 Review
Review

Affiliation:
1National Research Council, Institute for System Analysis and Computer Science “A. Ruberti”, 00185 Rome, Italy
2Experimental Neuroscience and Neurological Disease Models, Santa Lucia Foundation IRCCS, 00143 Rome, Italy
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0001-7362-8307
Affiliation:
3Department of Pharmacology and Experimental Neuroscience, University of Nebraska Medical Center, Omaha, NE 68198, USA
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-4792-0416
Affiliation:
4Department of Ophthalmology and Visual Sciences, Faculty of Medicine, University of British Columbia, Vancouver, BC V6T 1Z3, Canada
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-4796-140X
Affiliation:
5PeQuiM - Laboratory of Research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, MG, Brazil
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-7799-992X
Affiliation:
5PeQuiM - Laboratory of Research in Medicinal Chemistry, Institute of Chemistry, Federal University of Alfenas, Alfenas 37133-840, MG, Brazil
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0003-0324-2435
Affiliation:
6Instituto de Farmacologia e Neurociências, Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal
7Centro Cardiovascular da Universidade de Lisboa, CCUL (CCUL@RISE), Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-4258-9397
Affiliation:
6Instituto de Farmacologia e Neurociências, Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal
7Centro Cardiovascular da Universidade de Lisboa, CCUL (CCUL@RISE), Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0001-9030-6115
Affiliation:
8Neuroscience Theme, School of Medical Sciences, Faculty of Medicine and Health, The University of Sydney, Sydney NSW 2006, Australia
9Laboratory of Neurobiology and Pathological Physiology, Institute of Animal Physiology and Genetics, Academy of Sciences of the Czech Republic, 602 00 Brno, Czech Republic
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-9564-2799
Affiliation:
10Chief Medical Officer for Lutroo Imaging, LLC and Norroy North America, Ltd., Philadelphia, PA 19104, USA
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-1651-4212
Affiliation:
11Department of Neuroscience and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, 20149 Milano, Italy
12Department of Pathophysiology and Transplantation, “Dino Ferrari” Center, Università degli Studi di Milano, 20122 Milano, Italy
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-3977-6995
Affiliation:
11Department of Neuroscience and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, 20149 Milano, Italy
12Department of Pathophysiology and Transplantation, “Dino Ferrari” Center, Università degli Studi di Milano, 20122 Milano, Italy
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-7698-3854
Affiliation:
13Department of Biochemistry, Institute of Chemistry, University of São Paulo, Cidade Universitária, São Paulo CEP 05508-000, SP, Brazil
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-3217-4166
Affiliation:
14International Joint Research Center on Purinergic Signalling, School of Health and Rehabilitation, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, Sichuan, China
15Tianfu Jincheng Laboratory, Chengdu 610212, Sichuan, China
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-2543-066X
Affiliation:
13Department of Biochemistry, Institute of Chemistry, University of São Paulo, Cidade Universitária, São Paulo CEP 05508-000, SP, Brazil
14International Joint Research Center on Purinergic Signalling, School of Health and Rehabilitation, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, Sichuan, China
†These authors contributed equally to this work.
Email: henning@iq.usp.br
ORCID: https://orcid.org/0000-0002-2114-3815
Affiliation:
16Centro de Investigación Biomédica en Red Enfermedades Neurodegenerativas (CiberNed), National Institute of Health Carlos iii, 28031 Madrid, Spain
17Departament de Biochemistry and Molecular Biomedicine, University of Barcelona, 08028 Barcelona, Spain
18Institut de Química Teòrica i Computacional (IQTCUB), School of Chemistry, University of Barcelona, 08028 Barcelona, Spain
†These authors contributed equally to this work.
Email: rfranco123@gmail.com; rfranco@ub.edu
ORCID: https://orcid.org/0000-0003-2549-4919
Explor Neuroprot Ther. 2026;6:1004136 DOI: https://doi.org/10.37349/ent.2026.1004136
Received: August 17, 2025 Accepted: November 24, 2025 Published: January 04, 2026
Academic Editor: Shile Huang, Louisiana State University Health Science Center, USA
Neurodegenerative diseases, including Alzheimer’s, Parkinson’s, Huntington’s, and Amyotrophic Lateral Sclerosis, are characterized by multifactorial pathologies that extend beyond neuronal loss to include neuroinflammation, oxidative stress, mitochondrial dysfunction, and glial dysregulation. Despite extensive research, disease-modifying therapies remain elusive, hindered by late diagnosis, limited availability of specific biomarkers, and the persistent dominance of reductionist, single-target strategies. This comprehensive and informative review provides a critical synthesis of integrated neuroprotective strategies, with particular focus on glial mechanisms and biomarker-guided interventions. Therapeutic emphasis is placed on coordinated mechanisms targeting both neurons and non-neuronal cells, such as astrocytes, microglia, and oligodendrocytes. Emerging strategies are reported to include modulation of synaptic plasticity and neurotransmission, delivery of neurotrophic factors, activation of intrinsic cytoprotective pathways (e.g., Nrf2 signaling), restoration of proteostasis, and induction of regeneration via cellular reprogramming. Glial cells are discussed as therapeutic targets involved in inflammation, metabolism, myelination, and neuronal survival. Advances in predictive, preventive, personalized, and participatory (P4) medicine, supported by genomics, multi-omics, imaging, and real-world data, are presented as accelerating biomarker discovery and enabling earlier and more precise stage-specific interventions. Future success in combating neurodegeneration will depend on integrated approaches that combine protective, supportive, and regenerative strategies, appropriate for disease stage and patient profile. By reframing neuroprotection as a systemic, multicellular endeavor, this review highlights the potential to not only extend life expectancy, but also preserve meaningful quality of life in individuals affected by neurodegenerative diseases.
Neuroprotection remains a formidable challenge, as every central nervous system (CNS) insult, be it stroke, trauma, or neurodegenerative disease, activates multiple, overlapping injury pathways. This mechanistic complexity, coupled with the translational gap between animal models and clinical practice, has repeatedly undermined single-target strategies and continues to limit the development of effective therapies.
Factors such as age, genetic background, and pre-existing health conditions can significantly influence not only an individual’s response to CNS injury but also the efficacy of neuroprotective interventions. Despite promising outcomes in animal models, only a few neuroprotective treatments have gained approval for clinical use in humans. This underscores the urgent need for improved research strategies, careful selection of drugs, and suitably designed clinical trials. Moreover, the absence of reliable biomarkers to assess neuroprotection in humans further complicates progress in this field.
While every strategy has an intrinsic and often unavoidable limitations, at present we should keep focusing on the overall realization of our commitment to neuroprotection by: i) further enhancing the natural repair mechanisms and regenerative capacity of the CNS; ii) intervening earlier in the course of CNS injury to limit damage and improve outcomes; iii) adopting simultaneous application of a range of selective agents instead of single-targeted strategies, or by the provision of a single multi-target agent; iv) providing comprehensive supportive care crucial for neuroprotection beyond pharmacological interventions.
Under this perspective, as reported in a systematic analysis for the Global Burden of Disease Study 2021 [1, 2], the age-standardized rates of deaths per 100,000 individuals attributed to 37 unique conditions affecting the nervous system that now include neurodevelopmental disorders, late-life neurodegeneration, and emergent conditions such as cognitive impairment following coronavirus disease 2019 (COVID-19), has decreased by 33.6% from 1990 to 2021; age-standardized rates of Disability-Adjusted Life Years (DALYs) attributed to these conditions has decreased by 27%. Nevertheless, the medical community and the society continue to face a staggering burden: In 2021, an estimated 3.4 billion people, representing 43.1% of the global population, were living with a neurological condition, which accounted for 11.1 million deaths and 443 million DALYs, making neurological diseases the leading cause of disability worldwide.
This review provides a comprehensive overview of major neurological disorders, moving beyond a neuron-centric view to incorporate the critical part that glial cells play in disease mechanisms and treatment. Additionally, we discuss emerging biomarkers, current challenges, and future directions in research. The structure of the text progresses from an analysis of specific diseases and their common pathways to a discussion of rationally designed neuroprotective strategies, aiming to bridge scientific knowledge and clinical applications and, hopefully, contribute to better patient outcomes in the future.
Parkinson’s disease (PD) is primarily characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta and subsequent depletion of dopamine in the nigrostriatal pathway. Clinically, PD presents with hallmark motor symptoms such as bradykinesia, resting tremor, rigidity, and postural instability. However, non-motor symptoms, ranging from cognitive impairment, mood disorders, sleep disturbances, autonomic dysfunction, to anosmia, are increasingly recognized as integral to the disease and often precede motor onset by years. This complex symptomatology reflects the widespread and multisystem nature of PD pathology, which extends far beyond the basal ganglia to involve cortical, limbic, and peripheral autonomic structures [3, 4].
Despite decades of research, PD remains incurable, and currently approved treatments are largely symptomatic. Dopaminergic therapies such as levodopa, dopamine agonists, and monoamine oxidase B (MAO-B) inhibitors provide meaningful relief, particularly during the early stages of the disease, but their efficacy wanes over time [5]. Long-term use is frequently associated with debilitating motor complications, such as dyskinesias and fluctuations in symptom control [6]. Importantly, none of the available treatments address the underlying neurodegeneration, and disease progression continues unabated.
This lack of disease-modifying therapies highlights an urgent and unmet need for effective neuroprotective strategies capable of halting or slowing the loss of vulnerable neuronal populations. Multiple molecular mechanisms have been implicated in PD pathogenesis, including mitochondrial dysfunction, oxidative stress, impaired protein degradation pathways (ubiquitin-proteasome and autophagy-lysosomal systems), excitotoxicity, calcium dysregulation, neuroinflammation, and abnormal alpha-synuclein aggregation. These processes do not act in isolation but instead converge and amplify one another in a complex interplay that underlies neuronal vulnerability and death. Parkinsonism can be linked to genetics in only a small subset of patients (for a recent review and examples of such conditions, see [7]). The synaptic accumulation and misfolding of alpha-synuclein is considered a pathological hallmark and propagates in a prion-like fashion through interconnected neural circuits [8, 9]. Except for a few isolated cases, no definitive factors capable of triggering the pathological process have been identified. Potential suspects, including environmental or industrial toxins, heavy metals, and illicit drugs, have been considered, but none have been conclusively implicated.
Neuroinflammation plays a key role in the progression of PD. Activated microglia release reactive oxygen and nitrogen species, pro-inflammatory cytokines, and other neurotoxic mediators that exacerbate oxidative stress and mitochondrial damage. In parallel, astrocytes and peripheral immune cells contribute to a sustained pro-inflammatory environment within the CNS. These chronic inflammatory responses are now understood not simply as bystanders but as active drivers of neurodegeneration. Targeting dysfunctional glial responses, restoring microglial homeostasis, and modulating peripheral immune infiltration represent promising avenues for intervention. Similarly, enhancing endogenous mechanisms of neuronal resilience, such as antioxidant defenses, trophic support, and mitochondrial biogenesis, may provide additional protection against ongoing insult [10–12].
Mitochondrial impairment also occupies a central role in PD. Dysfunction of mitochondrial complex I in the electron transport chain has been observed in post-mortem PD brains and in several experimental models. This dysfunction compromises cellular energy metabolism, increases oxidative stress, and triggers apoptotic cascades. In genetic forms of PD, mutations in genes such as PINK1, PARKIN, DJ-1, and LRRK2 further highlight the vulnerability of mitochondrial and proteostatic systems, offering potential therapeutic targets [7, 13, 14].
However, attempts to develop neuroprotective treatments have so far met with limited success. Several promising compounds have failed in clinical trials due to a range of challenges, including inadequate disease models, late intervention timing, insufficient biomarker validation, and difficulty in distinguishing symptomatic from neuroprotective effects. Furthermore, the clinical heterogeneity of PD, along with the absence of definitive diagnostic biomarkers in early stages, complicates patient stratification and endpoint definition in trials (see details in [15]).
Given these challenges, there is a growing consensus around the need for multimodal approaches to neuroprotection in PD. Rather than targeting isolated mechanisms, future therapies must consider the convergence of mitochondrial stress, protein aggregation, and neuroinflammation. Combination therapies, or single agents with pleiotropic actions, are increasingly being explored to interrupt the pathological cascade at multiple levels. In parallel, efforts are underway to identify early biomarkers, including those based on cerebrospinal fluid (CSF), peripheral blood, imaging modalities, and multi-omic signatures, to enable earlier intervention, before extensive neuronal loss has occurred.
In sum, PD exemplifies the critical importance of neuroprotection in chronic neurodegenerative disorders. Its multifactorial pathophysiology, progressive trajectory, and lack of disease-modifying treatments underscore the need for early, multi-targeted, and personalized approaches. Advancing neuroprotective therapies in PD will not only impact millions of patients worldwide but also yield valuable insights applicable to other disorders marked by neuronal loss. This review will comprehensively explore these multifaceted pathological features from the perspective of neuroprotection.
