 Review
Review

Affiliation:
Department of Dental Pharmacology, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Okayama 700-8525, Japan
Email: trantienmanh1508@gmail.com
ORCID: https://orcid.org/0000-0003-3266-2773
Explor Target Antitumor Ther. 2021;2:249–265 DOI: https://doi.org/10.37349/etat.2021.00045
Received: February 22, 2021 Accepted: May 06, 2021 Published: June 28, 2021
Academic Editor: Zui Pan, The University of Texas at Arlington, USA
The article belongs to the special issue Calcium Signaling Apparatus in Cancers
Intracellular Ca2+ ions that are thought to be one of the most important second messengers for cellular signaling, have a substantial diversity of roles in regulating a plethora of fundamental cellular physiology such as gene expression, cell division, cell motility and apoptosis. It has been suggestive of the Ca2+ signaling-dependent cellular processes to be tightly regulated by the numerous types of Ca2+ channels, pumps, exchangers and sensing receptors. Consequently, dysregulated Ca2+ homeostasis leads to a series of events connected to elevated malignant phenotypes including uncontrolled proliferation, migration, invasion and metastasis, all of which are frequently observed in advanced stage lung cancer cells. The incidence of bone metastasis in patients with advanced stage lung cancer is estimated in a range of 30% to 40%, bringing about a significant negative impact on both morbidity and survival. This review dissects and summarizes the important roles of Ca2+ signaling transduction in contributing to lung cancer progression, and address the question: if and how Ca2+ signaling might have been engaged in metastatic lung cancer with bone metastasis, thereby potentially providing the multifaceted and promising solutions for therapeutic intervention.
Intracellular Ca2+ [(Ca2+)i] signaling is implicated in regulation of a variety of physiological processes deciding either cell survival or death. In unexcited states, (Ca2+)i ions are maintained at a very low level (in a range of 50–150 nM) nonetheless, a transient elevation of (Ca2+)i, obtained through either (Ca2+)i efflux from intracellular organelles into cytosol or through (Ca2+)i influx into cytosol from extracellular milieu, could mediate activation of various downstream signaling cascades [1]. Specifically, (Ca2+)i fluctuation is tightly regulated by a series of Ca2+ channels, pumps and/or exchanges. Dysregulation of (Ca2+)i homeostasis is a cause of the certain diseases such as developmental disorders, hypertension, cardiovascular disease, diabetes, Alzheimer’s disease, and cancer [2, 3]. In the context of cancer, whether dysregulated (Ca2+)i homeostasis is necessary for malignant initiation has been disputable; however, there have been increasing cues proposing that dysregulation of (Ca2+)i homeostasis might be a central point of defects in mechanisms upon tumor promotion.
The Ca2+ channels, pumps and/or exchanges at plasma membrane (PM) predominantly mediate the intermittent Ca2+ flux from outside to inside of cells such as voltage-gated Ca2+ channels (VGCCs), specific receptor-operated channels (ROCs) and store-operated Ca2+ channels (SOCs), which are stimulated by membrane depolarization, by the external agonists and by depletion of internal Ca2+ stores, respectively. Meanwhile, the inositol-1,4,5-trisphosphate (IP3) receptor (IP3R) and the Ryanodine receptor (Ca2+-induced Ca2+ release channels-RyR) are two key Ca2+ receptors releasing Ca2+ from the internal stores such as endoplasmic reticulum (ER). Mechanistically, binding of IP3 ligand to IP3R triggers IP3R activation, resulting in Ca2+ release from ER into cytosol [4] whereas RyRs, whose activity is dependent upon (Ca2+)i concentration, release (Ca2+)i from ER into cytosol in different cell types such as neurons, muscle cells, and epithelial cells [5, 6]. In addition, there are two leading systems responsible for Ca2+ extrusion across PM, including (1) the plasmalemmal Ca2+-ATPase (PMCA), a calmodulin (CaM)-dependent Ca2+ ATPase regulating contractility in vascular, bladder and uterine smooth muscle [7], and (2) the electrochemically driven Na+/Ca2+ exchanger (NCX), which is a bi-directional transporter exchanging three Na+ for one Ca2+ critically regulating (Ca2+)i in heart [8]. Besides, (Ca2+)i accumulation into ER could be mediated by the sarco-ER Ca2+-ATPase (SERCA), ubiquitously present in ER of all eukaryotic cells. For instance, SERCA played a role in promoting relaxation via pumping (Ca2+)i into the lumen of sarcoplasmic reticulum (SR) that is a major subcellular pool of Ca2+ [9].
