lymphoid cells (ILC-2), the bronchial epithelial cells release alarmins like IL-33, IL-25, and thymic stromal lymphopoietin (TSLP) upon stimulated by allergens, bacteria, viruses, or fungi that induces the activation of ILC-2 which triggers the type 2 immune response progressively and also release excessive quantities of cytokines including IL-5 and IL-13 that, respectively, cause severe eosinophilic inflammation and airway hyper-reactivity [77–80]. The preclinical studies reported from the steroid resistance murine model of asthma established the connection between the TSLP-signal transducer and activator of transcription 5 (STAT5) axis [81]. They have determined that IL-7 may be a moderator of corticosteroid sensitivity because it activates the STAT5 pathway, which increases the production of the anti-apoptotic proteins B-cell lymphoma-2 (Bcl-2) and Bcl-extra-large (Bcl-xL) and protects cells from apoptosis. Because IL-7 promotes the STAT5 pathway, which raises the synthesis of the anti-apoptotic proteins Bcl-2 and Bcl-xL and shields cells from apoptosis, they have concluded that IL-7 may be a moderator of corticosteroid sensitivity. The TSLP, a cytokine released from the epithelial cells, which is regulated by the ligand IL-7Rα induces the steroid resistance condition in ILC-2.
SRA shows a complex and heterogeneous phenotype, the exact mechanism of resistance to steroids is not elucidated completely. Infection-induced exacerbation, bacterial and viral respiratory infections, a high-fat diet, and obesity have been associated with steroid resistance in asthma. Understanding molecular and cellular mechanism pathophysiology of the disease phenotype enables to discover the new therapeutic approaches and the development of effective treatments [82–84].
AKT: protein kinase B
Bcl-2: B-cell lymphoma-2
DBD: DNA-binding domain
FEV1: forced expiratory volume in one second
GC: glucocorticoid
GREs: glucocorticoid response elements
GRs: glucocorticoid receptors
HDAC: histone deacetylase
Hsp90: heat shock protein 90
IL: interleukin
ILC-2: group 2 innate lymphoid cells
JNK: c-Jun N-terminal kinase
LBD: ligand binding domain
LTB4: leukotriene-B4
MAPK: mitogen-activated protein kinase
mRNA: messenger RNA
NETs: neutrophil extracellular traps
NF-κB: nuclear factor kappa B
NLRP3: nucleotide-binding domain, leucine-rich repeat, and pyrin domain-containing protein 3
PBMCs: peripheral blood mononuclear cells
PI3K: phosphoinositide 3-kinase
PTEN: phosphatase and tensin homologue
SRA: steroid-resistant asthma
STAT5: signal transducer and activator of transcription 5
Th2: T helper type 2
TLR2: Toll-like receptor 2
TSLP: thymic stromal lymphopoietin
The authors would like to acknowledge the infrastructure and support given by the JSS Academy of Higher Education & Research, Mysore, CSIR-Indian Institute of Chemical Biology, Translational Research Unit of Excellence TRUE campus, Kolkata, and the Swedish Research Council, Karolinska Institute, Sweden.
PAM, UM, and SVM: Conceptualization, Writing—review & editing. MVG: Conceptualization, Writing—original draft, Writing—review & editing. MKU: Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
MKU was supported by the Indian Council of Medical Research (ICMR) for Senior Research Fellowship (SRF) award [45/13/2022/TRM/BMS]. This work was partially supported by the Council of Scientific and Industrial Research (CSIR), India through grant [MLP137]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