Like Parkinson’s, Alzheimer’s disease (AD) is marked by progressive neuronal degeneration, but with distinct molecular signatures and therapeutic challenges. AD is the most common cause of dementia worldwide and a leading contributor to disability and dependency among the elderly. It is clinically defined by progressive cognitive decline, particularly in memory, language, executive function, and orientation, which in time culminates in significant loss of independence and diminished quality of life. Behavioral and neuropsychiatric symptoms, including apathy, agitation, depression, and hallucinations, often accompany the cognitive decline and contribute significantly to disease burden [2].
Neuropathologically, AD is characterized by extracellular accumulation of amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein. These hallmark lesions appear years, if not decades, before symptom onset and are associated with synaptic dysfunction, progressive neuronal loss, and widespread cortical and hippocampal atrophy. However, AD pathology extends beyond amyloid and tau, encompassing a multifaceted array of pathophysiological mechanisms, including chronic neuroinflammation, oxidative stress, mitochondrial dysfunction, impaired proteostasis (the homeostasis of protein folding, stability, and degradation), calcium dysregulation, excitotoxicity, and breakdown of the blood-brain barrier (BBB) [16, 17].
Despite substantial advances in our understanding of these mechanisms, effective therapeutic options remain extremely limited. Current approved treatments, including acetylcholinesterase inhibitors and the N-methyl-D-aspartate receptor (NMDAR) antagonist memantine, offer only modest symptomatic relief without altering disease progression. Even recent anti-amyloid immunotherapies, while targeting one of the core pathologies of AD, have shown limited clinical benefit and raised concerns regarding safety, efficacy, and applicability across the disease spectrum. These limitations reinforce the urgent need for neuroprotective strategies that can preserve neuronal integrity, delay progression, and extend the functional lifespan of patients [18, 19].
A recent metallomic study has found significantly lower cortical lithium (Li) in mild cognitive impairment and AD with selective sequestration of Li within amyloid plaques, implying an early disturbance of endogenous Li homeostasis in vulnerable cortex while serum levels remain unchanged; this shift is supported across cohorts and fractionation analyses that show reduced Li in non-plaque parenchyma and correlations with memory performance [20]. In mouse models, dietary Li deficiency accelerates Aβ deposition, tau phosphorylation, microglial reactivity, synaptic and myelin loss, and memory decline; single-nucleus RNA-seq indicates broad, cell-type–specific transcriptomic changes that overlap human AD signatures, and several of these effects are partly mediated by increased GSK3β activity, as pharmacologic GSK3β inhibition reverses Li-deficiency phenotypes. As a replacement strategy, lithium orotate (LiO), reported to bind Aβ less avidly than lithium carbonate, raised parenchymal Li, reduced Aβ and phospho-tau, and improved synaptic/myelin markers and behavior at physiological Li levels in AD mice, though these salt-specific advantages and translational implications remain preclinical [21–23]. Despite mechanistic interest and some preliminary signals, clinical trials in AD have not provided evidence of therapeutic benefit from lithium, and its toxicity profile limits its practical use in patients. Taken together, disturbed Li homeostasis may contribute to early AD biology (potentially via GSK3β/β-catenin signaling and microglial/myelin vulnerability), but lithium is not an established AD therapy; rigorous human validation of brain target engagement, salt selection, and risk-benefit evaluation at clinically practical exposures is still required before efficacy claims are warranted.
The need for neuroprotection in AD is underscored by the disease’s long preclinical phase, during which silent pathological processes accumulate before any measurable symptoms emerge. This extended prodromal window offers a critical opportunity for early intervention, provided reliable biomarkers and predictive tools are in place. Neurodegeneration in AD follows a characteristic trajectory, starting in the entorhinal cortex and hippocampus and gradually spreading to the associative neocortex. The degeneration of neurons in the ventral tegmental area, accompanied by reduced dopamine release and impaired connectivity in target regions, is recognized as a key feature of the early stages of AD, preceding even the formation of Aβ plaques. These alterations contribute to cognitive decline as well as to neuropsychiatric symptoms such as apathy and depression, which are frequently observed in patients [24–26]. Targeting early degenerative changes, such as synaptic loss, axonal transport disruption, mitochondrial compromise, and glial dysfunction, may prove more effective than attempting to reverse advanced neuronal death [27, 28].
Chronic inflammation, mediated by microglia and astroglia, is now recognized as a central driver of neuronal damage in AD. Notably, several risk genes associated with AD, including TREM2, CD33, CD36 and CR1, encode immune-related proteins, suggesting that immune dysfunction is not merely secondary but causally linked to disease pathogenesis. There is also increasing interest in neuroimmune modulation, with therapeutic strategies aiming to rebalance microglial activation states, prevent astrocyte-induced neurotoxicity, and limit peripheral immune cell infiltration. Additionally, efforts to enhance intrinsic neuronal protective mechanisms, through upregulation of neurotrophic factors, restoration of calcium homeostasis, or stabilization of synaptic plasticity, offer further promise [29, 30].
Neurons rely heavily on mitochondrial energy production, and in AD, early defects in mitochondrial dynamics, respiratory chain activity, and calcium buffering impair synaptic function and render neurons more vulnerable to stress. These deficits are compounded by oxidative damage, reduced antioxidant capacity, and accumulation of oxidized lipids, proteins, and nucleic acids. Protein misfolding and failure of clearance systems, including the autophagy-lysosome and ubiquitin-proteasome pathways, further compromise the cellular environment and lead to toxic intracellular accumulations [31, 32].
Despite the intricate interplay of pathological mechanisms in AD, therapeutic efforts have long prioritized Aβ targeting as a singular strategy. However, the consistent clinical failures of these reductionist approaches have prompted a paradigm shift toward multi-target interventions designed to concurrently modulate multiple facets of neurodegeneration. Therapy approaches include: i) novel small molecules and biologics with designed polypharmacology; ii) strategically repurposed drugs with established pleiotropic benefits; and iii) optimized lifestyle interventions. Particularly promising are agents that integrate complementary mechanisms, simultaneously mitigating neuroinflammation, counteracting oxidative stress, and preserving synaptic integrity, to address the disease’s multifactorial nature [33, 34].
One major challenge in advancing neuroprotective therapies in AD lies in trial design. Patient heterogeneity, long disease course, and variability in clinical presentation complicate recruitment, stratification, and outcome measurement. Furthermore, distinguishing genuine neuroprotective effects from symptomatic improvement requires the use of robust, disease-relevant biomarkers and long-term longitudinal studies. Nevertheless, advances in neuroimaging, fluid and blood-based biomarkers, and multi-omic profiling are beginning to enable more accurate staging, prognosis, and therapeutic targeting [35].
In conclusion, AD exemplifies the complexity and unmet clinical needs of neurodegenerative disorders. The progressive neuronal loss, limited treatment efficacy, and enormous social and economic burden underscore the critical importance of early, targeted, and multi-mechanistic neuroprotective approaches. A shift in focus from end-stage pathology to early intervention and neuronal preservation will be essential to transform the management of AD and improve outcomes for millions of affected individuals. Later, we will provide an in-depth analysis of these complex mechanisms, always from the perspective of neuroprotection.
Huntington’s disease (HD) is a rare, inherited neurodegenerative disorder with a known genetic etiology: the production of mutant huntingtin (mHTT) protein. Onset typically occurs in mid-adulthood, presenting a complex clinical picture. The motor syndrome encompasses involuntary choreiform movements, dystonia, and impaired coordination, often evolving into bradykinesia. This is accompanied by a range of psychiatric symptoms, including depression, irritability, and apathy, as well as progressive cognitive deficits in attention, executive function, and memory. Collectively, these symptoms lead to a progressive decline in the ability to perform daily activities and a significant deterioration in quality of life [36, 37].
The expansion of CAG trinucleotide repeats in the HTT gene results in the production of an mHTT protein containing an expanded polyglutamine sequence. Misfolded mHTT forms intracellular aggregates, disrupts cellular homeostasis, and triggers widespread neuronal dysfunction. The striatum, particularly the medium spiny neurons of the caudate nucleus and putamen, is the earliest and most severely affected brain region. As the disease progresses, cortical atrophy and white matter loss become more pronounced. Beyond the toxic gain-of-function effects of mHTT, HD pathogenesis involves a range of interconnected mechanisms, including mitochondrial dysfunction, transcriptional dysregulation, impaired autophagy, proteostasis failure, excitotoxicity, oxidative stress, and chronic neuroinflammation [38–43].
Despite a detailed understanding of the genetic basis of HD, effective therapeutic options remain extremely limited. Currently approved treatments such as tetrabenazine and deutetrabenazine target only chorea and offer symptomatic relief without altering disease progression. Antisense oligonucleotides and gene-silencing approaches that directly target HTT expression are under investigation, yet have shown mixed results in clinical trials, with concerns about efficacy, safety, and delivery. These limitations highlight the need for broader neuroprotective strategies that can delay neurodegeneration, support neuronal survival, and preserve motor and cognitive function [44–46].
The need for neuroprotection in HD is particularly urgent given the protracted presymptomatic phase, during which subtle cognitive and psychiatric changes may precede overt motor symptoms by years. This long prodromal window presents a crucial opportunity for early therapeutic intervention, especially in individuals with a known genetic diagnosis. Neurodegeneration in HD follows a relatively stereotyped progression, beginning in the striatum and extending to other cortical and subcortical structures. Early changes include synaptic loss, dendritic spine retraction, mitochondrial fragmentation, and alterations in gene expression, all of which contribute to neuronal dysfunction before irreversible cell death [39, 41, 43].
Chronic neuroinflammation is increasingly recognized as a significant contributor to HD pathogenesis. Microglia in HD adopt a reactive, pro-inflammatory state, releasing cytokines, reactive oxygen species, and complement proteins that exacerbate neuronal injury. Astrocytes also exhibit dysfunctional phenotypes, including impaired potassium and glutamate buffering, which further compromise neuronal health. Notably, mHTT expression in glial cells may independently drive inflammatory responses and metabolic disturbances. Peripheral immune system alterations, including increased cytokine levels and immune cell infiltration, suggest a systemic component to the inflammatory process in HD [47–49].
Mitochondrial dysfunction is another hallmark of HD. mHTT impairs mitochondrial biogenesis, disrupts calcium handling, and promotes fission over fusion, leading to fragmented and inefficient mitochondria. These alterations reduce ATP production, increase oxidative damage, and render neurons more vulnerable to metabolic stress. In parallel, defective autophagic clearance and ubiquitin-proteasome system dysfunction result in the accumulation of toxic protein species and damaged organelles, perpetuating cellular toxicity [50–52].
Although much of the therapeutic focus in HD has centered on reducing mHTT levels, single-target approaches have yielded limited success, prompting interest in multi-modal strategies. These include i) small molecules with antioxidant, anti-inflammatory, or neurotrophic properties, ii) lifestyle interventions such as exercise and diet and iii) cell-based therapies aimed at restoring lost neuronal populations or modulating the disease environment. Neuroprotective compounds that enhance mitochondrial function, reduce oxidative stress, and support proteostasis are also being explored in preclinical and early clinical studies [39, 53, 54].
A major barrier to progress in HD treatment development is the lack of robust biomarkers for disease progression and therapeutic response. However, recent advances in neuroimaging, body fluid biomarkers (such as neurofilament light chain—NfL), and digital phenotyping are beginning to enable more precise tracking of disease dynamics and individualized treatment approaches. Additionally, gene editing and RNA-targeting technologies offer hope for transformative therapies, though challenges remain in ensuring specificity, safety, and long-term efficacy [55–58].
In conclusion, HD illustrates the devastating consequences of single-gene mutations triggering a cascade of pathological events. The convergence of synaptic dysfunction, mitochondrial failure, neuroinflammation, and impaired protein clearance underscores the complexity of the disease and the need for early, multi-mechanistic neuroprotective interventions. As research advances, integrating genetic, molecular, and clinical insights will be critical to developing effective therapies that not only alleviate symptoms, but also modify the course of the disease and improve the lives of patients and their families.
Amyotrophic Lateral Sclerosis (ALS) is a devastating multisystem neurodegenerative disease characterized by the degeneration of both upper (cortical) and lower (brainstem and spinal) motor neurons, leading to progressive voluntary muscle weakness and paralysis. Despite its rarity, ALS represents the most prevalent and studied motor neuron disease, with clinical onset and progression varying significantly across individuals. Symptoms typically begin with muscle weakness, cramping, or dysarthria, and progressively extend to involve swallowing and respiratory muscles. Both familial (fALS, 5–10% of cases) and sporadic (sALS) forms show similar clinical and pathological features, with overlapping cognitive and behavioral changes, particularly frontotemporal-like symptoms, as well as progressive bulbar and limb motor dysfunction.