Lung cancer, also known as lung carcinoma, is the most frequently diagnosed malignancy and the leading cause of cancer death globally. Two major types of lung cancer best characterized include small cell lung carcinoma (SCLC) and non-SCLC (NSCLC), the latter accounting for approximately 85% of all lung cancers spreads locally to the thoracic cavity and to distant organs including bone [10]. Specifically, a range of 30%–40% of patients diagnosed with advanced stage lung cancer might have developed bone metastasis in a course of their etiological progression, bringing about a significant negative impact on both morbidity and survival [11]. Neoplastic bone formation is primarily derived from dysregulation of bone remodeling and homeostasis, tightly controlled by two functionally interrelated types of cells, (1) osteoblasts (OBs), which account for bone formation and (2) osteoclasts (OCs), which are responsible for bone resorption. It has been demonstrated that the “horrific consequence” of bone metastasis occurs as metastatic cancer cells enable to stimulate bone-resorbing activity of OCs, thereby leading to enhanced bone resorption [12]. Importantly, Ca2+ ions and cytokines released from osteoclast-triggered bone resorption promote tumorigenesis, contributing towards augmentation of tumor-propagating capacity of cancer cells and osteoclast-triggered bone resorption as well [13].
In summary, understanding of causes and consequences of regulatory mechanisms of (Ca2+)i signaling associated with lung cancer progression and development of metastatic lung cancer with bone metastasis may shed a light on the potential therapeutic targets or prognostic biomarkers for treatment of lung cancer patients with advanced stage lung cancer with bone metastasis.
As abovementioned, whether or not homeostatic disturbance of (Ca2+)i signaling, either transient or sustained, is one of major causes to initiate malignant events, comprising cell cycle, apoptosis, and metastasis, is still questionable. Nevertheless, followed by such malignant events, dysregulated (Ca2+)i signaling is frequently observed to contribute towards tumor progression. In this review article, the in-depth mechanisms upon contribution of dysregulated (Ca2+)i signaling towards lung cancer progression as well as metastatic lung cancer with bone metastasis were discussed.
At early stage of tumor progression, cancer cells normally acquire a vast number of biological alterations that sustain their uncontrolled replicative capacity. Over last few decades, upon the development and technical modernization allowing to probe (Ca2+)i transient oscillation. The functional importance of (Ca2+)i signaling in regulation of cell cycle has been progressively unveiled. As a consequence of the greater than several thousand-fold gradient between (Ca2+)i and extracellular Ca2+ [(Ca2+)e] levels [14], the opening of cell surface Ca2+ channels leads to an immediate influx of (Ca2+)e across PM. Besides, the transient elevation of (Ca2+)i could be mediated through Ca2+ efflux from the internal Ca2+ stores such as ER, Golgi complex and the others.
Recently Ca2+ signals have emerged to be the hub of controlling G1 phase, G1/S and G2/M phase transitions [15]. In reality, cells are frequently sensitive to depletion of (Ca2+)e in G1, in which Ca2+ is critical for the expression of specific genes required for cell division such as FOS, JUN and MYC. Specifically, FOSL1, also known as aka FRA-1, a member of Fos family, is required for Kras-induced lung tumorigenesis in vivo, and promotes human lung adenocarcinoma proliferation and survival [16]. Besides, C-myc functions as a downstream signal of several growth factor receptors such as epidermal growth factor receptor (EGFR), transforming growth factor alpha (TGFα), transforming growth factor beta (TGFβ) receptor, interleukin (IL)-6 receptor, Notch receptor, and Frizzled receptor [17]. Importantly, C-myc also serves as one of the master transcription factors of many target genes that encode for proteins essential for regulation of cell growth and proliferation such as p15, p21, CDK4, CDC25A, E2F1 [18–22].
One of most important pathways regulated by (Ca2+)i signaling towards cell cycle progression is mitogen-activated protein kinase-renin-angiotensin system (MAPK-Ras) pathway. MAPK [rat sarcoma virus (Ras), rapidly accelerated fibrosarcoma (Raf) and mitogen-activated protein kinase (MEK)] pathway keeps a major role in regulating a variety of cellular processes such as proliferation, differentiation and surivial. The abnormal expression of MAPKs is frequently observed in NSCLC [23]. It is best characterized that MAPK pathway is initiated by the external stimuli such as hormones, growth factors (GFs), cytokines and intracellular molecules, following the activation of the RAS upstream receptors including receptor tyrosine kinases (RTKs) and EGFRs. Activation of MAPK-Ras signaling pathway promoting cell cycle progression [24] by retinoblastoma (RB1) phosphorylation, which then triggers upregulation of cyclin D1-induced CDK4 or CDK6, eventually driving G1-to S-phase transition [25–32]. Deregulation of EGFR, also called ErbB-1, was found in a range of 40–89% of NSCLC [33]. Furthermore, (Ca2+)i is also crucial for regulation of several Ca2+-dependent cascades such as calciuneurin (CaN) and CaM-kinase. CaN, a Ca2+- and camodulin-dependent serine/threonine protein phosphatase, plays a key role in promoting cell cycle progression at G1/S phase transition through cyclin D1 stabilization [34]. Liu et al. [35] identified that CaNAα, an isoform of CaN, which was overexpressed in lung cancer tissues, promoted cell proliferation through accelerating G1-to S-phase transition in SCLC cells in vitro. CaN inhibition by cyclosporin A (CsA) blocked the transcriptional activity of CREB binding protein (CBP) and the nuclear factor of activated T cells (NFAT), leading to alleviate the expression of pro-inflammtory cytokine genes [36, 37]. Noticeably, activation of transcription factors such as CREB and myocyte enhancer factor-2 (MEF-2) was regulated by (Ca2+)i elevation by the (Ca2+)e influx across PM via L-type voltage-gated channels (LTCs) [38]. CsA-triggered CaN inhibition declined CDK2 activity by diminishing the expression of cyclin D1 during G1 [39], cyclin E and cyclin A [40]. Increases in (Ca2+)i concentration result in activation of CaN, which subsequently dephosphorylates NFAT proteins, allowing them to translocate to the nucleus to regulate the expression of the target genes [41].
