 Review
Review

Affiliation:
1Department of Physical Therapy, Rehabilitation Science, and Athletic Training, University of Kansas Medical Center, Kansas City, KS 66160, USA
2Department of Orthopedic Surgery and Sports Medicine, University of Kansas Medical Center, Kansas City, KS 66160, USA
Email: zliu2@kumc.edu
ORCID: https://orcid.org/0009-0007-8626-4558
Affiliation:
1Department of Physical Therapy, Rehabilitation Science, and Athletic Training, University of Kansas Medical Center, Kansas City, KS 66160, USA
Affiliation:
1Department of Physical Therapy, Rehabilitation Science, and Athletic Training, University of Kansas Medical Center, Kansas City, KS 66160, USA
ORCID: https://orcid.org/0000-0002-6075-1223
Affiliation:
1Department of Physical Therapy, Rehabilitation Science, and Athletic Training, University of Kansas Medical Center, Kansas City, KS 66160, USA
Email: wliu@kumc.edu
ORCID: https://orcid.org/0000-0002-4762-3104
Explor Neuroprot Ther. 2023;3:235–257 DOI: https://doi.org/10.37349/ent.2023.00049
Received: February 28, 2023 Accepted: June 22, 2023 Published: August 27, 2023
Academic Editor: Abdelhamid Benazzouz, Bordeaux University, France
Parkinson’s disease (PD) is a common neurodegenerative disorder affecting aged population around the world. PD is characterized by neuronal Lewy bodies present in the substantia nigra of the midbrain and the loss of dopaminergic neurons with various motor and non-motor symptoms associated with the disease. The protein α-synuclein has been extensively studied for its contribution to PD pathology, as α-synuclein aggregates form the major component of Lewy bodies, a hallmark of PD. In this narrative review, the authors first focus on a brief explanation of α-synuclein aggregation and circumstances under which aggregation can occur, then present a hypothesis for PD pathogenesis in the peripheral nervous system (PNS) and how PD can spread to the central nervous system from the PNS via the transport of α-synuclein aggregates. This article presents arguments both for and against this hypothesis. It also presents various non-pharmacological rehabilitation approaches and management techniques for both motor and non-motor symptoms of PD and the related pathology. This review seeks to examine a possible hypothesis of PD pathogenesis and points to a new research direction focus on rehabilitation therapy for patients with PD. As various non-motor symptoms of PD appear to occur earlier than motor symptoms, more focus on the treatment of non-motor symptoms as well as a better understanding of the biochemical mechanisms behind those non-motor symptoms may lead to better long-term outcomes for patients with PD.
Parkinson’s disease (PD) is an age-related neurodegenerative condition affecting about 3% of people 65 years or older [1, 2]. PD commonly presents with multiple motor and non-motor symptoms [3, 4]. It was estimated in 2020 that more than 930,000 people had a diagnosis of PD in the United States [5]. Current clinical diagnosis of PD primarily relies on the key motor symptoms [6], and dopamine replacement treatments are used to compensate for striatal dopamine deficiency in managing motor symptoms [7]. However, progress in scientific and clinical research has revealed that many brain areas and the peripheral autonomic nervous system are involved in the PD related neuropathology that are not directly involved in motor control [8]. Therefore, most patients with PD present with a variety of non-motor symptoms including neuropsychiatric symptoms, disorders of sleep, autonomic dysfunction, olfactory dysfunction, pain, fatigue, and others [9]. In fact, non-motor signs or symptoms may precede motor symptoms or diagnoses of PD by years and are very prevalent in PD patients [10, 11]. A recent hypothesis has suggested a different theory of neuropathological stages of PD, from which the Lewy body pathology is assumed to affect the lower brainstem areas and olfactory system before the nigrostriatal system [12]. This model has led to many scientific and clinical studies to examine potential risk factors of olfactory dysfunction [13] or rapid eye movement sleep behavior disorder (RBD) [6, 7]. In addition, the non-motor symptoms of PD are a major determinant of quality of life (QOL) and progress over time in people with PD.
Similarly, some past studies examined the effect of rehabilitation approaches on non-motor symptoms with outcomes related to PD pathological progress. Those studies offered promising results and new opportunities for slowing down the progress of the pathological process [14, 15]. This narrative review provides a summary of relevant basic science research findings on α-synuclein pathology in PD; a review of past studies on motor and non-motor symptoms with outcome measurements relevant to α-synuclein pathology; as well as discussion on future directions of rehabilitation research in managing motor and non-motor symptoms in PD and potential impact on α-synuclein pathology.
The protein α-synuclein plays a central role in neurodegenerative diseases, particularly PD, Lewy body dementia, and Lewy body associated diseases [16–20]. Surguchov and Surguchev [21] in their review point out that α-synuclein “exists in a dynamic equilibrium between soluble monomers and a variety of oligomers”. Soluble α-synuclein can form beta sheet-like oligomeric structures, which act as precursors to the signature insoluble fibrils that aggregate forming Lewy bodies [22]. This suggests that α-synuclein is capable of naturally forming aggregates, and that an overaccumulation of α-synuclein could be the cause of pathology leading to PD [21].