Pathologically, ALS is characterized by motor neuron degeneration and a constellation of co-occurring molecular abnormalities, including excitotoxicity, mitochondrial dysfunction, oxidative stress, necrosis, altered proteostasis, impaired cytoskeletal trafficking, DNA damage, and dysfunctional RNA metabolism. Neuroinflammation may have an important role, sustained by aberrant crosstalk between neurons and glial cells. Microglia, astrocytes, and infiltrating macrophages remain chronically activated and secrete neurotoxic factors, while dysfunctional oligodendrocytes and Schwann cells fail to uphold myelin and metabolic support. This glial dysregulation perpetuates motor neuron death, contributing to the rapid and irreversible nature of the disease [59, 60].
Potential triggers range from environmental and industrial toxins [61] to the confluence of factors such as strenuous physical exercise and intermittent, high-intensity sound [62]. Associations with certain occupations (farmers, truck drivers, airline pilots/cabin crew, professional soccer players) have been proposed, though none are particularly strong [63–68]. Epidemiological studies are limited by small sample sizes and uncertain outcomes. It has recently been noted [69] that researching ALS faces formidable challenges, and overcoming these barriers will be essential for developing truly effective strategies for prevention and treatment.
Genetically, over 40 risk genes have been associated with ALS, with key mutations including those in C9ORF72, SOD1, TARDBP, and FUS. These mutations result in toxic gain- and loss-of-function effects, which impair essential cellular processes. Still, the initial molecular trigger, or “primum movens”, remains undefined, and it is likely that no single causative mechanism can account for the clinical and molecular heterogeneity observed in ALS patients. This multifactorial etiology supports the view of ALS as a “point of no return” disease, where the failure of therapies to halt disease progression reflects the deep complexity of its biology [59, 60].
Epidemiologically, the burden of ALS is rising worldwide. According to the Global Burden of Disease Study 2021, ALS prevalence has increased by nearly 68% between 1990 and 2021, with incidence rising by 74.5% and associated DALYs by 105.5% [1]. Bayesian models forecast that although prevalence may plateau or modestly decline by 2040, mortality and overall disease burden will continue to rise, driven largely by population aging, especially in high-income countries [1] and increased environmental toxic compounds. This rising global impact underlines the urgency for better treatments and earlier diagnosis.
From a therapeutic standpoint, current interventions are limited. Riluzole remains the main approved therapy for ALS, despite yielding only modest survival gains through partial attenuation of glutamatergic excitotoxicity [70–73]. Edaravone (RADICAVA®), a free-radical scavenger, was approved in several countries after showing efficacy in a specific subgroup of early-stage ALS patients [74, 75]. More recently, tofersen (QALSODY®), an antisense oligonucleotide targeting mutant SOD1, has shown potential in reducing NfL levels and slowing functional decline, though concerns remain regarding inflammatory side effects and the failure of initial phase III trials [76–78].
Emerging candidates such as PrimeC, a combination of ciprofloxacin and celecoxib, have shown promising results in slowing progression and improving survival in phase II trials [79, 80]. Similarly, dazucorilant, a selective glucocorticoid receptor modulator, was granted “fast track” status after showing improved survival despite missing its primary endpoint [81]. Another promising molecule, Usnoflast (previously ZYIL1), targets the NLRP3 inflammasome, a key component of neuroinflammation, and has been successfully tested in phase IIa trials for ALS [82, 83]. The compound is currently being evaluated in phase IIb trials.
Altogether, ALS exemplifies the profound complexity of neurodegenerative diseases characterized by neuronal loss. It underscores the limitations of single-targeted treatments and highlights the urgent need for multi-target approaches, search for clinical markers to enable early diagnosis, and robust clinical trial designs. Continued advances in omics technologies and biomarker discovery are essential to understand the intricate pathophysiology of ALS and to develop more effective and personalized neuroprotective strategies.
Stroke, particularly ischemic stroke, is a leading cause of adult disability and death worldwide, characterized by the sudden interruption of cerebral blood flow and the subsequent cascade of energy failure, excitotoxicity, oxidative stress, and neuronal death [84–87]. Unlike chronic neurodegenerative diseases such as PD, AD, HD or ALS, which involve slowly progressive and multifactorial neuronal loss, stroke represents an acute insult where the timing of intervention is critical. In this context, neuroprotection primarily depends on the rapid restoration of cerebral perfusion, typically through thrombolytic or endovascular therapies. Time-sensitive management is therefore the most effective strategy to limit irreversible damage and preserve neuronal integrity [84]. Given these distinct pathophysiological dynamics and therapeutic priorities, stroke will not be discussed further in this review, which focuses instead on chronic neurological diseases characterized by progressive neuronal loss, complex molecular interactions, and the need for sustained neuroprotective strategies.
While reshaping our way to connect basic science to clinical needs, we are currently witnessing new trends in drug discovery, sustained by several recent achievements obtained, for instance, in the field of “predictive-preventive-personalized-participatory (P4) medicine”. These trends have profound roots dating back to antiquity and the philosopher and skillful physician of the classical Greek period, Hippocrates, sometimes considered the father of Western medicine. In his “Hippocratic Corpus”, compiled centuries before the advent of precision medicine, Hippocrates had set up the foundations of what we would now call a personalized approach to healthcare, having done so even in the absence of the sophisticated diagnostic tools that are available today. Hippocrates was indeed the first to emphasize concepts such as individual patient needs, the role of lifestyle, and the body’s natural functions and development. Maintaining that diseases are a combination of environmental factors, diet, and living habits, Hippocrates appears to have anticipated the four principles of P4 medicine [88], which seeks to individualize care according to each patient’s distinctive biological and sociocultural profile.
P4 medicine strives to improve treatment effectiveness and clinical outcomes by a more holistic approach that carefully considers individual differences that contrasts with more traditional “one size fits all therapy”. The ongoing transformation is already evident; in 2020, 42% of the clinical trials in the USA were biomarker-based, compared to only 5% in 2005. The accelerated drug approval introduced by the FDA in 2024 was probably made possible by growing acceptance of precision medicine and genetic screening in targeted therapies, for instance, those for non-small-cell lung cancer and pancreatic adenocarcinoma, by approving zenocutuzumab-zbco (brand name Bizengri®, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-zenocutuzumab-zbco-non-small-cell-lung-cancer-and-pancreatic), and for transthyretin-mediated amyloidosis causing cardiomyopathy and heart failure, by approving acoramidis (brand names Attruby and Beyonttra, https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-drug-heart-disorder-caused-transthyretin-mediated-amyloidosis).
The P4 medicine paradigm continues to demonstrate its value across chronic diseases, even as attention increasingly shifts toward rare disorders. Common conditions such as diabetes, obesity, mental health disorders, and metabolic dysfunctions are poised to reap the greatest rewards from this new era in drug discovery. At the same time, rapid advances in genomics, electronic health records, digital health technologies, and data-driven research are leading a parallel surge in neurotherapeutics, an area with many urgent but unmet needs.
Neuroscientists can use genomic and proteomic data to identify individuals at risk long before the appearance of symptoms. Research continues for identifying molecular triggers and environmental factors that could predict the risk of developing diseases such as AD, PD, HD, ALS, Multiple Sclerosis (MS), epilepsy and others. Using predictive data, P4 medicine [88] can guide lifestyle changes and tailor treatments to each patient’s genetic background, environment, medication response, and disease course. These personalized strategies aim to prevent or slow neurological decline, improve symptom control, reduce adverse effects, and enhance overall outcomes. Looking ahead, patients will be better informed, encouraged to provide feedback, and closely monitored to optimize therapeutic results.
In addition to genetic data, large studies should look at the expression of relevant genes (proteomics) and the molecular pathways/processes in which the gene products are involved (e.g., metabolomics, receptor-associated cascades, growth factor effects, kinases, and many more), thus constantly improving our understanding of the neuropathological mechanisms and uncovering novel targets.
The future of clinical research in neuroscience, therefore, lies in collecting real-world data and using them as catalysts for clinical trial design and in further development of individually-tailored treatment procedures.
The nervous system is susceptible to diverse disruptions that impair homeostasis, whether locally or systemically. Below, we examine key mechanisms underlying neuronal degeneration, highlighting how they drive cellular damage (Figure 1). Like the entire review, this section concentrates on chronic, progressive diseases.
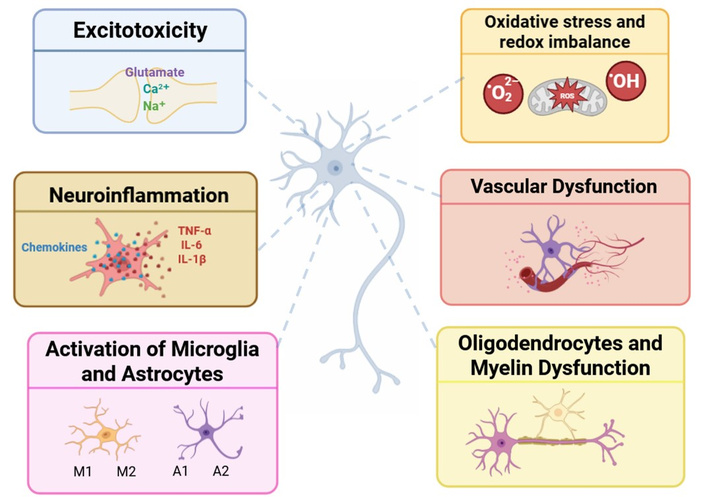
Schematic representation of the integrated mechanisms contributing to neuronal damage in neurodegenerative diseases. This diagram illustrates the main cellular and molecular mechanisms of primary neuronal damage in neurodegenerative conditions. Excitotoxicity: Excessive glutamate release resulting in sodium and calcium influx, which impairs neuronal viability. Neuroinflammation: Release of chemokines and cytokines, such as TNF-α, IL-6, and IL-1β. Activation of Microglia and Astrocytes: These cells can adopt phenotypes with distinct functions. Microglia M1 promotes inflammation and neurotoxicity, while the M2 phenotype supports tissue repair and anti-inflammatory signaling. A1 astrocytes show a neurotoxic profile, contributing to synaptic loss, whereas A2 astrocytes exhibit neuroprotective properties. Oxidative stress and redox imbalance: Accumulation of reactive oxygen species (ROS) and a decrease in antioxidant activity disrupts mitochondrial functions and promotes cellular damage. Vascular Dysfunction: Compromise of the blood-brain barrier integrity leads to impaired nutrient and oxygen supply, contributing to neuronal dysfunction. Oligodendrocytes and Myelin Dysfunction: Degeneration of oligodendrocytes disrupts myelin sheath integrity, leading to impaired axonal action potential conduction and neural integrity. Altogether, these interrelated mechanisms create a pathological environment and further progressive neuronal damage, forming the biological basis of neurodegenerative diseases. Cell illustrations were generated with the assistance of Sora, an AI-based image generation platform.
Excitotoxicity was one of the first mechanisms of neuronal damage that was identified. The word, allegedly coined by John Olney, refers to neuronal injury caused by excessive excitatory stimulation, particularly prolonged cellular depolarization [89]. This process commonly involves the main excitatory neurotransmitter of the nervous system, glutamate. This overstimulation results in excessive sodium and calcium influx, causing persistent neuronal depolarization [90]. Excitotoxicity may occur when, for instance, extracellular glutamate accumulates due to impaired uptake by astrocytes or excessive synaptic release. The glutamate-induced persistent depolarization affects mitochondrial membrane polarization, impairing mitochondrial function and energy metabolism (see section Oxidative stress and redox imbalance in the CNS, and [91]).
Three primary ionotropic glutamate, NMDA, alpha-amino-3-hydroxy-5-methyl-4-isooxazole-propionate (AMPA), and kainate receptors [92], can trigger excitotoxic cascades. While overstimulation of any of these receptors may contribute to excitotoxicity, excessive calcium influx via NMDA and AMPA receptors is particularly harmful. Ion influx activates enzymes such as endonucleases, phospholipases, and proteases, damaging both the plasma and mitochondrial membranes of the neuron. Excitotoxic mechanisms are implicated in various conditions, including ALS, AD, PD, epilepsy, and traumatic brain or spinal cord injury [91]. Other triggers include hypoglycemia that may affect brain energy metabolism [93].
β-methylamino-L-alanine (BMAA), a naturally occurring weak agonist of glutamate receptors, has been implicated in age-related neurological disorders, though its exact role remains debated due to insufficient evidence [94, 95]. Similarly, dietary intake of β-N-oxalyl-amino-L-alanine (BOAA), a plant-derived excitotoxin and potent agonist of ionotropic glutamate receptors, induces lathyrism, a neurodegenerative disease characterized by irreversible motor dysfunction due to spinal cord damage [96]. In ALS, the dysfunctional uptake of glutamate by astrocytic transporters contributes to excitotoxicity at least in a subset of ALS patients [97], however, simple inhibition of glutamate transport failed to produce an ALS-like condition in animals (see section The classical view: microglia as engines of inflammation for more details).