Orai3 channels, frequently overexpressed in NSCLC, mediate Ca2+ entry via store-operated Ca2+ entry (SOCE) and promote cell cycle progression via Atk pathway [42]. Orai3 silencing downregulated MAPK kinase pathway via diminishing the phosphorylation form of ERK1/2, and expression of C-myc, which triggered cell cycle arrest in G1 phase in breast cancer [43]. On the contrary, overexpression of Ca2+ release-activated Ca2+ channel protein 1 (Orai1), also known as CRACM1, triggered reduction of store-operated Ca2+ influx and attenuation of EGF-mediated proliferative signaling and driving cell cycle arrest in A549 lung cancer cells [44]. Furthermore, antigen-stimulated opening of Ca2+ release activated Ca2+ (CRAC) channel, a highly Ca2+-selective store-operated channel, enables the refilling of ER Ca2+ stores and maintain the persistency of Ca2+ oscillations which are first identified essential for T cell proliferation and cytokine production [45]. Conformational change and redistribution of stromal interaction molecule 1 (STIM1), the ER Ca2+ sensor, and Orai1, a key subunit of CRAC channel pore are required for activation of CRAC channel, which, in turn, triggers Ca2+ release from ER lumen into cytosol through activating IP3R and/or (Ca2+)e influx [46, 47]. Also, the depolarization-induced opening of VGCC Cav1.2 is directly suppressed by STIM1, causing a sustained internalization of VGCC Cav1.2 [48]. Heretofore, Wang, et al. [49] identified that STIM1 was significantly overexpressed in lung cancer tissues as compared to that of non-neoplastic lung tissues; furthermore, Ge, et al. [50] revealed that STIM1 knockdown induced cell cycle arrest at G2/M and S phases through alleviating expression of CDK1 and 2 in A549 and SK-MES-1 cells, and abolishing tumorigenesis and growth of lung cancer cells in nude mice xenograft. Upon reaching to cytosol, Ca2+ often forms complexes with the molecular components of “(Ca2+)i signaling molecular toolkit” specific for each cell type given. Among such direct effectors essential for (Ca2+)i signaling are CaM and Ca2+/CaM-dependent protein kinases II (CaMKII), protein phosphatase 2B (PP2B) and protein kinase C (PKC), modulating the transcriptional activity of various transcription factors for a large number of genes required for cell cycle progression [51]. Using 1-[N, O-Bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine) (KN-62), a specific CaMKII antagonist, Williams, et al. [52] observed that blockade of CaMKII activity inhibited the exponentially proliferative capacity of SCLC cells through ameliorating cell cycle arrest at S phase. Altogether, the Ca2+ channels/pumps/exchangers and Ca2+-handling proteins identified affect cell cycle progression in lung cancer cells (Table 1).
The summary of the roles of the major Ca2+ channel/pump/exchanger and Ca2+-handling proteins in regulation of cell cycle in lung cancer cells
| Ca2+-channel/pump/exchangerand Ca2+-handling proteins | Cell line | Expression | Described roles |
|---|---|---|---|
| Orai3 | NCI-H23 and NCI-H460 | Decreased SOCE, abolished cell proliferation and triggered cell cycle arrest at G0/G1 phase [42] | |
| Orai1/CRACM1 | A549 | Not determined | Orai1/CRACM1 overexpression attenuated EGF-mediated store-operated (Ca2+)e influx, and triggers G0/G1 cell cycle arrest [44] |
| STIM1 | A549 SK-MES-1 | STIM1 silencing inhibited colony formation, and induced cell cycle arrest at G2/M and S phases [50] | |
| CaMKII | NCI-H69, NCI-H128, NCI-H146 and NCI-H345 | Not determined | KN-62-induced inhibition of CaMKII activity triggered reduced DNA synthesis and cell cycle arrest at S phase [52] |
| CaNAα | SBC-3 | Promoted G1/S phase transition [35] |
: increased
Apoptosis, a programmed cell death (PCD), is important for removal of mutated or transformed cells from the body essential for embryogenesis, development and tissue homeostasis of multicellular organisms. Principally, apoptosis comprises two core pathways: (1) the extrinsic pathway and (2) the intrinsic pathway, which are sequentially referred to as death receptor (DR)-mediated pathway and mitochondrial pathway [53]. Though stimulated in different manners, these pathways commonly converge into the same destination. Both the intrinsic and extrinsic apoptotic mechanisms lead to the activation of caspase 8, an initiator of a series of the apoptotic events through activating caspase 3, 6 and 7, subsequently resulting in cell collapse, chromatin condensation, breakdown of nuclear DNA, formation of apoptotic bodies and recognition of apoptotic cells by phagocytic cells [54].