Formation of insoluble α-synuclein fibrils is influenced by many factors, beginning with the increase in intracellular concentration of soluble monomeric α-synuclein. Molecular crowding is another factor that accelerates fibril formation [23, 24]. External agitation of the α-synuclein solution has also shown a dramatic increase in fibrilization rate [25, 26]. Decreasing the temperature and/or increasing the pH of the protein solution have shown to drastically accelerate fibril formation [27, 28]. Pesticides and herbicides (rotenone, dieldrin and paraquat) have been shown to dramatically increase fibril formation rate of α-synuclein in solution in vitro [29]. Notably, epidemiological studies show an increase in risk of PD among populations exposed to pesticides and herbicides [30, 31]. There are also epidemiological explorations that suggest exposure to heavy metals is associated with a higher risk of developing PD [32–35].
Both in vivo and in vitro studies show rapid uptake of α-synuclein fibrils into neurons [36–39]. Evidence shows that α-synuclein is internalized by neurons via endocytosis [40]. Mouse model studies have shown that microglia, astrocytes and oligodendrocytes also take up α-synuclein from the extracellular space [41, 42]; and that α-synuclein is phagocytosed by microglia, monocytes, and astrocytes [43, 44], but that phagocytic activities seem to be reduced in human monocytes from elderly donors as well as in microglia from adult mice compared to those from young mice [43].
In vivo studies have shown that injecting pre-formed α-synuclein fibrils induces aggregation of α-synuclein both at the site of injection and at distant areas of the brain [45–58]. In vitro studies suggest that axonal transport in both the anterograde and retrograde directions (Figure 1) [37, 40, 59, 60] is responsible for the in vivo observed α-synuclein aggregation in distant areas of the brain.
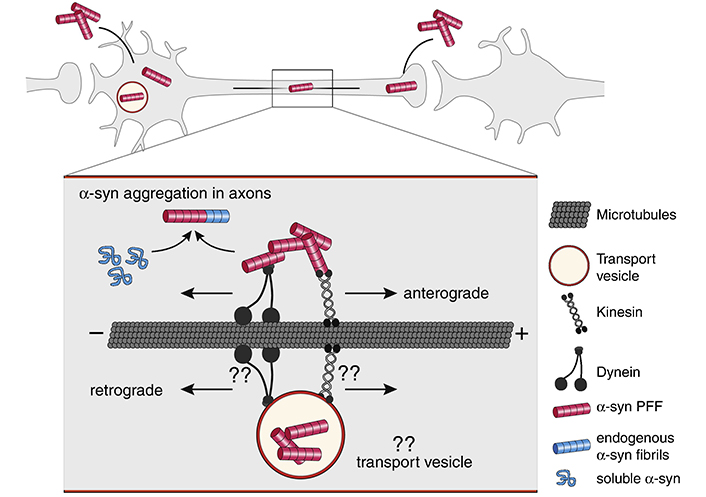
Axonal transport of α-synuclein (α-syn) fibrils. α-Syn fibrils can be internalized both in the dendrite/cell body compartment and in axons. α-Syn fibrils are actively transported along microtubules both in the anterograde and retrograde direction. It is currently not well understood if internalized α-syn fibrils are being transported directly in the cytoplasm or in transport vesicles following endocytosis. Kinesins and dynein families of proteins are the main types of molecular motors transporting cargos along microtubules in anterograde and retrograde directions. The motor and adapter proteins mediating α-syn fibril transport, both in the cytosol as well as in transport vesicles, are still unknown. Aggregation is thought to initially occur in axons, where α-syn fibrils can encounter and template the misfolding of soluble endogenous α-syn proteins that are transported along axons for delivery to synapses. “??” indicates unknown mechanisms and molecular players. The figure and the legend both borrowed from [40]. PFF: preformed fibrils
Note. Reproduced from “Internalization, axonal transport and release of fibrillar forms of alpha-synuclein,” by Bieri G, Gitler AD, Brahic M. Neurobiol Dis. 2018;109:219–25 (https://www.sciencedirect.com/science/article/pii/S0969996117300554). CC BY-NC-ND.
A few mechanisms have been proposed for how α-synuclein can transfer from cell to cell (Figure 2) [40]. Soluble α-synuclein has been known to be secreted by neurons into cerebrospinal fluid (CSF) and the central nervous system (CNS) interstitial fluid [61, 62]. One study has shown that a chaperone protein can mediate delivery of misfolded proteins to an endosome, which is then released into the extracellular space [63]. Exosomes may also play a role in α-synuclein release into the extracellular space, but the amount of α-synuclein in exosomes appear to be relatively low [64, 65], and though soluble and oligomeric α-synuclein have been found in exosomes, no α-synuclein fibrils have been detected in exosomes [40]. Another possible mechanism of α-synuclein cell-to-cell transfer includes tunneling nanotubes, open-ended channels formed by membrane protrusions connecting distant cells and allowing transfer of different types of cargo between connected cells [66]. An in vitro study has shown that α-synuclein fibrils transfer from donor to acceptor cells via lysosomal vesicles within the tunneling nanotubes [36]. One persistent question in PD is how α-synuclein initially forms aggregates inside the cells. Braak et al.’s [8] hypothesis posits that α-synuclein aggregates are transported from the peripheral nervous system (PNS) into the CNS, but it does not go into details as to how aggregates are formed in the PNS in the first place.