Experimental models frequently employ the natural neurotoxin kainic acid to trigger excitotoxic neuronal death, providing a well-established paradigm for studying neurodegeneration [98].
Notably, emerging evidence suggests that excitotoxicity may synergize with immune dysregulation, exacerbating neuronal vulnerability and accelerating disease progression [99].
Although present at appreciable levels in the brain, the role of aspartate remains poorly understood [100]. This amino acid is a potent excitatory agent acting primarily through NMDA receptors [101]. However, the risk of aspartate-induced excitotoxicity has not been fully evaluated. A critical gap is that aspartate does not appear to have been tested directly at metabotropic G protein-coupled glutamate receptors (mGluRs) [100]. Because activation of mGluRs is known to confer neuroprotection by limiting glutamate-induced excitotoxicity [102–107], aspartate’s apparent failure to activate these receptors would leave this protective mechanism unengaged. This absence of mGluR-mediated buffering could make aspartate a particularly potent excitotoxin, underscoring the need for further investigation.
Although excitotoxicity results from exacerbated excitatory transmission, it is important not to forget the relevance of GABAergic transmission and how GABAergic system modulators may affect neuronal plasticity and/or protect against cognitive decline after brain insults. Recent reviews have highlighted this issue [108, 109].
Inflammation is a protective immune response to infection or tissue injury. However, in the CNS, this process, termed neuroinflammation, differs fundamentally from peripheral inflammation. Unlike classical inflammation (characterized by heat, redness, and pain), neuroinflammation primarily involves the release of pro-inflammatory mediators (e.g., cytokines, chemokines, and ROS) by resident immune cells, particularly microglia, the CNS’s innate immune effectors [110, 111]. Notably, astrocytes also contribute by amplifying inflammatory signaling under pathological conditions.
In certain neurological disorders, T lymphocytes can infiltrate the CNS by crossing the BBB, where they exacerbate neuroinflammatory responses through pro-inflammatory cytokine release and glial cell activation. While this mechanism is most prominent in MS, growing evidence implicates T cell involvement in other neurodegenerative diseases as well [112].
Chronic neuroinflammation arises when persistent stimuli, including genetic mutations, pathological protein aggregates (e.g., Aβ and α-synuclein), traumatic injury, or neurotoxic exposure, convert an otherwise transient, protective inflammatory response into a sustained and maladaptive state. This chronic activation impairs neuronal homeostasis, worsens synaptic dysfunction, and ultimately accelerates neurodegenerative processes [113–116].
Glial cells, i.e., microglia, astrocytes, and oligodendrocytes, are essential for supporting and modulating neuronal function [117, 118]. Recent single-cell transcriptomic studies have revealed unexpected heterogeneity within glial populations [119]. Though traditionally viewed as immune-privileged, the CNS possesses its own immune defenses, with microglia acting as the primary immune effectors and constituting the predominant “immune” population in the CNS [120]. These cells constantly monitor the local environment and initiate inflammatory signaling in response to disruption [121]. Depending on stimuli, microglia can adopt distinct phenotypes: the M1 pro-inflammatory state (induced by lipopolysaccharide and characterized by production of cytokines such as TNF-α, IL-1β, and IL-6) and the M2 anti-inflammatory state (induced by IL-4 or IL-13, promoting repair) [122, 123]. While this M1/M2 classification oversimplifies microglial diversity, it remains useful for distinguishing functional profiles [124, 125]. More recent transcriptomic studies have described additional phenotypes, including disease-associated microglia (DAM), which are characterized by downregulated homeostatic genes and upregulated inflammatory ones [126]. The possibility of M2 microglia has led to new therapeutic opportunities that are covered in another section of this review.
Astrocytes are star-shaped glial cells critical for neuronal support, ionic homeostasis, neuroprotection, and synaptic regulation [127]. Despite being discovered alongside neurons, astrocytes remained understudied for decades. More recent advances have revealed their essential roles in CNS development, physiology, and disease pathogenesis [128]. These cells display intricate arborization patterns and form extensive gap junction-coupled networks that dynamically regulate synaptic activity, pH balance, and cerebral microcirculation. Astrocytes are fundamental components of both the BBB and the glymphatic waste clearance system [129–131]. In pathological conditions, astrocytes undergo significant molecular and morphological remodeling, adopting either a detrimental A1 (neurotoxic) or beneficial A2 (neuroprotective) reactive state that, respectively, drives injury progression or promotes tissue repair [132, 133]. The identification of these dichotomous astrocytic states, particularly the reparative A2 phenotype, has revealed promising therapeutic targets that will be examined in later sections of this review.
Astrocytes participate in glutamate clearance, converting it into glutamine, and play a key role in antioxidant defense. Under stress conditions, astrocytes may release toxic factors that amplify neuronal damage [134]. Dysfunctional microglia may fail to eliminate pathogens or apoptotic cells and may trigger maladaptive inflammatory responses [135, 136].
Oligodendrocytes, which make up about 20% of brain cells, produce and maintain myelin. Their dysfunction has been implicated in AD, where notable myelin alterations are reported [137]. Additionally, oligodendrocytes contribute to regulation of neuroinflammation, neuronal metabolic support, and stress response. They are active participants in neurodegenerative pathophysiology, as evidenced by transcriptomic studies [138].
Oligodendrocytes and Schwann cells ensure proper signal conduction by maintaining myelin and providing trophic support to axons. Damage or degeneration of oligodendrocytes is directly implicated in MS [139], and in ALS and frontotemporal dementia [140].
A critical initiating event in neuroinflammation is the detection of homeostatic disturbances within the CNS. This recognition is mediated by specialized molecular sensors, including pattern recognition receptors (PRRs), activated by damage-associated molecular pattern (DAMP) molecules produced upon exogenous threats and endogenous danger signals, subsequently activating inflammatory cascades [141, 142].
While glial dysfunction in neurodegenerative diseases (AD, PD, HD) primarily emerges as a consequence of underlying pathology, it nevertheless creates a self-perpetuating cycle that exacerbates disease progression. Through sustained release of pro-inflammatory mediators and impaired neuroprotective functions, dysfunctional glia amplify neuronal damage, effectively accelerating the neurodegenerative process [143].
Glial cells and neurons maintain dynamic communication via signaling molecules (cytokines, neurotransmitters, ROS, NO, glutamate) and extracellular vesicles containing proteins, mRNA, and miRNA. These vesicles can propagate toxic proteins like tau and β-amyloid and influence the progression of neurodegenerative diseases [144, 145].
Crosstalk between astrocytes and microglia has neuroprotective potential. In PD models, suppressing microglia-induced conversion of astrocytes to the A1 neurotoxic phenotype preserved dopaminergic neurons [146]. In vitro, astrocyte-to-microglia transfer of protein aggregates improved clearance [147]. However, in disease states, this interaction may turn maladaptive, promoting inflammation and neurodegeneration.
Neuronal death in neurodegeneration occurs via multiple mechanisms: apoptosis, necroptosis, pyroptosis, and ferroptosis. Apoptosis involves caspase-3-mediated protein degradation without inflammation [148]. Necroptosis, triggered when apoptosis is impaired, involves receptor-interacting protein kinases (RIPK) 1 and 3, and the mixed lineage kinase domain-like protein (MLKL), resulting in membrane rupture and inflammatory signaling [149]. Pyroptosis, in contrast, involves caspase-1 activation by inflammasomes like NLRP3, leading to cytokine release and membrane permeability increase [150, 151]. In PD and AD, pyroptosis is associated with elevated inflammatory cytokines and Aβ-mediated inflammasome activation [152, 153]. Ferroptosis is increasingly recognized as a distinct mechanism of regulated cell death contributing to age-related neurodegenerative disorders. Its involvement adds an epigenetic layer to neuronal vulnerability, potentially driving the progressive neuronal loss observed in conditions such as AD [154, 155]. Pathways leading to cell death release DAMPs, fueling further neuroinflammation. Modulating glial activity and these death pathways may offer therapeutic avenues. Current research is focused on targeting inflammasomes, glial signaling, and regulated cell death as strategies to mitigate neurodegeneration [156].
Oxidative stress is a key pathological feature of neurodegenerative diseases. It arises from an imbalance between the production of ROS and the body’s antioxidant defenses [157]. The CNS is particularly vulnerable to oxidative damage due to its high oxygen consumption, abundance of polyunsaturated fatty acids, and relatively low levels of antioxidant enzymes [158]. Understanding how oxidative stress contributes to disease mechanisms is essential for developing effective neuroprotective strategies [159], targeting neurons and glial cells.
Molecular oxygen is vital for energy production and cellular signaling. However, its metabolism can generate ROS, which include both radical (e.g., superoxide O2•–, hydroxyl radical •OH) and non-radical species (e.g., hydrogen peroxide H2O2) [160]. Nitrogen species such as the nitric oxide radical (NO•) and the peroxynitrite (ONOO–) also contribute to oxidative damage [161].
ROS are mainly produced in mitochondria during aerobic respiration, especially at complexes I and III of the electron transport chain [162, 163]. Other sources include nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, xanthine oxidase, peroxisomes, and enzymes associated with the endoplasmic reticulum [164, 165].
Physiologically, ROS act as signaling molecules involved in proliferation, immunity, and plasticity. They modulate redox-sensitive pathways such as those mediated by mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and some tyrosine phosphatases. For example, during infection, ROS activate immune cells and stimulate cytokine release [166]. Nitric oxide plays roles in blood flow regulation, neurotransmission, and immune responses [167]. Dysregulation of redox homeostasis leads to ROS accumulation, mitochondrial dysfunction, disrupted iron metabolism, and DNA damage, ultimately contributing to oxidative stress [168–170].
ROS can induce lipid peroxidation, particularly in the presence of iron, triggering ferroptosis, a regulated form of cell death [171]. Aβ oligomers exacerbate this process, producing reactive aldehydes like 4-hydroxy-2-nonenal (HNE) that damage proteins and contribute to neurodegeneration [172]. Iron, though essential for mitochondrial respiration and neurotransmitter synthesis, can generate ROS when dysregulated. Excess iron accumulation is linked to oxidative damage in AD, PD, and other neurodegenerative diseases [173, 174]. Compared to other organs, the brain has reduced levels of catalase and glutathione peroxidase activity, impairing its ability to neutralize H2O2 and other electrophilic compounds [175, 176]. DNA damage caused by ROS, such as base oxidation and strand breaks, disrupts genomic stability. When repair mechanisms fail, neuronal apoptosis may ensue [177]. Indeed, oxidative DNA damage markers like 8-hydroxy-2-deoxyguanosine (8-OHdG) are elevated in AD [178–180].
This vulnerability is compounded by an age-dependent decline in endogenous antioxidant defenses, contributing significantly to neuronal dysfunction and neurodegenerative pathogenesis. Critical roles in maintaining redox are exerted by antioxidant systems, including the thioredoxin/peroxiredoxin systems and the nuclear erythroid related transcription factor 2 (Nrf2), which regulates the expression of detoxification proteins such as Kelch-like ECH-associated protein 1 (Keap1) [181].
Notably, Nrf2 plays a dual protective role by both repressing neuroinflammatory signaling and promoting the transcription of antioxidant genes, establishing it as a compelling therapeutic target in neurodegenerative disorders [182, 183]. Intriguingly, antioxidant defense mechanisms in the CNS diverge substantially from those in peripheral tissues such as erythrocytes, where the glutathione–NADPH system is well characterized and robust. In contrast, the CNS’s antioxidant architecture remains only partially understood, and its complexity is heightened by neuronal heterogeneity and limited regenerative capacity. As highlighted elsewhere [184], neurons rely heavily on astrocytes for redox balance, creating a compartmentalized and interdependent defense system. This cell-specific reliance introduces unique vulnerabilities during oxidative stress episodes. Moreover, while the glutathione system is present in neural cells, its relative importance compared to other detoxification strategies remains unclear. Current therapeutic approaches aim to modulate these pathways, including direct Nrf2 activation and glial-targeted interventions, although significant challenges persist in delivering antioxidant agents across the BBB and in validating neuroprotection markers. These limitations underscore the need for further mechanistic insights and innovative delivery strategies to fully harness neuroprotective potential of targeting Nrf2-mediated pathways.
Although oxidative stress constitutes a unifying pathological feature among major neurodegenerative diseases, the molecular pathways through which it drives neuronal injury appear to be disease specific. A striking and still unresolved observation is that the most vulnerable neuronal populations differ markedly across disorders, affecting distinct brain regions such as the hippocampus in AD, the substantia nigra in PD, or the motor cortex and spinal cord in ALS. These differences likely reflect complex interactions among region-specific neuronal subtypes, glial responses, genetic predispositions, and disease-specific triggers, each contributing to unique patterns of redox imbalance. Deciphering these divergent mechanisms is essential for developing targeted, pathology-specific antioxidant therapies [185].