During carcinogenesis, cancer cells acquired specific mechanisms of protection against apoptosis [55]. Among these, Ca2+ has been emerged as an important element exploiting its specialized effects towards regulation of apoptosis [56]. Under specific conditions at the initial step of mitochondria-induced apoptosis, overload of mitochondrial Ca2+, a vital sensitizer of the mitochondrial permeability transition (MPT) triggers mitochondrial swelling, perturbation of the mitochondrial outer membrane [57]. MPT pore-triggered release of the pro-apoptotic factors such as cytochrome c, apoptosis inducing factor (AIF), procaspase-9, Smac/DIABLO, and endonuclease G into cytosol causes a massive activation of proteases (caspases) and phospholipases [58–61]. To resist to apoptosis, cancer cells typically acquire the highly protective mechanisms against abolishment of mitochondria-triggered Ca2+ signals.
ER-derived Ca2+ signals, critical for regulating the apoptosis-related events, are also engaged in the mitochondria-induced apoptosis via the mitochondria-associated membranes (MAMs) juxtaposed between ER and mitochondria [62, 63]. Ca2+ storage in the ER is accomplished by the action of SERCA and of the intraluminal ER Ca2+-binding proteins such as BiP, calreticulin and calnexin whereas the release of Ca2+ from the ER is virtually mediated by IP3Rs. The role of MAMs for regulation of (Ca2+)i homeostasis is mediated by IP3R3, RyR and SERCA [63]. Upon ER-derived Ca2+ signal-induced mitochondrial remodeling, B-cell lymphoma-2 (Bcl-2) proteins the first anti-apoptotic proteins identified regulate apoptosis via regulating Ca2+ transfer between ER and mitochondria [64]. These proteins are functionally categorized into the anti-apoptotic group (Bcl-2, BCL-XL, and Mcl-1) and the pro-apoptotic group (Bax, Bak, Bim, Bid, etc.). Anti-apoptotic Bcl-2 proteins regulates apoptosis by modulating the ER-mitochondrial Ca2+ transfer via the MAMs [65] while overexpression of pro-apoptotic Bcl-2 decreased both ER-Ca2+ release either by the direct control of IP3R3-mediated pore opening or by lowering the Ca2+ content of the ER, which weakens Ca2+-triggered MPT, and thus enables cancer cell to resist to apoptosis [66]. Abnormal upregulation of pro-apoptotic Bcl-2 is frequently observed in various types of cancer cells such as gastric, colon, breast and lung cancer [67–70]. Indeed, alleviation of ER-Ca2+ levels and signals has been observed in pro-apoptotic Bax and Bak-knockout murine embryonic fibroblasts (MEFs) [71]. Strikingly, enhanced ER Ca2+ levels by ectopic expression of SERCA2 rescue their sensitivity to death stimuli, suggesting the functional necessity of Bcl-2 proteins in regulating the ER-mitochondrial Ca2+ gateway and cell death in MEFs [71]. Bcl-2 mutant has also been reported to reduce ER Ca2+ by inhibition of SERCA2 as a consequence of a reduction of SOCE [72, 73]. Bergner, et al. [74] reported that a reduction of Ca2+ content correlated with a decreased expression of SERCA2 pumping Ca2+ into the ER, an increased expression of IP3R releasing Ca2+ from the ER in various types of lung cancer cell lines. In contrast, the detailed mechanisms underlying Bcl-2-mediated regulation of SOCE remains controversial. Depletion of Ca2+ in the ER causes translocation of the SOC channel activator, STIM1, to the PM [75]. Thereafter, binding of STIM1 to Orai1 and/or transient receptor potential channel 1 (TRPC1) forces them to open for allowing Ca2+ entry across the PM [75].