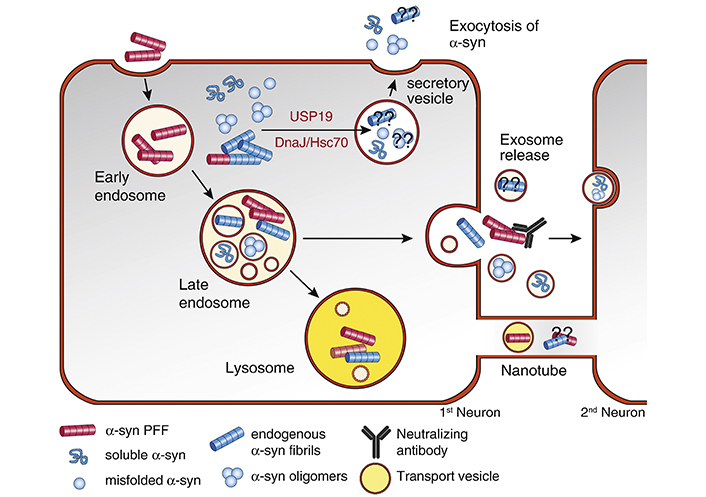
Cellular pathways mediating α-syn release. Following endocytosis, part of the internalized α-syn PFF can be secreted again from the endosomal compartment without being targeted to the lysosome for degradation. Neutralizing antibodies can block the secreted α-syn fibrils and prevent the cell-to-cell transmission. Cytosolic α-syn monomers, oligomers, and possibly fibrils can be targeted to secretory vesicles for release. Two molecular players with chaperone activity, including USP19 and DnaJ/Hsc70, can mediated the secretion of cytosolic α-syn. It is currently unknown if these two pathways can also mediate the secretion of α-syn fibrils as well. Small amounts of soluble and oligomeric α-syn are packaged into endosome-derived membrane vesicles called exosomes and can be secreted into the extracellular space. It is currently unclear if α-syn fibrils are present in exosomes. Tunneling nanotubes can also mediate the direct release and transmission of α-syn between cells, possibly in lysosome-derived transport vesicles. Neutralizing antibodies target α-syn fibrils released into the extracellular space and potentially reduce or prevent transmission to a second cell. “??” indicates unknown mechanisms and molecular players. The figure and the legend both borrowed from [40]. DnaJ/Hsc70: chaperone DnaJ/heat shock cognate 71kDa protein; USP19: ubiquitin specific peptidase 19
Note. Reproduced from “Internalization, axonal transport and release of fibrillar forms of alpha-synuclein,” by Bieri G, Gitler AD, Brahic M. Neurobiol Dis. 2018;109:219–25 (https://www.sciencedirect.com/science/article/pii/S0969996117300554). CC BY-NC-ND.
As referenced above, α-synuclein can be internalized into neuronal cells via endocytosis [40], and several studies have found specific proteases that degrade α-synuclein in the lysosome [67, 68]. An in vivo study found that an over-expression of α-synuclein in a mouse model resulted in an increased α-synuclein concentration in the lysosome [69]. Another study found that oligomeric α-synuclein is also cleared from the cell via the lysosome [70]. Yet another study found that proteasomal inhibitors applied to mouse cortical regions led to an accumulation of α-synuclein [71]. α-Synuclein aggregation itself can lead to lysosomal impairment, due to α-synuclein accumulation in lysosomes, which may result in neurons disposing of aggregates through the tunneling nanotubes [36, 72, 73]. In addition, astrocytes seem to play a role in degrading α-synuclein aggregates via lysosomal digestion [74]. Astrocytes that fail to degrade excess α-synuclein oligomers will develop mitochondrial disturbances and cellular stress. That stress will lead astrocytes to transfer their α-synuclein aggregates to nearby astrocytes as well as to the extracellular space [75]. These findings suggest the lysosome as an important factor in the intracellular regulation of α-synuclein, and that α-synuclein aggregation may be a result of the lysosomal dysfunction.
Braak et al. [8] (2003) first suggested a staging process for the spread of α-synuclein aggregation. Their original stage hypothesis and later observations of related symptoms by them and other investigators is reflected in Table 1. They hypothesized that the overall progression of PD begins in two places of the PNS: the enteric nervous system (ENS) of the gut, which progresses to the CNS via the vagus nerve and the dorsal motor nucleus of the vagus (DMV) in the medulla oblongata, and the neurons of the nasal cavity (Figure 3) [76–78]. Stages 1–3 are classified as prodromal, non-motor symptom phase of PD, whereas stages 4–6 signify the symptomatic somato-motor phase of PD. In stage 1, α-synuclein is present in the olfactory bulb and the dorsal nucleus of the vagus nerve, coinciding with a loss of smell, which is considered an early sign of PD [79, 80]. In stage 2, nuclei in the lower brainstem are affected, followed by the nuclei that project to the cerebral cortex, correlating with sleep problems, some movement issues, lowered blood pressure, constipation, and possible emotional problems. In stage 3, α-synuclein aggregates reach the midbrain, including the substantia nigra and the amygdala, resulting in the thermoregulation disorder and cognitive impairment. In stage 4, α-synuclein aggregates, composing Lewy bodies, continue to affect the amygdala and the cerebral cortex, leading to bradykinesia, resting tremor, rigidity, and postural instability—the hallmark motor symptoms of PD. In stages 5 and 6, the deep layers of the cerebral cortex are affected, resulting in motion fluctuation, fatigue, visual hallucinations, dementia, and other psychiatric symptoms [81–83].