The defining pathological feature of PD is the selective degeneration of dopaminergic neurons in the substantia nigra pars compacta [4, 186]. While the precise mechanisms remain incompletely understood, these neurons exhibit particular vulnerability to oxidative stress due to their high basal metabolic activity, autonomous pacemaking requiring intense calcium cycling, and ROS generation during dopamine metabolism [187]. This susceptibility is compounded by impaired redox homeostasis and multiple pathogenic factors: i) dysfunctional protein clearance mechanisms involving PTEN-induced kinase 1 (PINK1), Parkin, and α-synuclein proteostasis; ii) mitochondrial impairment leading to ROS accumulation; and iii) environmental exposures to toxins like pesticides and heavy metals [188–190]. Together, these elements create a self-reinforcing cycle of oxidative damage, protein aggregation, and neuronal degeneration that characterizes PD progression.
In AD, oxidative stress also plays a central role promoting Aβ accumulation and tau phosphorylation [191] and impairing proteostasis (the homeostasis of protein folding, stability, and degradation), synaptic plasticity and other processes critical for learning and memory [188, 192]. Based on findings from the CRND8 mouse model, in which amyloid deposits are associated with neuropathological changes, it is conceivable that similar mechanisms, potentially involving the accumulation of metal ions such as copper, iron, or zinc, could also occur within amyloid plaques in human patients [193, 194].
An informative account of the causes of neuronal-death-vulnerability that can be deduced from familial cases of Parkinson’s and Alzheimer’s diseases has been described elsewhere [195].
HD is due to a CAG repeat expansion in the HTT gene. While huntingtin folds and functions normally, the mutant form exhibits aberrant folding, leading to aggregation and neuronal toxicity. The mHTT protein interacts improperly with other cellular proteins, disrupting normal biological functions. ROS exacerbate the misfolding process, promoting the accumulation of protein aggregates near neuronal axons and dendrites, ultimately impairing synaptic communication [196]. Mitochondrial Ca2+ handling is also disrupted, leading to the opening of the mitochondrial permeability transition pore (mPTP) and the release of cytochrome c, which triggers apoptosis. This imbalance is also associated with excessive superoxide production and mitochondrial DNA damage [197].
Cytoplasmic protein aggregates, a pathological feature of ALS, disrupt key cellular degradation mechanisms, including both the ubiquitin-proteasome system and autophagy pathways, resulting in catastrophic failure of protein homeostasis. Mitochondrial dysfunction resulting from accumulation of trans-activation response element DNA binding protein 43 (TDP-43) increases the production of ROS, causing oxidative stress and establishing a vicious cycle that contributes to disease progression. Thus, oxidative stress emerges as a central mechanism in the disease pathogenesis, interconnected with proteostatic and mitochondrial dysfunction [198, 199].
A detailed understanding of the distinct pathophysiological mechanisms and redox imbalances underlying each neurodegenerative disorder is crucial for advancing personalized therapeutic strategies, enabling interventions tailored to the specific cellular vulnerabilities and molecular drivers of each disease.
Disruptions in cerebral perfusion, whether due to trauma, ischemia, or hemorrhage, are well-established drivers of acute neuronal injury. The neurovascular unit, composed of endothelial cells, pericytes, astrocytic end-feet, and vascular smooth muscle cells, plays a central role in regulating cerebral blood flow and maintaining the integrity of the BBB. These components are essential for ensuring nutrient delivery and shielding neural tissue from circulating toxins and inflammatory mediators [200]. Loss of BBB integrity in prodromal or early symptomatic phases may exacerbate neuroinflammation and accelerate neuronal loss [201].
However, despite this contributory role, it is not reasonable to propose direct therapeutic targeting of vascular dysfunction or BBB endothelial cells as a primary strategy for treating neurodegenerative disorders. These structures, while involved in disease progression, are not the root cause of neuronal degeneration and remain challenging to modulate without risking systemic side effects or impairing essential barrier functions. Therefore, therapeutic efforts are better directed toward neuronal and glial targets, which more directly govern disease onset and progression.
Independent of the trigger of neurodegeneration leading to AD, PD, HD or ALS, common pathological themes emerge: excitotoxicity, mitochondrial dysfunction, oxidative stress, glial and vascular dysfunction, often interacting in complex, cascading patterns. Understanding these interconnected processes is vital for developing strategies to halt or reverse neurodegeneration and protect neuronal integrity in aging and disease.
The convergence of excitotoxicity, oxidative stress, and immune dysregulation highlights the multifactorial nature of neuronal damage. A key advancement in addressing these mechanisms is the shift from a neuron-centric to a holistic view that encompasses the critical role of glial cells, a perspective that informs the diverse therapeutic toolkit explored in the following sections.
Neurons are the principal cellular targets in most neurodegenerative diseases, and their progressive loss accounts for the irreversible nature of clinical decline in conditions such as AD, PD, HD, and ALS. Neuron-focused therapeutic strategies seek to prevent degeneration, enhance survival, and restore function. However, these approaches face inherent challenges: Neurons have limited regenerative capacity, are highly susceptible to metabolic stress, and require preservation of precise synaptic connectivity for proper network integration.
This section presents current neuron-targeted therapeutic strategies, organized along a conceptual axis from extracellular modulation (e.g., receptor activation and trophic signaling) to intracellular mechanisms (e.g., signal transduction, transcriptional regulation and metabolic support). Particular attention is given to interventions that enhance synaptic plasticity, activate intrinsic neuroprotective pathways, and promote functional network regeneration (Figure 2).

Neuron-targeted therapeutic strategies for neurodegenerative diseases. This diagram summarizes four key classes of neuron-directed interventions designed to counteract neurodegeneration and support functional recovery. Enhancing Synaptic Plasticity (top left, blue): Modulation of AMPA and NMDA receptors boosts excitatory synaptic transmission and long-term potentiation (LTP). These strategies aim to restore synaptic strength essential for memory, learning, and network dynamics. Neurotrophic Factor Delivery (top right, orange): Administration of trophic molecules like BDNF and CDNF, either via receptor activation (e.g., TrkB) or exosome-based delivery, promotes neuronal survival, synaptic stability, and overall network resilience. Activating Intrinsic Neuronal Defense (bottom left, blue): Engaging cellular pathways such as Nrf2 and SIRT1 enhances antioxidant capacity, mitochondrial function, and autophagic clearance. These self-defense mechanisms help shield neurons from oxidative damage and protein misfolding. Stimulating Regeneration and Connectivity (bottom right, orange): Techniques like PTEN inhibition and neuromodulation (e.g., deep brain stimulation or chemogenetics) promote axonal growth and reconstitution of neural circuits. Together, these therapeutic domains represent a multifaceted approach to treating neurodegenerative conditions such as AD, PD, and ALS. Neuron icon by Vecteezy (https://www.vecteezy.com/).
One of the earliest consequences of neurodegeneration is the loss of synaptic strength and plasticity. Synaptic plasticity is a fundamental process underlying cognitive functions such as learning and memory [202]. Synaptic alterations are a hallmark of numerous neurological and psychiatric disorders, making the restoration of synaptic efficacy a central therapeutic goal [203, 204]. Enhancing plasticity can help stabilize circuit function, compensate for neuronal loss, and delay clinical deterioration. This subsection explores therapeutic strategies designed to boost synaptic adaptation, including receptor modulation, epigenetic tuning, and activity-based interventions.
Among pharmacological approaches, positive allosteric modulation of AMPA receptors using AMPAkines (e.g., aniracetam, CX516) has shown promise. AMPA receptors mediate fast excitatory neurotransmission, and their potentiation enhances excitatory postsynaptic potentials (EPSPs), promoting long-term potentiation (LTP), the cellular correlate of learning and memory [205, 206]. AMPAkines increase channel open time without directly activating the receptor, and may also elevate neurotrophic factors such as BDNF (brain-derived neurotrophic factor), contributing to neuronal survival and synaptic remodeling [207]. Consequently, AMPA receptor potentiation is being investigated as a therapeutic strategy for cognitive dysfunction in AD, schizophrenia, and depression.
NMDA receptors are likewise critical for calcium-dependent synaptic plasticity. Their activation permits calcium influx and initiates intracellular signaling cascades involving calmodulin kinase II (CaMKII), cAMP Response Element Binding Protein (CREB) and MAPK, culminating in the transcription of genes required for long-lasting synaptic enhancement [208]. Nonetheless, overstimulation of NMDA receptors can trigger excitotoxic damage, necessitating tightly controlled modulation. Therapeutic strategies under investigation include partial agonists (e.g., D-cycloserine) and modulators of the glycine co-agonist site, which may enhance plasticity while minimizing toxicity. NMDA receptor dysfunction seems implicated in schizophrenia, post-traumatic stress disorder, autoimmune cognitive syndromes, and age-related cognitive decline. In AD, subtype-specific NMDA receptor antagonists or allosteric modulators may protect against excitotoxic injury and support synaptic integrity [209–211]. Memantine, a currently approved NMDA receptor modulator, has demonstrated modest symptomatic benefit in AD patients but does not significantly alter disease progression [212–214].
In addition to receptor modulation, epigenetic regulation of plasticity-related genes offers another therapeutic frontier. Expression of key genes such as BDNF and Arc is governed by histone acetylation and DNA methylation, processes controlled by enzymes like histone acetyltransferases (HATs) and DNA methyltransferases (DNMTs) [215, 216]. Pharmacological or behavioral modulation of these pathways may reactivate transcriptional programs essential for synaptic restructuring.
Physical interventions such as aerobic exercise and intermittent fasting promote synaptic plasticity via metabolic reprogramming and increased BDNF expression [217–219]. Furthermore, non-invasive brain stimulation (NIBS) techniques, such as transcranial magnetic stimulation (TMS) and transcranial direct current stimulation (tDCS), have demonstrated potential in reversing cortical atrophy and enhancing plasticity [220–222].
While synaptic plasticity defines the adaptability of neural circuits, neurotransmission ensures signal fidelity and integration across brain networks. Neurodegenerative diseases often disrupt neuromodulatory pathways, further impairing cognitive and motor functions. This section focuses on pharmacological strategies that enhance neurotransmitter signaling and modulate network activity to support function and recovery.
Neuromodulatory systems constitute key therapeutic targets for cognitive enhancement [223]. The cholinergic system, essential for attention, learning, and cortical plasticity, is profoundly disrupted in AD [224]. Early therapeutic efforts using muscarinic receptor agonists like xanomeline were limited by peripheral side effects. However, recent approaches that pair xanomeline with trospium, a peripherally acting muscarinic antagonist, have improved tolerability and clinical efficacy, leading to regulatory approval [225, 226]. Additionally, acetylcholinesterase inhibitors (e.g., donepezil) and both muscarinic and nicotinic receptor agonists support cholinergic tone and enhance cognitive performance [227].
Dopaminergic signaling plays a central role in working memory, motivation, and goal-directed behavior. Dopamine D1 receptor agonists and reuptake inhibitors such as methylphenidate are used to enhance prefrontal cortex function and are effective in conditions such as PD and attention-deficit/hyperactivity disorder (ADHD) [228]. The noradrenergic system, particularly through α2A-adrenergic receptor signaling, modulates executive function and emotional regulation. Agents such as guanfacine (an α2A adrenergic agonist) and atomoxetine, a norepinephrine reuptake inhibitor, have demonstrated clinical efficacy in improving attention and working memory, and are used in the treatment of ADHD and post-traumatic stress disorder [223, 229].
In summary, the therapeutic modulation of neuromodulatory systems provides a diverse toolkit to enhance synaptic function. These approaches, ranging from receptor-targeted pharmacology to systemic neuromodulation, can be integrated with molecular and epigenetic interventions. Future strategies will likely emphasize combinatorial regimens, optimized in terms of target hierarchy, temporal dynamics, and dosage to maximize benefit while minimizing adverse effects.
Building on the modulation of neurotransmitter systems, another crucial dimension of neuroprotection involves sustaining the cellular environment that allows neurons to survive, adapt, and thrive. This includes not only neurotransmission but also the support of neurotrophic factors that promote neuronal health and synaptic maintenance. The section explores how such trophic support, alongside intrinsic resilience mechanisms and regenerative capacity, can be leveraged therapeutically.
BDNF plays a pivotal role in enhancing synaptic efficacy and promoting neurogenesis, particularly in glutamatergic circuits [230, 231]. BDNF signaling is frequently downregulated in neurodegenerative conditions, and strategies to restore its levels have demonstrated improvements in synaptic integrity and cognitive outcomes [129, 130]. Cleavage of the signaling receptor for BDNF, the tropomyosin receptor kinase B-full length (TrkB-FL), occurs in human tissue and in animal models of brain diseases [232]. Prevention of that cleavage may, therefore, prove a way to go towards novel therapies to halt neurodegeneration. Important in this context is the recent development of drug, a small TAT-TrkB peptide, that prevented TrkB-FL cleavage, improved cognitive performance, ameliorated synaptic plasticity deficits and prevented Tau pathology progression in vivo in a mouse model of AD [233].