Oncogenic K-RAS, which degenerates ER Ca2+ dynamics [76], and Akt, which not only phosphorylates and inactivates several pro-apoptotic Bcl-2 such as Bad, Bax and hexokinase-2, but more importantly diminishes mitochondrial Ca2+ overload via alleviating IP3R opening, also contribute towards inhibition of the intrinsic apoptosis inhibition [77, 78]. Concomitantly, downregulation of protein phosphatase and tensin homolog (PTEN) in NSCLC tumors [79] antagonized F-box and leucine rich repeat protein 2 (FBXL2)-induced ubiquitination of IP3R3, thereby stabilizing IP3R3 in ER [80]. Besides, EGFRs with its aberrant expression and constitutive activation in in NSCLC [81] stimulate three of the most well-characterized signaling branches such as Ras-MAPK, phosphoinositol 3 kinase (PI3K)-protein kinase B (PKB)/Akt and phospho lipase C (PLC)-PKC pathways, enhancing ER-mitochondria Ca2+ transfer, thereby abolishing mitochondria-induced apoptosis. Also, loss of promyelocytic leukemia protein (PML) isoform IV, a suppressor of transcriptional activity of EGFR, for instance, on cyclin D1 gene promoter in lung cancer cells [82] also contributed to the EGFR-mediated mitochondria-induced apoptosis and cell cycle arrest [81].
Metastasis, a term used to describe the spread of cancer cells from the primary tumor to surrounding tissues and to distant organs, is the major cause of morbidity and mortality in cancer patients. Among all solid tumors, SCLC is one of the most aggressive malignancy associated with a majority of patients diagnosed with metastatic disease [83]. Metastatic SCLC cells easily dissociate from lungs to disseminate throughout bloodstream and/or lymph system to anatomically distant organs such as lymph nodes, brain, liver and bone [83].
Progress of metastatic cascades primarily begins with the loss of cell-extracellular microenvironment [extracellular matrix (ECM)] as well as cell-cell attachment. Cells are connected to the ECM at focal adhesion points by structural complexes linking membrane spanning integrins to the cytoskeleton. Therefore, migratory capacity of cancer cells are principally assessed by the rate of focal adhesion assembly and disassembly. Noticeably, Ca2+ pulses promote the association of focal adhesion kinase (FAK), which regulates focal adhesion turnover, with the focal adhesion complex (FAC). More detailed, Ca2+ pulses strengthen the FAK at the specific sites where it is phosphorylated in a CaMKII-dependent manner. The movement of migrating cells is initialized by the extension of the protrusive front edge, which is known as lamellipodia. For cell protrusion, actin polymerization in lamellipodia and filopodia is required [84]. The attachment of lamellipodia to the substratum and contraction of the rear edge enable cells to move towards the lamellipodia. Establishment of a gradient difference of (Ca2+)i levels, which was lower in the front, and higher in the rear of the migrating, polarized cells, caused rear retraction, focal adhesion (at the rear) and protrusion (at the front) [85, 86]. Following protrusion, the cell front starts to retract and locally adhere to ECM in lamella [87], which plays a pivotal in actomyosin contractility and F-actin disassembly in a treadmill-like manner [88]. Furthermore, actin and myosin, two of the important structural constituents, are regulated indirectly by Ca2+ signaling via the activation of the cyclic element-mediated Ca2+-dependent kinases, named calpain [89], and regulation of Rac1, RhoA, Cdc42, protein kinase A (PKA) [90, 91], and local Ca2+ signals between lamellipodia and lamella [92]. For retraction of the rear edge, Ca2+ signaling play a vital role in maintaining contractivity and stabilizing the directional movement via modulating the Ca2+ influx through L-type Ca2+ channels [93]. Ca2+-dependent MLC kinase (MLCK)-mediated phosphorylation of myosin light-chain (MLC) triggers myosin II-induced actomycin contractility [94], promoting the retraction and adhesion more efficiently [95, 96].
It is well-characterized that “local Ca2+ pulses” in the front of migrating cells are released from ER via IP3-induced activation of IP3Rs [97], which is generated through activation of RTK-PLC-dependent signaling pathways. It is therefore proposed that ER-derived Ca2+ release by the axis of RTK-PLC-IP3R would be major source of Ca2+ pulses in the front of migrating cells. Indeed, EGFR, also known as HER1, belonging to the ErbB family of structurally related RTKs that comprises four isoforms: ErbB2 (HER2), ErbB3 (HER3) and ErbB4/HER4 [98], is overexpressed and constitutively activated in 62% of NSCLC cases [99]. It is clear that EGFR is the key activator of the ERK/MAPK, Akt-PI3K, and PLCγ-PKC signaling cascades [100]; furthermore, Tsai, et al. [101] reported RTK and PLC were enriched at the leading edge of migrating cells, in correlation with intensity of local Ca2+ pulses in the cell front.