Stages in PD pathology and α-synuclein aggregate locations
| PD stages | α-Synuclein aggregate location | Signs/symptoms |
|---|---|---|
| Stage 1 | Olfactory bulb, dorsal nucleus of vagus nerve, medulla oblongata, ENS, gut | Olfactory dysfunction, constipation, RBD |
| Stage 2 | Lower brainstem, cerebral cortex | Lowered blood pressure, continued stage 1 symptoms |
| Stage 3 | Midbrain, substantia nigra, amygdala | Cognitive impairment, thermoregulation disorder, continued stage 2 and previous symptoms |
| Stage 4 | Amygdala, basal prosencephalon, mesocortex | Bradykinesia, resting tremor, rigidity, postural instability, continued stage 3 and previous symptoms |
| Stage 5 | High order sensory association areas of neocortex, prefrontal neocortex | Motion fluctuation, fatigue, visual hallucinations, dementia, continued stage 4 and previous symptoms |
| Stage 6 | First order sensory association areas of neocortex, premotor areas, primary sensory areas, primary motor field | Continued stage 5 and previous symptoms |
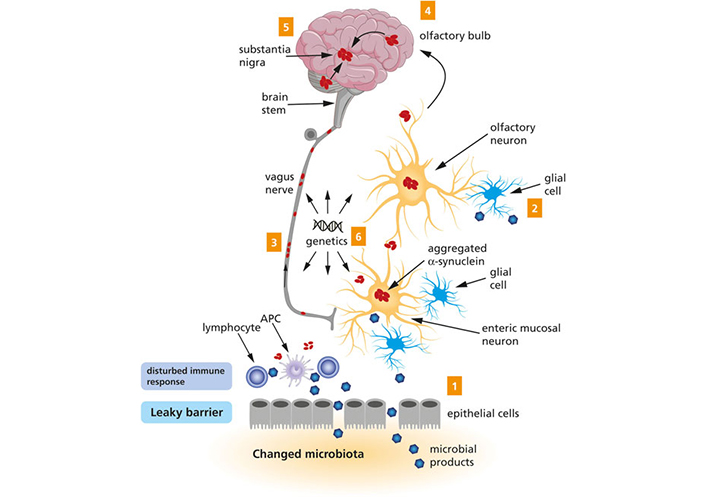
A schematic representation of the Braak’s hypothesis of PD. Microbial products come into contact with olfactory and/or enteric neurons, which trigger the aggregation of α-syn (1 and 2). The aggregated α-syn spreads toward the CNS via the olfactory bulb and the vagus nerve (3 and 4). Eventually, the aggregated α-syn arrives at the substantia nigra (5). Genetic factors are likely to contribute to PD, but the exact mechanism remains to be elucidated (6). The figure and the legend both borrowed from [78]. APC: antigen-presenting cell
Note. Reproduced from “Exploring Braak’s hypothesis of Parkinson’s disease,” by Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJA, Kraneveld AD. Front Neurol. 2017;8:37 (https://www.frontiersin.org/articles/10.3389/fneur.2017.00037/full). CC BY.
Many in vivo studies demonstrated α-synuclein movement and transport [45–58] supporting Braak’s hypothesis for α-synuclein spread from the ENS to the CNS. Braak’s hypothesis states that PD progression begins with inflammation in the gut. PD patients have been found to suffer from increased permeability of the intestinal barrier [84], leading to increased inflammation and oxidative stress, both possibly triggering α-synuclein aggregation [78]. In PD patients, Lewy body pathology has been found in the vagus nerve [85], providing further evidence in support of Braak’s hypothesis that Lewy body pathology begins in the ENS and spreads via the vagus nerve [86]. Epidemiological studies have shown lower risks for PD development in patients who have undergone vagotomies [87, 88], suggesting that the vagus nerve may play an important role in α-synuclein transport to the CNS. Steiner et al. [89] presented the concept of α-synuclein behaving like a prion based on in vivo studies of prion-like movement of α-synuclein. The prion-like hypothesis resulted from an interpretation of findings by Kordower et al. [90] and Li et al. [91] showing Lewy body pathology not only in the host neurons of PD patients, but also in grafted neurons from healthy donors. This hypothesis fits well with Braak’s hypothesis in that if α-synuclein behaves like prions, then misfolded α-synuclein will act as seeds for further misfolding of nearby α-synuclein proteins, leading to widespread pathology. While Braak’s hypothesis is based on the spread of Lewy body pathology via α-synuclein spread, it suggests that the greater the spread of the Lewy bodies the more severe disease ensues [78].