Another key neurotrophic factor, glial cell line-derived neurotrophic factor (GDNF), has demonstrated significant neuroprotective effects in preclinical models of PD. GDNF exerts its protection by binding to the GDNF family receptor α-1 (GFRα) and c-Ret receptor complex, thereby maintaining the function and survival of dopaminergic neurons [234–236]. More recently, endoplasmic reticulum stress-regulating neurotrophic factors such as mesencephalic astrocyte-derived neurotrophic factor (MANF) and cerebral dopamine neurotrophic factor (CDNF) have emerged as promising agents. These proteins help maintain proper proteostasis and attenuate endoplasmic reticulum stress, thereby supporting dopaminergic neuron viability [237, 238].
Additional neurotrophic factors, including insulin-like growth factor 1 (IGF-1) and nerve growth factor (NGF), exert complementary effects by regulating cellular metabolism, promoting anti-apoptotic pathways, and enhancing myelination [239, 240]. Despite their therapeutic promise, clinical translation has been hindered by the difficulty of achieving effective delivery into the brain. The BBB remains the major obstacle, and even molecules capable of crossing it often do so inefficiently or without reaching therapeutically relevant concentrations. To overcome this limitation, new delivery platforms are under development. Extracellular vesicles, constituted by exosomes derived from mesenchymal stem cells, can encapsulate BDNF, GDNF, and related factors, facilitating their transport across the BBB with minimal immunogenicity [241, 242]. In parallel, vectors consisting of adeno-associated viruses (AAV) provide a gene therapy-based approach for sustained, localized expression of neurotrophic factors in target brain regions [243, 244]. Other approaches include small-molecule mimetics and lifestyle interventions, such as phenolic-rich diets, which may enhance endogenous neurotrophin production [245–247].
A novel and increasingly compelling area of research involves the neuroimmune interface, particularly the role of regulatory T cells (Tregs). In preclinical neurodegeneration models, Tregs provide dual benefits: Suppression of neuroinflammatory cascades and induction of neurotrophic factor release, including CDNF, via crosstalk with neural substrates [248–250]. This immunomodulatory mechanism highlights a promising therapeutic avenue that bridges the immune and nervous systems.
Emerging paradigms consist of a convergence of molecular, cellular, and systemic strategies aimed at optimizing neurotrophic support. The integration of gene therapy, targeted delivery vehicles, and immune-based modulation offers a sophisticated framework to counteract neuronal loss and functional decline in neurodegenerative disorders.
While neurotrophic factor therapies focus on enhancing extrinsic support mechanisms, neurons also possess internal defense pathways. These intrinsic mechanisms, ranging from oxidative stress control to autophagy regulation, form the next layer of neuron-targeted interventions.
Intrinsic neuroprotection refers to the activation of endogenous cellular programs that bolster neuronal survival under stress. These mechanisms often intersect with pathways influenced by trophic factors, creating a continuum of extrinsic and intrinsic support. One of the principal molecular regulators of such defense is the Nrf2–antioxidant response element (ARE) signaling axis, which orchestrates antioxidant and cytoprotective gene expression programs [251]. Pharmacological activators of Nrf2, including dimethyl fumarate and sulforaphane, mitigate oxidative stress and improve mitochondrial efficiency in preclinical disease models [252].
Sirtuins, particularly SIRT1 and SIRT3, have also emerged as key modulators of neuronal resilience. These NAD+-dependent deacetylases regulate mitochondrial biogenesis through transcriptional players such as the peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α), as well as pathways involved in DNA repair and redox balance [253, 254]. Sirtuin activation has demonstrated neuroprotective effects in models of neurodegeneration and neuroinflammation [255].
Given their extended lifespan, neurons rely heavily on autophagy-lysosomal mechanisms to clear misfolded proteins and damaged organelles. Pharmacological enhancers of autophagy such as rapamycin and metformin, act broadly on this system, while more targeted strategies, such as urolithin A to stimulate mitophagy, are under active investigation [256, 257].
A novel and increasingly studied structure in this context is the primary cilium, a microtubule-based organelle that functions as a signaling hub for pathways including Sonic Hedgehog (Shh), Wnt/Wingless, and mechanistic target of rapamycin (mTOR). Through this compartment, neurons are able to coordinate oxidative stress responses, survival signaling, and proteostatic control [258–260]. Structural disruption of the primary cilium impairs these signaling processes and is associated with exacerbation of neurodegenerative pathology [261, 262].
Emerging therapeutic strategies have begun to target ciliary signaling directly. These include the use of Smoothened (Smo) agonists to activate Shh signaling, as well as small-molecule modulators of intraflagellar transport (IFT) proteins to restore ciliary trafficking and structural integrity [263–268]. Such interventions hold promise for enhancing neuronal stress resistance by reinforcing endogenous cytoprotective mechanisms [262, 266].
Purine nucleosides, namely adenosine and guanosine, have relevant neuroprotective roles by acting both centrally and peripherally, via activation of their receptors and intracellular signaling mechanisms [269, 270]. A main problem with adenosine receptor-based therapies, in particular those directed towards the ubiquitous neuroprotective receptor, the adenosine A1 receptor, is the side effects. Less problematic in this respect are the antagonists of the pro-excitotoxic adenosine A2A receptor [271] as well as drugs directed towards the relatively disregarded low expressed adenosine A3 receptor [272, 273]. A way to go may be to selectively target receptors overexpressed in diseased tissue, as it has been recently highlighted for a drug with A3 receptor agonist properties that selectively boosts GABAergic transmission [274].
Yet even when intrinsic defenses succeed in delaying damage, functional recovery remains limited unless structural connectivity is restored. This highlights the importance of regenerative strategies to rebuild disrupted networks.
Regeneration-based therapies are designed to overcome the inherently limited axonal regenerative potential. Utilizing a combination of molecular interventions and bioengineering, these approaches seek to reconstruct functional neural networks lost to injury or disease. This is possible because, despite the traditional classification of adult CNS neurons as non-regenerative, they maintain a latent capacity for growth. Strategic manipulation of intrinsic pathways, exemplified by Phosphatase and Tensin homolog (PTEN) inhibition, can suppress growth constraints and re-activate robust axonal extension [267, 268]. Likewise, transcriptional reprogramming with factors such as SRY-Box Transcription Factor 11 (SOX11), Krüppel-Like Factor 7 (KLF7), or c-Jun transcription factor induces pro-regenerative phenotypes in CNS neurons [267, 275].
Eventually, extrinsic factors in the neural environment also pose major barriers. Nogo-A, a myelin-associated neurite outgrowth inhibitor, along with chondroitin sulfate proteoglycans in the extracellular matrix, actively suppress axonal outgrowth. Interventions such as chondroitinase ABC and neutralizing antibodies against myelin inhibitors have demonstrated modest success in facilitating axonal sprouting and synaptic reorganization [276].
A conceptually transformative strategy involves cellular reprogramming. In animal models, glial cells such as astrocytes and NG2 oligodendrocyte precursor cells can be directly converted into functional neurons in vivo via ectopic expression of neurogenic transcription factors like NeuroD1 and Ascl1. This approach bypasses the complexities of cell transplantation and enables region-specific neuronal replacement within the native brain environment [277].
However, translating these findings into clinical applications presents significant challenges [278]. While proof-of-concept is firmly established in rodent models, issues such as delivery efficiency, long-term integration, and avoidance of maladaptive circuitry must be resolved to enable therapeutic implementation in the human CNS.
Ultimately, meaningful recovery depends not only on regenerating individual neurons but also on reconstructing functional network architecture. Neuromodulatory techniques, such as optogenetics, chemogenetics using designer receptors exclusively activated by designer drugs (DREADDs), and clinical interventions like deep brain stimulation and vagus nerve stimulation (VNS), are being utilized to reestablish circuit synchrony. Evidence suggests that restoring coordinated activity, rather than simply increasing cell number, is key to improving behavioral outcomes [279].
Translation of these regenerative and network-based therapies into clinical practice will require careful calibration. For example, mTOR signaling must be modulated to promote autophagy and clearance of toxic protein aggregates (e.g., Aβ or tau) without compromising synaptic maintenance [280]. Similarly, regenerative outgrowth must be guided to avoid aberrant rewiring and potential excitotoxicity. The rapid advancement of tools in molecular reprogramming, gene delivery, and circuit-level modulation may shift the paradigm, transforming the idea of functional brain restoration from speculative to increasingly attainable.
Still, successful regeneration requires not just structural growth but functional integration into existing circuits. Emerging neuromodulation techniques may help bridge this gap.
Despite significant advances in understanding neuronal dysfunction, translating these insights into effective treatments remains challenging. As a result, combating neurodegenerative diseases now demands a multidisciplinary approach aimed at developing therapies that not only slow progression but also preserve and restore cognitive and functional capacity.
Pharmacological approaches targeting AMPA and NMDA receptors, cholinergic and dopaminergic systems, as well as epigenetic and lifestyle-based interventions, offer measurable symptomatic relief and provide valuable tools for modulating synaptic function. Nonetheless, their long-term disease-modifying potential remains limited, in part due to the complexity of underlying pathophysiological processes and compensatory mechanisms within the human brain.
Enhancing intrinsic neuronal resilience through pathways such as Nrf2–ARE signaling, sirtuin activation, and the regulation of autophagy reflects a promising avenue grounded in the reinforcement of cellular homeostasis. Yet even these strategies face limitations in terms of delivery specificity, metabolic variability among patients, and potential off-target effects. Similarly, the restoration of neurotrophic support, whether through exogenous administration, gene therapy, or immune-mediated modulation, illustrates a growing sophistication in therapeutic design, although questions regarding long-term efficacy, immune tolerance, and BBB penetration remain unresolved.
One of the most conceptually ambitious directions in neurotherapeutics is the shift from protective to regenerative paradigms. Experimental techniques that aim to reprogram glial cells into neurons or stimulate latent axonal growth pathways have yielded promising results in preclinical models. However, their translation to human applications is far from imminent. The CNS in humans presents considerable anatomical, immunological, and functional challenges, and ensuring that regenerated neurons integrate meaningfully into pre-existing circuits without causing maladaptive rewiring or excitotoxicity is a task of extraordinary complexity. Neuromodulatory technologies such as deep brain stimulation, optogenetics, and chemogenetics offer tools to partially guide and refine network-level activity; however, regenerative medicine for the human brain remains largely conceptual in the clinical domain.
In summary, the therapeutic landscape for neurodegeneration is moving toward a multimodal, multi-tiered approach. No single intervention will suffice. Rather, success will depend on the strategic combination of protective, supportive, and regenerative strategies tailored to the temporal and pathological stage of disease. The future of neuron-targeted therapies lies in their integration, molecular precision, circuit-level modulation, and personalized timing, working together to restore lost function in the human brain. The most ambitious and promising advances on targeting neurons still belong to the realm of experimental research, and their successful translation into safe, effective, and scalable interventions for human neurodegenerative disease will require a careful balance of innovation, mechanistic understanding, and clinical restraint.
Having examined neuron-focused therapeutic strategies, we now turn to astrocytes, a glial population increasingly recognized as active players and potential therapeutic targets in neurodegeneration. Astrocytes contribute to neuronal communication and the maintenance of brain homeostasis, supporting a wide array of physiological processes (Figure 3). Their discovery is closely tied to the work of Camillo Golgi, whose staining techniques first made glial cells visible. The name “astrocyte”, derived from their characteristic star-shaped morphology, was coined later by Mihály Lenhossék. A major advance came in 1913, when Santiago Ramón y Cajal, using the gold-sublimate method, described two key astrocyte subtypes: Protoplasmic astrocytes, found in grey matter and marked by fine, elaborate processes, and fibrous astrocytes, present in white matter and distinguished by longer, fiber-like extensions. He further showed that astrocytic processes envelop neurons and associate with blood vessels, foreshadowing their functional diversity. These foundational observations laid the groundwork for today’s understanding of astrocyte heterogeneity and specialization in the CNS [281].
Major roles of astrocytes in the CNS. Astrocytes play essential roles in the CNS by performing multiple interconnected functions. These include inter-astrocyte communication; the formation and maintenance of the BBB; regulation of ion and water homeostasis; and energy metabolism by providing energetic substrates to neurons. Astrocytes also interact with microglia to induce innate immune responses, contribute to injury responses and protect healthy tissue. They support oligodendrocytes and promote axonal myelination through both secreted factors and direct astrocyte–oligodendrocyte or astrocyte–axon interactions. Furthermore, astrocytes regulate synaptic function by modulating synaptic transmission and plasticity, buffering extracellular K+, taking up neurotransmitters, and releasing gliotransmitters. Therapeutic integration across these domains holds potential for multi-modal intervention in AD, PD, HD, ALS and other neurodegenerative disorders. Neuron icon was designed by Inkscape (https://inkscape.org/da/).