In addition to RTK, G-protein coupled receptors (GPCRs) on local Ca2+ pulses in the cell front via activating PLC, which hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2) to release IP3, which binds to IP3R to trigger transient Ca2+ release [102, 103]. In a meanwhile, depletion of ER luminal Ca2+ sensitizes SOC channels located at PM to infux (Ca2+)e across PM [104]. Chantôme, et al. [105] once reported that the interaction of Orai1 with SK3 channel, a potassium channel belonging to the small conductance Ca2+-activated potassium (KCa) channel family, regulated the constitutive (Ca2+)e entry through Orai1 localization within the lipid raft, which affected the migratory ability of breast cancer cells. Disruption of interaction between SK3 and Orai1 from lipid rafts weakened SK3-mediated Ca2+ entry, migration and bone metastasis [105]. Moreover, STIM/Orai-mediated SOCE is also essential for elevation of (Ca2+)i level. STIM1, the ER Ca2+ sensor, and Orai3, constituent a native SOC entry essential for NSCLC progression [42, 50]. Increasing evidence implied that STIM1 assisted the turnover of cell matrix adhesion complexes, thereby enhancing cell migration by maintaining local Ca2+ pulses in the front of migrating cells [106]. In migrating cells, local Ca2+ pulses near its leading edge cause depletion of Ca2+ in the front ER, resulting in activation of STIM1 at the cell front [101]. More specifically, STIM1 was remarkably translocated to the ER-PM junction in cell front rather than cell rear during cell migration, thereby promoting maintenance of cell polarity and motility [101].
In addition, inhibition of the activity of Ca2+ permeable channels, PMCA and NCX, by the specific blockers, which are vanadate (V5+) and KB-R7943, respectively, led to a decrease in migratory capacity of MDCK-F cells, suggesting these channels were of significant importance for cell migration [107]. Furthermore, inactivation of ER membrane-located SERCA, which is responsible for pumping (Ca2+)i into the ER lumen, triggering a leak of the ER lumical Ca2+ into cytosol [101]. The high (Ca2+)i concentration caused MLCK saturation and myosin contractility [101]. Indeed, Atousa Arbabian [108] once reported that dysfunctional SERCA diminishes the ER luminal Ca2+, thereby disabling further Ca2+ signaling through IP3Rs, suggesting a physiological importance of SERCA on lung cancer progression, invasion and metastasis.
Bone is one of the most common metastatic sites for lung cancer, in which 36% of patients with bone lesions, and a range of 20%–60% with bone marrow micrometastasis [109, 110]. Metastasis lung cancer with bone metastasis is a major source of morbidity and mortality; however, it is not frequently detected in the patients until pain, skeletal-related events (SREs) in spine, ribs, pelvis and proximal long bones, pathological fractures and nerve compression syndromes occur [111]. Therefore, comprehension of why and how the various specific features of bone microenvironment, associated with spatio-temporal fluctuations of Ca2+ signaling network preferentially towards bone metastasis is essential for development of the efficacious drug program.
Bone is a dynamic organ included a variety of embryo-derived cells such as hematopoietic, stromal, endothelial, adipocytes, OCs, OBs and osteocytes [112]. Two important mediators of the hematopoietic stem cell (HSC) environment are (1) the chemo-attractant stromal derived factor-1 (SDF-1) or C-X-C motif chemokine ligand 12 (CXCL12) and (2) the cell adhesion factor (Annexin2 or ANXA2) [113]. CXCL12 regulates HSC homing to the bone marrow, while ANXA2 is likely involved in HSC binding to the osteoblastic niche, and may act as an anchor of CXCL12 and aid in localization to the niche [113]. The disseminated tumor cells (DTCs) could survive in a quiescent state in bone marrow of cancer patients for years. Increasing evidence suggests that DTCs gain access to the bone marrow using homing mechanisms similar to those of HSCs. The interaction of CXCL12, which is secreted by bone marrow stromal cells including fibroblasts and endothelial cells, to C-X-C motif chemokine receptor 4 (CXCR4), which is aberrantly expressed in tumor cells, allows tumor cells to directionally migrate to bone [114] mainly through upregulating the two most crucial downstream pathways comprising IP3K and MAPK pathways. Nonetheless, the detailed mechanisms of how CXCR4 /CXCL12 interaction stimulates metastasis and/or tumor growth and their complete implications on metastatic lung cancer in bone are unknown.
Bone homeostasis is maintained by two major types of bone cells, consisting of OBs and OCs, which are responsible for bone formation and bone resorption, respectively [115]. Of these, OBs, differentiated from mesenchymal stem cells, participate in regulating bone remodeling by generating ECM and calcium phosphate crystals, which are deposited into the interstitial space of the matrix [116]. OCs, the polarized, multinucleated myoleoid lineage cells, adhere to the bone surface through αvβ3 integrin, form ruffled borders, and secrete acid to solubilize calcium phosphate crystals as well as secret the collagenases and proteinases such as tartrate-resistant acid phosphatase (TRAP), matrix metallopeptidase 9 (MMP9), and cathepsin K (CTSK) that demineralize and degrade extracellular proteins such as type I collagen [117].