There is also evidence disputing Braak’s hypothesis [8]. Studies examining autopsies from PD patients have found that 7–11% of PD patients do not exhibit Lewy body pathology in the DMV but they do exhibit Lewy body pathology in higher regions of the brain [92, 93]. Additionally, 27–33% of PD patients do not show Lewy body pathology in the ENS [78]. The Arizona Study of Aging and Neurodegenerative Disorders conducted over 460 autopsies of patients with incidental Lewy body disease [94], believed to be a prodromal stage of PD. While Braak’s hypothesis suggests that the olfactory bulb may be one of the first-affected regions of the body in α-synucleinopathy, the Arizona study found no case where α-synuclein inclusions were present in a peripheral region but not in the brain [94]. The authors believe this result argues against Braak’s [8] contention that PD is a PNS-first disease because one would expect to see prodromal presentations of PD in the PNS in at least some autopsies. There are also studies that do not support gut-to-brain propagation of α-synuclein pathology in vivo. Specifically, injection of α-synuclein fibrils into the colon walls of mice and non-human primates yielded pathology in the ENS, but only transient pathology into upper levels of the PNS, which disappeared after a month [95]. Engelender and Isacson [96] proposed the “threshold theory”, which posits that widespread expression of α-synuclein throughout the body can simultaneously damage many different neurons, and that any dysfunction would come from the inability of reserve neurons to compensate for the damaged neurons. The researchers argue that PNS neurons are less inter-connected than CNS neurons, and therefore have fewer neurons in reserve. Damage to the PNS neuronal network will result in symptoms faster than damage to the CNS neuronal network because a greater percentage of PNS neurons is required to be functional in order to maintain the physiological function [96]. This “threshold” number of neurons required to maintain function is Engelender’s and Isacson’s [96] explanation as to why prodromal non-motor symptoms of PD occur prior to motor symptoms. Engelender and Isacson [96] also argue that somewhat contrary to Braak et al.’s [8] finding, up to 47% of PD cases do not follow an ascending progression of the disease, suggesting that the ascending pathway may not be the only possible direction of the disease progression, and that the Lewy bodies do not travel from the ENS or other peripheral structures to the brain [92, 97]. Horsager et al. [98] argue that there may be two subtypes of PD, the “brain-first” and the “body-first” subtypes, with the “body-first” subtype strongly associated with RBD. RBD is also a non-motor symptom of PD [99]. The two-subtype theory of PD progression may explain why Braak’s hypothesis of the PD progression may be representative of only a subset of PD cases. One review points to Braak’s hypothesis as representing 51–83% of all PD cases and highly representative of a young onset and long duration of the disease, but does not represent disease progression for all PD cases [78].
Neuroinflammation is strongly linked to PD and neurodegeneration [100]. Two possible mechanisms to describe neurodegeneration exist: “cell-autonomous” and “non-cell-autonomous” [101]. Cell-autonomous neuron death refers to accumulated damage within neurons resulting in neuronal death, possibly contributed by aggregated α-synuclein and an inability of the neurons to clear the α-synuclein via lysosomes. The non-cell-autonomous mechanism refers to indirect neuronal degeneration caused by interactions between the neuron and its surrounding cells, such as glial cells. Specifically, activated microglia while releasing cytokines and starting a neuroinflammatory response triggers neurotoxicity [102]. There is ample evidence showing the cytokine production and microglial activation are elevated in PD patients [103–106]. A post-mortem study on excised brains showed higher concentrations of cytokines in the striatum and CSF of PD patients compared to people who did not have PD [107]. Activated microglia have also been detected in transgenic mouse models of PD [108–110]. An in vitro study has shown that activated microglia slows down degradation of α-synuclein and results in α-synuclein accumulation in the microglial cytoplasm [44]. A mouse study showed that both a decreased amount of microglia and an increased amount of activated microglia result in greater cell-to-cell transfer of α-synuclein between neurons, as the microglia are unable to clear α-synuclein from the extracellular space [111].
α-Synuclein behaves like an endogenous agonist for toll-like receptors (TLRs) on microglia membranes [40, 112]. TLRs are cell surface proteins on immune and non-immune cells and are a part of the innate immune response. Agonists to TLRs activate microglia, initiating inflammation, and leading to microglia releasing pro-inflammatory cytokines, such as tumor necrosis factor α (TNFα) and interleukins. Increased amount of a specific TLR, neuronal TLR2 was found postmortem in the brains of patients with PD, and TLR2 activation leads to an increase in the neuronal cytosolic levels of α-synuclein and decreased autophagy of α-synuclein, which would promote α-synuclein accumulation in the cell [113, 114]. TLR2 mainly responds to oligomeric α-synuclein, but not to monomeric α-synuclein [115]. Therefore, the general consensus is that oligomeric α-synuclein inhibits phagocytosis, while monomeric α-synuclein promotes it [116].
In vivo studies have shown that injection of α-synuclein fibrils into the substantia nigra of mice induces recruitment of peripheral innate immune cells to the brain [117]. An in vitro study showed that pathological forms of α-synuclein, such as its fibrillar form, activate human monocytes, and their activation is greatly increased by extracellular vesicles carrying α-synuclein [118]. When mutated forms of α-synuclein that cannot form fibrils were injected into mouse brains, aggregates were still observed [119]. The mice did exhibit heightened neuroinflammation, linking inflammation with α-synuclein aggregation. Drug-induced mouse models of PD have shown that exercise may induce anti-inflammatory effects that can partially protect dopaminergic neurons from α-synuclein aggregation induced toxicity [120–125].