To understand their therapeutic potential, it is first important to explore how astrocytes sustain neural homeostasis and how this balance can shift from protective to detrimental in disease context. Astrocytes exert both neuroprotective and neurotoxic effects, depending on environmental cues. From the earliest accounts of AD, Alois Alzheimer documented changes in non-neuronal cells near amyloid plaques, a phenomenon now known as reactive astrogliosis [282]. This response is not exclusive to AD [283], but is also a hallmark of PD, ischemic stroke, and ALS, in which astrocyte reactivity is critically involved in disease development.
In AD, astrocytes assist in Aβ removal via autophagy pathways [284] and activation of the urea cycle, notably through enhanced expression of ornithine decarboxylase 1, a key enzyme in polyamine biosynthesis [285]. This leads to increased GABA and H2O2 levels through MAO-B–mediated conversion of putrescine [286]. While initially protective, rising H2O2 levels disturb redox balance, promote oxidative stress, and contribute to neuronal death and brain shrinkage. Ultimately, this cycle impairs antioxidant defenses and drives a self-reinforcing process that hastens AD progression.
Reactive astrocytosis is a consistent pathological hallmark not only in AD, but also in PD, ischemic stroke, and ALS. Astrocytes contribute to neurodegeneration by releasing neurotoxic molecules and disrupting neuronal equilibrium [287–290]. Their influence on neuronal communication and brain homeostasis is largely exerted via gliotransmitters such as GABA, glutamate, D-serine, lactate, and ATP/adenosine, in addition to neurotrophic factors like proBDNF and BDNF. Astrocytes also regulate oxidative stress and cytokine production through complex pathways that are still being unraveled.
Transcriptomic techniques, including RNA sequencing of region-specific samples, have enabled a nuanced understanding of the astrocyte transcriptome in rodents and humans. These studies have revealed that astrocytes express a rich, cell-specific repertoire of genes. Moving beyond classical markers like glial fibrillary acidic protein (GFAP), this transcriptome includes genes for extracellular matrix components, glutamate transporters, and various transcription factors. This diversity reflects the pronounced spatial and temporal heterogeneity of astrocytes [291].
Astrocytes actively participate in synaptic signaling through the release of gliotransmitters, modulation of ion balance, and cytokine control [292]. Under pathological conditions, they may initially exert neuroprotective effects, yet prolonged activation often results in metabolic disturbances and neurotoxic gliotransmission, highlighting their dual role in CNS pathology and their potential in therapy [293].
CNS injury-induced astrocyte activation has strengthened the view that many neurodegenerative diseases involve non-cell-autonomous mechanisms, where glial cells, especially astrocytes, actively shape disease progression. Research on ALS has been particularly informative in establishing this concept [294].
Beyond homeostatic support, astrocytes serve as key regulators of fundamental CNS physiology, influencing metabolic supply, neurotransmission, and synaptic plasticity.
Metabolic support is mediated by the astrocyte–neuron lactate shuttle, which fuels neuronal activity under both normal and stress conditions.
Astrocytes take up glucose via GLUT1 and metabolize it via glycolysis, generating lactate through lactate dehydrogenase (LDH). This lactate is then exported by MCT1 and MCT4 transporters and imported into neurons via MCT2, where it fuels neuronal ATP production [295, 296]. Altered LDH expression impacts neuronal excitability and synaptic function [297]. Calcium-dependent regulation of this astrocyte-neuron lactate shuttle allows flexible adaptation to neuronal energy needs. Lactate also protects neurons during metabolic stress and influences gene expression related to plasticity [298].
In addition to energy metabolism, astrocytes contribute to GABAergic signaling by synthesizing GABA through the MAO-B and diamine oxidase pathways, and possibly through glutamate decarboxylation [299–302]. GABA is released via Best1 and VRACs, fine-tuning extrasynaptic GABAA receptor activity and modulating network excitability [303, 304]. Sirtuin 2 (SIRT2) and aldehyde dehydrogenase 1 family member A1 (ALDH1A1) are critical for astrocytic GABA production in AD; upregulation of these enzymes occurs in hippocampal astrocytes of patients and of a transgenic AD model [305]. Members of this astrocytic GABA production system are increasingly recognized as therapeutic targets.
In addition to GABAergic signaling, astrocytes play a fundamental role in maintaining glutamate homeostasis, which is essential for modulating tonic activation of N-methyl-D-aspartate receptors (NMDARs) [284, 306], typically composed of two GluN1 subunits and either two GluN2A or two GluN2B subunits [307].
Astrocytes regulate extracellular glutamate through uptake via EAATs [308] and release via channels like TREK1 and Best1, vesicular pathways, and the cystine/glutamate antiporter [4, 30–32, 284, 309–311]. Glutamate synthesis involves the TCA and glutamate-glutamine cycles, supporting neurotransmitter recycling and synaptic plasticity [312, 313]. These actions modulate NMDA and AMPA receptor activity, ensuring a balanced excitatory-inhibitory environment. Interestingly, the activity of these ionotropic receptors in turn regulates cell metabolism [314].
L-serine is converted to D-serine by serine racemase in astrocytes, regulating NMDA receptor activation via GluN1 subunits [315]. D-serine release occurs via exocytosis, ASC transporters, and Best1 channels [284, 306, 316–319]. Dysregulation of D-serine contributes to cognitive and neuropsychiatric symptoms in neurodegenerative conditions [320, 321].
Astrocytes release ATP through both vesicular and non-vesicular routes, with subsequent conversion to adenosine by ectonucleotidases [322–326]. Via purinergic receptors these molecules modulate neurotransmission by inhibiting excitatory input and enhancing GABAergic tone [326–330]. They also influence firing rates and axonal conduction, shaping plasticity and behavioral state transitions [326, 331]. Effects vary by receptor subtype. Activation of adenosine A1 receptors by astrocytic ATP-derived adenosine generally suppresses synaptic activity across multiple brain regions, including the hippocampus [332–337], cortex [338], cerebellum [339], retina [340], amygdala [341], and nucleus accumbens [342]. In contrast, activation of adenosine A2A receptors tends to facilitate synaptic transmission, particularly in the hippocampus [343, 344]. They are also expressed in the hypothalamus [345]. Remarkably, astrocytes can exert opposing effects on different synaptic inputs depending on the adenosine receptor subtype.
Astrocytes synthesize and secrete proBDNF in response to diverse stimuli, possibly internalizing and processing neuronal proBDNF into mature BDNF [64–67, 346–349]. Vesicular release, mediated by VAMP3, is regulated by Ca2+ signaling [350]. While BDNF supports plasticity via TrkB, proBDNF can activate p75 receptors and trigger apoptosis [67, 349]. TrkB-T1 signaling in astrocytes also influences GABA uptake, calcium dynamics, and cell morphology [351–355].
The functional impact of astrocyte-derived proBDNF appears to be highly context-dependent. Under physiological conditions, it supports synaptic plasticity [346], whereas under pathological states, such as during astrocytic necroptosis, proBDNF may exert neurotoxic effects and promote apoptotic signaling [349].
In cultured astrocytes, the truncated isoform of the TrkB receptor (TrkB-T1, also known as TrkB-Tc) mediates rapid intracellular Ca2+ transients in response to brief BDNF exposure. This effect is dependent on phospholipase C activation and subsequent calcium release from IP3-sensitive intracellular stores [351]. Beyond calcium signaling, BDNF–TrkB-T1 interactions also regulate astrocytic function by modulating the activity of the GABA transporter GAT-1, thereby influencing GABA uptake dynamics [352]. Additionally, TrkB-T1 has been implicated in structural modulation, including the regulation of dendritic filopodia formation in hippocampal neurons [353], the morphology of neocortical layer I astrocytes in adult brain slices [354], and astrocyte shape via Rho GTPase signaling in primary culture systems [355].
Astrocytes release cytokines like IL-1β, IL-6, TNF-α, CCL2, and CXCL10 in response to injury and inflammation [128, 356]. Cytokine expression is typically induced by the activation of Toll-like receptors (TLRs), which initiate downstream signaling cascades involving NF-κB and MAPK pathways [357, 358].
Astrocytes are capable of producing both pro-inflammatory and anti-inflammatory cytokines, such as IL-6 [358], and the bidirectional communication between astrocytes and microglia is central to shaping the inflammatory milieu within the CNS. Notably, microglia can induce a neurotoxic astrocytic phenotype by releasing IL-1α, TNF-α, and complement component C1q. These factors collectively reprogram astrocytes to secrete additional neurotoxic mediators, further amplifying neuroinflammatory responses [357].
Astrocytes generate H2O2 and NO via mitochondrial respiration and enzymes such as MAO-B, diamine oxidase, NADPH oxidases, and inducible nitric oxide synthase (iNOS) [359–364]. Dysregulation leads to oxidative stress, promoting inflammation and neurodegeneration. Antioxidant capacity, including catalase activity, may be impaired in pathological conditions [365]. H2O2 can diffuse or be transported via aquaporins, while NO passively permeates membranes [366]. Astrocytes generate ROS through multiple enzymatic pathways, including mitochondrial sources, particularly the electron transport chain, and cytosolic enzymes such as MAO-B, diamine oxidase, and NADPH oxidases. In reactive astrocytes, MAO-B activity is notably elevated, leading to increased production of H2O2, while NADPH oxidase 2 is frequently upregulated, further amplifying ROS generation [359, 363].
ROS and reactive nitrosylation species (RNS) also act as critical signaling molecules that modulate synaptic strength, neurotransmitter release, and cerebral blood flow [367]. For example, the NO molecule plays a well-established role in vasodilation and synaptic communication. However, when ROS and RNS levels become dysregulated, they can cause oxidative damage to lipids, proteins, and DNA, triggering inflammation, impairing synaptic function, and ultimately contributing to neuronal degeneration [368]. This state of oxidative and nitrosative stress is a hallmark of numerous neurodegenerative diseases, underscoring the central role of astrocytic redox regulation in maintaining CNS homeostasis and preventing neuropathology.
Astrocytes in the CNS may dynamically shift between neuroprotective and neurotoxic states. This functional polarity is finely modulated by membrane-bound proteins, receptors, channels, and transporters that govern essential processes such as ion homeostasis, neurotransmitter clearance, and neuroimmune signaling (Figure 4). Given this critical regulatory capacity, astrocytes are now increasingly considered direct therapeutic targets, with several strategies emerging to modulate their activity and counteract neurodegenerative cascades.
Astrocyte-targeted therapeutic strategies for neurodegenerative disease intervention. Dysfunction of key astrocytic molecular systems, such as those mediated by the EAAT2, AQP4, and purinergic receptors, contributes to neurodegenerative and neurological diseases, including AD, ALS, epilepsy, MS, and sleep fragmentation–associated pathology. Top row: In healthy conditions, the excitatory amino acid transporter 2 (EAAT2; known as GLT-1 in rodents) maintains glutamate homeostasis by clearing extracellular glutamate in exchange for Na+, K+, and H+ ions. In AD, EAAT2 downregulation leads to overactivation, Ca2+ overload, tau hyperphosphorylation, and dendritic spine collapse. In ALS, reduced astrocytic EAAT2 levels and altered microRNA regulation (e.g., increased miR-218 and decreased miR-124a) result in the accumulation of EAAT2-derived toxic species, accumulation of glutamate, and motor neuron degeneration. Middle row: Aquaporin-4 (AQP4) regulates astrocytic water transport and is essential for glymphatic clearance. In AD, decreased AQP4 activity impairs Aβ and tau clearance, partly via reduced LRP1-mediated transport. In epilepsy, AQP4 upregulation contributes to altered ionic and osmotic homeostasis that promotes hyperexcitability. Sleep fragmentation induces AQP4 mislocalization from astrocytic endfeet, reducing CSF clearance efficiency. Bottom row: Purinergic signaling, mediated by adenosine (A1, A2A) and P2-type (P2X7, P2Y1) receptors, coordinates astrocytic inflammatory and metabolic responses. In AD, altered A2A and A1 receptor-mediated signaling modulates EAAT2 expression, AQP4 polarization, and inflammatory states, while P2X7 receptor hyperactivation exacerbates neuroinflammation and astrocyte-mediated cell death. In epilepsy, upregulated P2Y1 receptor signaling disrupts Ca2+ dynamics and enhances glutamate release. In MS, elevated P2X7 receptor activation promotes cytokine release and oligodendrocyte injury, contributing to demyelination and neurodegeneration. Therapeutic integration across these domains holds potential for multi-modal intervention to prevent neurodegeneration.