Bone has several particular characteristics such as acidified milieu, hypoxia (O2 deficiency) and high level of (Ca2+)e, enabling tumor cells to establish an acidic microenvironment via production of a large amount of lactic acid, which then creates the local areas inside bone, thereby accelerating tumor cell dormancy and promotes osteolysis [118]. The release of bone resorption-derived (Ca2+)e triggers activation of Ca2+-sensing receptor (CaR), a G protein-coupled receptor, on PM of tumor cells and OBs [119], OCs [120] and especially tumor cells [121], including lung adenocarcinoma [122]. Activation of Ca2+-sensing receptors enhances the secretion of parathyroid hormone-related peptide (PTHrP), which subsequently binds to its receptor, PTHR1, to increase receptor activator of nuclear factor kappa-B (RANK) ligand (RANKL) expression in bone marrow stromal cells, thereby promoting osteolysis [121]. Furthermore, RANKL-mediated osteoclast differentiation triggers IP3R-induced local Ca2+ release, inducing activation of one of the master transcription factors of osteoclastogenesis, the NFATc-1 [123], subsequently entering nuclei to bind to the promoters of specific genes required for osteoclast differentiation [123]. In addition, it is unknown whether bone resorption-derived (Ca2+)e might have been responsible for activating the Ca2+ channels, pumps and exchangers to promote differentiation and growth of metastatic lung cancer cells (MLCCs) in bone.
In regardless of RANKL, TGFβ, a bone resorption-derived factor, enhances the PTHrP expression in tumor cells and OBs, thereby promoting osteolysis [124]. Specifically, TGFβ-mediated signaling pathway activating a couple of important intracellular cascades, consisting of MAPK, PI3K/Akt, and Rho-like GTPase signaling cascades [125], critically acts as a driver of tumor progression and metastasis [126]. Importantly, the tumor cells could significantly produce not only the ILs such as IL-6, IL-8, and IL-11, required for osteoclastogenesis [127], but also strengthen the expression of CXCR4 and CXCR7, establishing a “fertile soil” that accelerates tumor cells to adhere to bone matrix and thrive in bone [128, 129] (Figure 1).
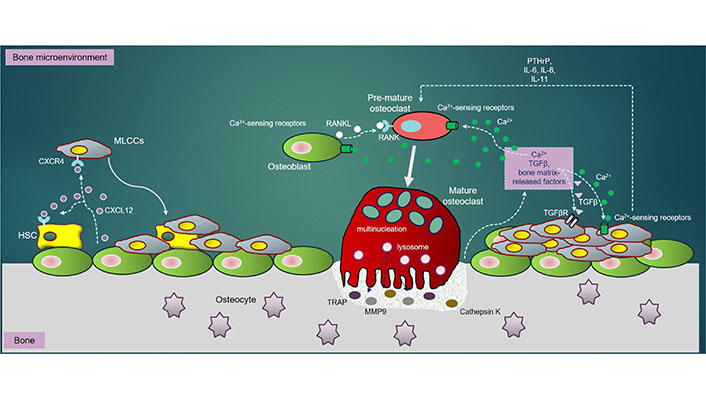
The MLCCs in bone: once in bone extracellular matrix (BEM), MLCCs encounters with the bone marrow stromal cells. The CXCR4/CXCL12 interaction enables MLCCs to attach to the osteogenic niches, strengthening MLCCs to survive, proliferate and metastasize. RANKL, secreted by OBs, directly binds to RANK receptor on PM of pre-OCs, triggering differentiation of pre-OCs into mature (multinucleated) OCs. Mature OCs secrete bone-resorbing elements including TRAP, CTSK and MMP9, all of which resorb bone to release Ca2+ ions, TGFβ and other bone matrix-release factors into BEM. Ca2+ ions released subsequently activate OC differentiation through NFATc-1 downstream signaling pathways. Besides, bone resorption-derived Ca2+ ions interact with Ca2+-sensing receptors highly expressed in OBs, OCs and MLCCs, which further promotes survival, proliferation, differentiation and metastasis of MLCCs in bone. Moreover, the interaction of bone resorption-derived TGFβ to its receptor, TGFβR highly expressed in MLCCs, activates several important downstream signaling cascades such as MAPK, PI3K/Akt, and Rho-like GTPase, which synergistically enhance metastatic properties of MLCCs in bone. Additionally, the ILs (IL-6, IL-8 and IL-11) and PTHrP secreted by MLCCs also contribute towards augmentation of OC differentiation and bone resorption
In this article, I have reviewed the role of Ca2+ as a key regulator of lung cancer progression and bone metastasis with the metastatic lung cancer. Principally, Ca2+ signals are intrinsic to all aspects of cancer biology, especially in the metastatic lung cancer with bone metastasis. Therefore, identification of the key Ca2+ channels, pumps and/or exchangers would be beneficial for development of treatment strategies for lung cancer. Unfortunately, the exact mechanisms underlying Ca2+ signaling-mediated regulation of lung cancer progression has been incomprehensively understood. Basically, three major steps of bone metastasis required include (1) migration, (2) adhesion and invasion to bone, and (3) proliferation, growth and metastasis in bone. However, it is unclear whether aberrant changes in Ca2+ signals are one of the primary causes of initiation of lung cancer progression.