Inflammation in the gut may also contribute to PD risk and progression. Most PD patients exhibit non-motor symptoms such as constipation and gastrointestinal (GI) reflux [86, 126, 127]. There are many factors that could trigger PD pathology in the GI tract: disturbance of the gut microbiota [128], certain gut infections that could lead to inflammation [129–131], or inflammatory bowel disease (IBD) [132]. Increased intestinal permeability or “leaky gut” has also been shown to present in PD patients [133]. One study showed increased levels of endotoxins as well as α-synuclein in the intestinal walls of PD patients compared to healthy patients [84]. Mouse models have shown that gut inflammation can exacerbate neuroinflammation and lead to neuronal loss in the substantia nigra making it a potential risk factor for PD [134]. Furthermore, clinical studies have shown bowel inflammation evidence in PD patients [135–137]. In addition, many cohort studies showed an increased risk for PD in IBD patients [132, 138–141]. Moreover, some of those studies reported that anti-inflammatory medication for IBD lead to decreased risk of developing PD [139, 140]. Of interest, the Lewy body pathology is found in the GI tract in people with PD [142–144]. A study on pediatric patients showed a positive correlation between α-synuclein expression in the enteric neurites of the upper GI tract and inflammation severity in the intestinal wall [130]. However, more research is needed to elucidate possible molecular mechanisms of inflammation in the gut and how it may lead to PD progression.
Past animal studies have explored whether physical exercise could decrease α-synuclein aggregation and lead to halting PD progression since α-synuclein is considered one of the main pathogenic proteins of PD [145]. In a transgenic mouse model of PD, mice that were pre-symptomatic exercised in a running wheel over the span of 3 months and their motor and cognitive functions were assessed using the Rotarod and Morris Water Maze, respectfully. The mice that ran on the wheel had significantly better outcome measures and had significantly less α-synuclein aggregation in the brain when compared to the controls [146]. Similar findings were obtained using a chemically induced mouse model of PD with animals exercised by running on a treadmill. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-injected mice in the endurance exercise group showed a reduction in α-synuclein in the striatum, leading to restoration of their motor function when compared to control mice, sedentary mice, and MPTP-injected sedentary mice [147]. It was suggested that exercise prevented aggregation of α-synuclein by increasing the level of the neuroprotective protein deglycase DJ-1 known to defend cells from abnormal protein aggregation by upregulating a chaperone protein heat shock protein 70 (Hsp70) [146]. The downregulation of α-synuclein by exercise may add to neuroprotection by decreasing neuroinflammation, due to diminished pro-inflammatory cytokine levels and prevented activation of the TLR2 signaling cascade, and reducing cell death resulting in neuronal cell preservation and restored levels of tyrosine hydroxylase [147]. The precise mechanism of exercise downregulating α-synuclein is not known; three possible pathways have been suggested: inducing antioxidative capacity; enhancing apoptosis of cells containing an excess amount of α-synuclein; and increasing autophagy and ubiquitin-proteasome pathway activity to remove overabundance of α-synuclein [147]. Further clinical studies into exercise and its effects on α-synuclein aggregation may gain valuable insight into possible treatment regimens for PD [146, 147].
By definition, PD is a progressive neurodegenerative disorder that causes the death of dopaminergic neurons in the basal ganglia [148], resulting in primary motor symptoms including bradykinesia, resting tremor, rigidity of muscles and postural instability [149]. Other major motor symptoms also include various impairments in functional activities such as gait, speech, and daily living [3]. As disease progresses, motor impairments worsen as shown in the increased episodes of freezing of gait, leading to decreased independence and QOL [150, 151].
Without a known cure, treatment of PD has traditionally focused on managing motor symptoms. Pharmacological therapy or surgery can improve some of motor symptoms of PD, but the effect of dopaminergic drugs usually diminishes over time and side effects (e.g., dyskinesias or hallucinations) emerge [152]. Gait and balance impairment make a significant contribution to the decrease of QOL in people with PD [153]; however, current treatment options have limited effects on the progressive deterioration of gait and balance function [152, 154, 155].
Rehabilitation therapies, primarily using various physical exercises, are gaining increased attention in the treatment of PD [156]. Rehabilitation therapies are typically utilized to maximize functional abilities and minimize secondary complications [157, 158]. Randomized controlled trials and systematic reviews have extensively investigated the effects of conventional exercise-based therapies, including aerobic (walking/running) and strength training programs. They have shown evidence in improving cardiovascular fitness, muscle strength and endurance, and motor function in people with PD [158–164]. Specifically, a randomized clinical trial of high-intensity treadmill exercise in individuals with new-onset PD observed significantly less worsening of motor function [164]. These conventional exercises are used primarily to reverse and/or prevent deconditioning (due to lack of physical activity), muscle weakness, or various health conditions [165]. The less common forms of aerobic exercises including cycling [166] and aquatic exercise [167] have also been tried in PD patients. Based on the findings of past animal studies, conventional exercises may stimulate the brain to increase the production of growth factors that can protect dopamine-producing neurons [123, 168] and consequently, slow down the deterioration of the motor function [169–171]. The findings of these promising animal studies need to be confirmed by clinical trials in people with PD.
Motor learning through neuroplastic adaptation is considered the underlying mechanism of many exercise interventions in people with PD. The basal ganglia play an important role in motor learning. The diminished capacity of motor learning in individuals with PD may be due to damage of neurons in the basal ganglia. However, there has been sufficient evidence supporting the remaining motor learning capacity in people with PD when performing upper limb motor tasks [172], or gait and postural control tasks [163]. Recent studies have uncovered promising health benefits following less conventional forms of exercises, including therapeutic dance [173–175], Tai Chi [176], Nordic walking [177, 178], cueing [179, 180], Lee Silverman Voice Treatment (LSVT)-Big [181–183], attention strategies [180, 184] and boxing [185], to counteract the functional deterioration induced by PD. These exercises have been proven with evidence of statistically significant but small short-term benefits in patients with PD. For example, individuals with PD often step or reach at abnormally small amplitude without feedback due to their misperception. The LSVT-Big program, designed to address such deficits by teaching patients to sustain bigger movements in performing daily activities, has been shown to improve the motor function of PD patients at various stages of the disease [181–183].
On average eight non-motor symptoms were reported in each patient with PD and 98.6% of patients had at least one of the non-motor symptoms [186]. Depression occurs in 10–45% of patients with PD [187, 188]. Psychosis and visual hallucinations are strongly related to the necessity of placement in a nursing home; visual hallucinations have been estimated to affect 40% of patients with PD [189]. People with PD have a six times higher risk of developing dementia than the general population, and about 40% of people with PD are diagnosed with dementia [190]. Autonomic dysfunction includes excessive drooling, orthostatic hypotension, urinary dysfunction, erectile and sexual dysfunctions, constipation and GI dysfunctions [187]. Constipation is one of the most common non-motor symptoms that commonly precedes motor symptoms in people with PD [191, 192]. Disorders of sleep and wakefulness include sleep fragmentation and insomnia, RBD, restless leg syndrome, sleep disordered breathing and excessive daytime sleepiness [187, 193]. It has been speculated that nearly all patients with PD have some sleep disturbances that started early in the disease progression [193–195]. Other non-motor symptoms in people with PD include olfactory dysfunction, pain, fatigue, and voice and speech disorders [187]. Olfactory dysfunction, which has been investigated as a potential preclinical maker of PD, can affect up to 90% of people with PD [187].
The presence of non-motor symptoms significantly diminishes the QOL, and most of these symptoms are resistant to the current therapies aimed at resolving the motor symptoms of PD [196]. Non-motor symptoms can precede the motor symptoms by decades and be present in all stages of the disease, with the severity of non-motor symptoms ranging from mildly debilitating in the early stages to severely debilitating in the later stages [197]. Since non-motor symptoms are correlated to aging, and life expectancy has steadily been increasing, the management of these non-motor symptoms have become increasingly important [187].
A meta-analysis of by Fang et al. [198] (2018) analyzed eight past studies and found a statistically significant reduction in risk of PD when comparing those who partook in physical activity very frequently versus those who exercised little to none. Furthermore, the risk of PD decreased by 9% with each increase of 10 metabolic equivalents (METs)/h per week; the risk of PD decreased by 17% for men specifically with each increase of 10 METs/h per week in moderate to vigorous physical activity [199]. The results of a cohort study of male veterans exhibited a significant negative relationship between high levels of fitness (greater than 12 METs) and incidence of PD [200]. This cohort study used the maximal exercise testing to objectively measure fitness level, and the onset of PD was taken from the Veterans Affairs computerized program. A possible mechanism behind a reduction in risk of PD caused by physical activity is based on the assumption that long-term, low-to-moderate intensity exercise negatively regulates the inflammatory response [201]. Exercise stimulates skeletal muscle myocytes to release multiple myokines with anti-inflammatory effects including the inhibition of microglial activation. Additional effects of exercise include synthesis and release of neuroprotective heat-shock proteins; increase in leptin sensitivity; upregulation of antioxidants; enhancement of the glymphatic system; downregulation of expression of proinflammatory cytokines such as inteleukin-1β and TNFα; downregulation of the TLR signaling pathway; and changes in the microbiome [201]. These individual pathways and mechanisms are important areas for future research.
Another study examined the effects of a balance training program and found that moderate-intensity balance training could reduce inflammation and increase the level of anti-inflammatory cytokines to promote neuroprotection against inflammation [202]. After 12 weeks of moderate-intensity balance training, the balance-trained PD patients significantly improved their balance control ability, increased the plasma concentration of interleukin 10 and decreased TNFα concentration when compared to a PD control group. Evidence suggests that these changes can lead to improved brain plasticity and reduced neuronal injury [201–203].
Some investigators have studied the effect of complementary therapies, especially mind-body practices, in terms of outcomes in cognition, depression and QOL in people with PD. The mind-body practice exercises involve the specifically designed gentle body movements in combination with deep breathing and meditation, including Tai Chi, Qigong, and Yoga. The practice of mind-body exercises has shown to be safe and cost-effective for chronic health conditions [204, 205].