To what extent can therapeutic strategies exploit these Ca2+-regulated processes? Accumulating preclinical and clinical evidence has elucidated the relationship between aberrant Ca2+ signaling and tumor progression. Using the specific blockers of Ca2+ channels, pumps and/or exchangers has demonstrated the significant antitumor effects on lung cancer progression (Table 2), indicating that Ca2+ signaling would be a promising target for novel lung cancer treatments. However, before contemplating such efficacious therapeutic interventions based on pharmacological modulation of Ca2+-regulators, it is crucial to design more potent and specific, but less off-target drugs targeting Ca2+-regulators, including Ca2+ channels, pumps and/or exchangers. Therefore, further studies are required to verify the toxicity and pharmacokinetic of such modulators prior to the clinical tests.
Summary of the major compounds targeting Ca2+ channels/pumps/exchangers in lung cancer progression
| Ca2+ channel/pump/exchanger | Drug Candidates | Pharmacological effects |
|---|---|---|
| TRPCs | SKF-96365 | Cell cycle arrest at S/G2M phase, and invasive ability in A549 cell line [130] |
| ATRA2-ABP | Proliferative inhibition in A549 cells line [131] | |
| Carvacrol | Degeneration of cell morphology, and apoptosis in A549 cell line [132, 133] | |
| Capsaicin | Apoptosis in SCLC cell lines, NCI-H82, NCI-H69 [134] | |
| Tetrahydrocannabinol and cannabidiol | Inhibition of proliferation, epithelial-mesenchymal transition (EMT) and migration in A549, H460 and H1792 lung cancer cell lines [135] | |
| Dexamethasone | Growth suppression in NSCLC cell lines, A549 and H1299 [136] | |
| RyR | Compound K | ER-mediated apoptosis in A549 and SK-MES-1 cell lines [137] |
| Paclitaxel | Cell cycle arrest at G2/M phase in A549 cell line [138] | |
| IP3R3 | Α-Lipoic acid (LA) | Apoptosis in A549 cell line [139] |
| Curcumin | Apoptosis in NSCLC cell lines, A549 and H1299 [140] | |
| VGCCs | Verapamil, Diltiazem, and Nifepine | Cell death in chemoresistant lung cancer cells derived from A549 cell line [141] |
| SERCA | 2-deoxy D-glucose and metformin | Apoptosis in A549 cell line [142] |
| Voltage-dependent anion channel (VDAC) | R-Tf-D-LP4 | Apoptosis and inhibition of tumor growth HepG2 and Huh-7 cell lines [143] |
(Ca2+)e: extracellular Ca2+
(Ca2+)i: intracellular Ca2+
Bcl-2: B-cell lymphoma-2
CaM: calmodulin
CaMKII: calmodulin-dependent protein kinase II
CaN: calciuneurin
CRAC: Ca2+ release-activated Ca2+ channels
CXCL12: C-X-C motif chemokine ligand 12
CXCR4: C-X-C motif chemokine receptor 4
ECM: extracellular matrix
EGFR: epidermal growth factor receptor
ER/SR: endoplasmic/sarcoplasmic reticulum
ER: endoplasmic reticulum
HSCs: hematopoietic stem cells
IL: interleukin
IP3: inositol-1,4,5-trisphosphate
IP3R: IP3 receptor
MAMs: mitochondria-associated membranes
MAPK: mitogen-activated protein kinase
MLCC: metastatic lung cancer cell
MPT: mitochondrial permeability transition
NFAT: nuclear factor of activated T cell
NSCLC: non-small cell lung cancer
OBs: osteoblasts
OCs: osteoclasts
Orai1: Ca2+ release-activated Ca2+ channel protein 1
PI3K: phosphoinositol 3 kinase
PKC: protein kinase C
PLC: phospho lipase C
PM: plasma membrane
PTHrP: parathyroid hormone-related peptide
RANKL: receptor activator of nuclear factor kappa-B ligand
RyR: ryanodine receptor
SCLC: small lung cell lung cancer
SERCA: SR/ER Ca2+-ATPase
SOC: store-operated Ca2+ channel
SOCE: store-operated Ca2+ entry
STIM1: stromal interaction molecule 1
TGFβ: transforming growth factor beta
TRP: transient receptor potential
VGCC: voltage-gated Ca2+ channel
The author contributed solely to the work.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Garhima Arora ... Samrat Chatterjee
Chaochu Cui ... Xianwei Wang
Wen-Li Hsu ... Etsuro Ito
Alana L. Cutliffe ... John J. Mackrill
Kathryn A. Skelding ... Lisa F. Lincz
Tianying Xie ... Jianru Xiao