Tai Chi and Qigong have shown the potential in improving cognition, depression, and QOL in people with PD. Tai Chi and Qigong are defined as mind-body exercise interventions with a foundation based in Chinese medicine, martial arts and the Asian lifestyle; both have been practiced for thousands of years for maintaining health and preventing illness [14, 206]. These mind-body interventions are used to train individuals in their abilities to improve body awareness, focus mental attention and enhance planned thought processes [206, 207]. The studies show that QOL scores significantly improved with Tai Chi or Qigong exercise. One review reported that Tai Chi and Qigong improved QOL in patients with PD by 38%, which is comparable to the 4–38% increase in QOL produced by deep brain stimulation [206]. There were slight improvements with cognition (specifically, attention and working memory), warranting further investigation [207, 208]. Significant improvements were reported on the depression scales, i.e., Beck Depression Inventory, Geriatric Depression Scale, and Montgomery-Asperg Depression Rating Scale [207]. Improvements in depression are hypothesized to be due to moderate improvements in cardiovascular system function (lowering heart rate and deepening breaths), improvements in focusing thought processes that lead to decreased rumination and catastrophizing, and secondary improvements in self-confidence and self-efficacy [206].
Qigong has been shown to decrease the levels of pro-inflammatory cytokines, which are closely related to the pathogenesis of PD. Previous studies indicate that TNFα contributes to sleep-wake regulation, and feelings of sleepiness and fatigue [14, 209, 210]. A pilot study assessed TNFα levels after a six-week Qigong intervention [14]. After the intervention, plasma TNFα levels were significantly reduced, and the changes in the TNFα level were associated with the changes in scores of the PD Sleep Scale 2. More research in this direction is needed. There were high levels of adherence and no serious adverse events that occurred in any of the included reviews and studies [14, 206, 207] suggesting that Tai Chi and Qigong are likely safe interventions for people with PD.
Yoga is an ancient Indian mind-body approach combining specific posture, regulated breathing and meditation to improve health and wellbeing [211]. It has been widely used as a complementary therapy for individuals with neurological disorders. A recent systematic review reported that Yoga practices were safe and well-accepted in patients with PD; and that these interventions might benefits patients in ameliorating both motor and non-motor symptoms [212, 213]. Yoga is a cost-effective intervention to reduce anxiety and depression [211, 214, 215]. Studies of Yoga intervention in PD have also reported improvement in executive function and working memory compared to an intensive stretching exercise or passive control [216–218]. A few studies have also indicated that Yoga may reduce stress and inflammation, enhance neuroplastic processes and increase antioxidant levels [219, 220]. A recent study reported an implementation of tele-Yoga as safe, feasible, and enjoyable intervention in people with PD, with preliminary results of improvements in some of the clinically relevant outcome measures [221]. However, the study by Cheung et al. [15] showed improvement in motor function and QOL after a 12-week Yoga training protocol but did not find significant differences in changes of either non-motor symptoms or markers of oxidative stress in people with PD. More studies are needed on non-motor symptoms and biomarkers in PD population.
In this review, we focus on non-pharmaceutical approaches, such as physical exercise or mind-body movement practices, which have shown promising results in managing non-motor symptoms of PD. However, there were limitations in the past studies. A major limitation is the small sample sizes inherent in those studies and thus, an increased risk of biased results. In addition, in the home or community settings, adherence to physical exercise or mind-body practice programs is often low. Nevertheless, non-pharmaceutical treatments are generally safe for people with PD, and they offer benefits in at least some aspects of general health such as cardiovascular fitness. Future clinical trials with large sample sizes may determine the effect of non-pharmaceutical approaches on non-motor symptoms in people with PD. Furthermore, there has been a lack of mind-body practice trials that examined biomarker changes related to α-synuclein aggregates or neuroinflammation in PD patients, except a few pilot studies [14, 15]. Future research is needed to focus on this area to fully understand the role and underlying biological pathways of mind-body practice intervention on the non-motor symptoms of PD.
This review addressed both the importance of α-synuclein to the pathology of PD, as well as the importance of a renewed focus of rehabilitation therapy on the non-motor symptoms of PD. In addition, this review presented a compelling hypothesis of PD pathogenesis originating in the PNS and spreading to the CNS, as well as research supporting and rebutting that hypothesis. Finally, this review further suggested the connections between inflammation and PD progression, as well as the potential of various rehabilitation therapies, especially the mind-body practices, on inflammation and α-synuclein pathology. If PD pathogenesis is predicated in the PNS as suggested by some, the prodromal non-motor symptoms of PD would take on a greater significance. Any therapies aimed at alleviating these prodromal symptoms may yield a preventative effect on PD pathogenesis, especially if any therapies could be seen to affect α-synuclein aggregation or transport. Future studies are needed to examine the non-motor prodromal symptoms of PD and the connection between inflammation and PD, both from a therapeutic and a biochemical standpoint.
CNS: central nervous system
ENS: enteric nervous system
GI: gastrointestinal
IBD: inflammatory bowel disease
METs: metabolic equivalents
PD: Parkinson’s disease
PNS: peripheral nervous system
QOL: quality of life
RBD: rapid eye movement sleep behavior disorder
TLRs: toll-like receptors
TNFα: tumor necrosis factor α
ZL: Conceptualization, Visualization, Writing—original draft, Writing—review & editing. JL: Writing—original draft, Writing—review & editing. IVS: Conceptualization, Visualization, Writing—review & editing. WL: Conceptualization, Supervision, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was partially supported by the
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