Among these regulatory systems, three have emerged as particularly compelling therapeutic targets due to their pivotal involvement in both physiological and pathological contexts: The excitatory amino acid transporter 2 (EAAT2, also known as GLT-1 in rodents), aquaporin-4 (AQP4), and purinergic receptors, particularly the P2X7 and P2Y1 subtypes. Modulating EAAT2, AQP4, and purinergic signaling offers a unique opportunity for targeted intervention, selectively disrupting neurotoxic cascades while preserving the essential homeostatic functions of astrocytes. This therapeutic strategy holds significant translational promise for neurodegenerative diseases such as AD, ALS, and PD [128, 360].
EAAT2, which mediates over 90% of synaptic glutamate uptake [369], is the primary mechanism for clearing extracellular glutamate; EAAT2 prevents excitotoxicity by maintaining extracellular glutamate concentrations at ~25 nM [308, 370–373].
In AD, reactive astrocytes show significantly diminished EAAT2 expression due to a dual mechanism involving ROS and pro-inflammatory cytokines. These factors not only suppress EAAT2 transcription but also accelerate transporter degradation. Post-mortem analyses reveal a striking 60–70% loss of EAAT2 protein levels in the hippocampus and cortex of AD patients [283, 374]. Glutamate accumulation to micromolar levels [375] results in GluN2B-mediated NMDA receptor overactivation, Ca2+ overload, and subsequent tau hyperphosphorylation and dendritic spine collapse [376], linking excitotoxicity to amyloidogenesis [28].
The role of glutamate transport is particularly intriguing in ALS. Initially, severe loss of astrocytic EAAT2 was detected in spinal cords of several patients who had died with sporadic ALS [97]. Subsequently. the finding was corroborated in a transgenic model of ALS where rats or mice express the mutant Cu2+/Zn2+-dependent superoxide dismutase (SOD1G93A) originating from a form of human familial ALS; the levels of EAAT2/GLT1 as well as the efficiency of glutamate transport were significantly reduced in the transgenic animals [377–380]. Expression of EAAT2 is also altered (decreased) in the motor cortex of ALS patients, apparently in a post-transcriptional manner [381, 382]. Apart from the SOD1G93A transgenic animals, other models have been produced and shown to be deficient in EAAT2/GLT1 [383, 384]. Induction of Astrocyte Elevated Gene-1 (Aeg-1), which codifies for metadherin, reduces EAAT2 expression, while Aeg-1 silencing restores transporter levels and improves neuronal viability [385]. Additionally, EAAT2 is subject to caspase-3-mediated cleavage, generating toxic C-terminal fragments, the appearance of which precedes the manifestations of ALS neurodegeneration [386]. Moreover, pharmacological inhibition of this cleavage delays neurodegeneration [386, 387]. At the post-transcriptional level, motor neuron-derived miR-218 suppresses EAAT2 expression in astrocytes, establishing a detrimental feedback loop. Loss of miR-124a further exacerbates EAAT2 downregulation [388–390]. Strikingly, selective deletion of EAAT2 in spinal cord astrocytes is sufficient to induce motor neuron degeneration and impairs motor learning and coordination in mice [391]. On the basis of the above data [97, 377–379, 381–391], it is tempting to conjecture that EAAT2 is a key functional protein on which the health and survival of motor neurons depend. Accordingly, it could be singled out as a prime target for the development of effective ALS therapies. However, simple crossing of the SOD1G93A mice with mice overexpressing EAAT2 (to about a double of the normal level) resulted in an ALS model where the appearance of ALS signs and the onset of the motor neuron loss were only marginally delayed, with no effect on the final outcome [392]. Nevertheless, therapeutic strategies aimed at enhancing EAAT2 expression are still pursued.
Early attempts included the use of a beta-lactam antibiotic, ceftriaxone, which delayed the disease progression in animal models by upregulating EAAT2/GLT1 [393]. However, ceftriaxone failed to demonstrate efficacy in clinical trials [394]; this created a controversy that arguably slowed progress toward establishing EAAT2 as a therapeutic target in ALS [395, 396]. One should have perhaps heeded warnings from contemporary studies suggesting that a simple inhibition of glutamate transport in spinal cord in vivo, even if done over a period of time, does not necessarily produce the loss of motor neurons [397].
One unresolved question in ALS is why the neurons most susceptible to the altered/deficient EAAT2 (and neurodegeneration) come from two different populations which do not have much in common: The lower motor neurons in the spinal cord are cholinergic and project peripherally while the upper motor neurons are glutamatergic, excitatory, reside in the cerebral neocortex and target the brain stem and spinal cord (corticobulbar and corticospinal tracts). The answer may lie in their size (including the long axons) and activity, leading to high metabolic demands [398]. This can make them, as well as the adjacent astrocytes and their metabolism, particularly vulnerable to deficient EAAT2; indeed, inhibition of glutamate transport in the guinea pig cerebral cortex in vitro has been shown to result in characteristic changes in the tissue metabolome [399]; it is not only what happens outside (increased glutamate in the extracellular space) but also inside the cells (metabolism) when glutamate transport falters. Persistent higher activity (neuronal excitation) caused by chronically increased extracellular glutamate, combined with higher metabolic/energy needs, could then produce a critical state potentially resulting in neuronal death. This would initially affect the most “metabolically” exposed upper and lower motor neurons. Maintenance of normal brain metabolism, at least in patients with known vulnerabilities to ALS, could thus represent a possible preventative strategy (not that we would advocate using currently over-the-counter available nootropics or supplements; they have not been tested for the purpose, are likely to be ineffective, and could carry significant risks in ALS patients).
In the meantime, attempts at upregulation of EAAT2 have continued, with encouraging [400, 401] or, sometimes, mixed/negative outcomes [402]. More promising, perhaps, are multi-target approaches that simultaneously enhance EAAT2 function and reduce oxidative stress, potentially offering a more robust neuroprotective effect [403].
AQP4 is the primary water channel in astrocytes, densely localized at perivascular end feet and critical for maintaining brain water homeostasis. It facilitates glymphatic clearance, a convective exchange of CSF and interstitial fluid (ISF) that removes metabolic waste, but also Aβ and tau [359, 366]. AQP4’s polarized distribution depends on its anchoring to the dystrophin complex via α-syntrophin, this interaction is relevant for its physiological function [404].
In AD, impaired function of AQP4 disrupts Aβ clearance. Genetic deletion of Aqp4 in mouse models reduces Aβ efflux by approximately 55%, accelerating plaque deposition [405, 406]. This dysfunction is closely associated with the loss of astrocyte polarity, marked by a redistribution of AQP4 from perivascular end feet membranes into the neuropil. Such mislocalization impairs glymphatic clearance and alters water and potassium homeostasis, potentially exacerbating cognitive decline in AD [407]. Age-related reductions in arterial pulsatility further compromise AQP4’s perivascular localization, intensifying Aβ retention [408]. Although elevated perivascular AQP4 immunoreactivity is reported in AD brains, this likely reflects a maladaptive compensatory response rather than restored function [409]. Tau clearance is similarly impaired; glymphatic dysfunction strongly correlates with the burden of neurofibrillary tangles [410].
AQP4 dysregulation contributes to several other CNS disorders. In temporal lobe epilepsy, AQP4 is paradoxically upregulated despite the loss of α-syntrophin, suggesting maladaptive redistribution and impaired buffering [411]. In models of traumatic brain injury, AQP4 depolarization impedes tau clearance, and global Aqp4 knockout worsens neuropathology [412]. Sleep fragmentation in animal models of AD diminishes AQP4 expression, reducing CSF clearance to cervical lymph nodes [413, 414]. In neuromyelitis optica, pathogenic autoantibodies target AQP4 by binding to orthogonal arrays of particles, thereby disrupting astrocyte function [415, 416].
Given AQP4’s narrow pore and lack of canonical ligand-binding domains, direct pharmacological targeting remains challenging. Consequently, therapeutic strategies have shifted toward indirect, receptor-mediated modulation.
Additionally, metabotropic glutamate receptor 5 (mGluR5) physically associates with AQP4, modulating its permeability properties [417], while circadian regulators such as Per2 contribute to AQP4 polarization and glymphatic flow [418]. In neuromyelitis optica, Aquaporumab, a monoclonal antibody targeting AQP4-IgG, has demonstrated therapeutic potential [419].
Recent advances in imaging, including contrast-enhanced MRI with intrathecal gadolinium tracers, allow in vivo assessment of glymphatic clearance efficiency [420]. Collectively, these findings underscore the promise of receptor-based modulation of AQP4 function in treating neurological disorders characterized by impaired solute drainage.
Astrocytes play a central role in maintaining CNS homeostasis through purinergic signaling, wherein extracellular ATP and its degradation product adenosine modulate synaptic activity, inflammation, and neuronal excitability via purinergic P2X, P2Y, and adenosine receptors [324, 326]. Among these, the purinergic P2X7 receptor, activated by high extracellular ATP levels, promotes neuroinflammation and cell death in neurodegenerative conditions such as PD, AD, and MS [331, 421]. Conversely, adenosine A1 receptor activation exerts neuroprotective effects, while adenosine A2A receptor activation has been associated with synaptic dysfunction. The balance between these opposing outcomes is governed by ectonucleotidases, which control the conversion of ATP to adenosine [326]. Importantly, A2A receptors also modulate AQP4 polarization. Antagonism of A2A receptors can prevent stress-induced AQP4 mislocalization and preserve glymphatic clearance. Notably, deletion of the equilibrative nucleoside transporter 1 (ENT1) leads to reduced AQP4 expression, further linking purinergic signaling to astrocytic water channel regulation [422]. A2A receptor antagonism also has potential in reducing neuroinflammation and promoting solute clearance following CNS injury [423].
Astroglial P2Y1 and P2X7 receptors are key mediators in epilepsy and MS. In epilepsy, P2Y1 receptor overactivation perturbs intracellular calcium dynamics and increases glutamate release, thereby intensifying seizure activity. Pharmacological blockade using MRS2179, an antagonist of the P2Y1 receptor, restores synaptic balance and may represent a promising strategy for treating drug-resistant epilepsy [424, 425]. Inflammatory cytokines like TNF-α further exacerbate this pathological loop [426]. In MS models, reactive astrocytes upregulate P2X7 receptors, driving cytokine release and oligodendrocyte injury [427, 428]. P2X7 signaling also facilitates harmful interactions with infiltrating immune cells [429]. Inhibition of this receptor reduces gliosis, protects axons, and improves clinical outcomes in preclinical MS models [430].
Multiple studies have reported increased adenosine levels in the CSF of ALS patients [431], along with increased adenosine A2A receptors expression in their lymphocytes [432]. In the SOD1(G93A) ALS animal model, decreased expression of adenosine A1 receptors and increased A2A receptors levels have also been observed, even before the onset of clinical symptoms [433]. Notably, hippocampal synaptic dysfunction in SOD1(G93A) mice was rescued by pharmacological blockade of A2A receptors, suggesting a pathogenic role for adenosinergic signaling in early disease stages [434]. Although these studies do not specifically differentiate between neuronal and astrocytic contributions, it is possible that the modulation of astrocytic A1 and A2A receptors may have an impact on the onset and progression of ALS, given the well-established importance of astrocytes in the disease [435].
Altogether, astrocytic purinergic receptors represent attractive therapeutic targets due to their early involvement in disease pathogenesis and in regulating key processes such as inflammation, neuronal excitability, and interstitial solute clearance. Future research should prioritize the development of receptor-selective modulators with proven long-term efficacy, capitalizing on astrocyte-specific biology to optimize neuroprotective therapeutic outcomes.
Although EAAT2, AQP4, and purinergic receptors each fulfill distinct roles in astrocytic physiology, their functional interdependence presents a compelling opportunity for synergistic therapeutic interventions. Crosstalk among these systems is mediated by converging pathways involving calcium signaling, redox homeostasis, inflammatory cascades, and extracellular nucleotide metabolism.
For example, reduced EAAT2 activity leads to elevated extracellular glutamate concentrations [309, 310], which trigger excessive calcium influx into both neurons and astrocytes. This calcium overload promotes ATP release from astrocytes, activating P2X7 receptors and intensifying neuroinflammatory responses [421]. Simultaneously, reactive astrocytes often upregulate AQP4; when this channel becomes mislocalized, it may facilitate the spread of ROS, further exacerbating oxidative stress [436, 437]. Purinergic signaling, meanwhile, modulates both excitatory and inhibitory neurotransmission by regulating glutamate and GABA release [323]. The degradation of ATP into adenosine, controlled by ectonucleotidases, affects astrocytic tone via adenosine A1 and A2A receptors [438]. These receptors, in turn, influence EAAT2 expression, AQP4 polarization, and the astrocytic potential to produce cytokines.
Therapeutic strategies that concurrently target these interrelated systems may yield superior outcomes compared to monotherapies. Enhancing EAAT2-mediated glutamate uptake, while simultaneously inhibiting P2X7-driven inflammation or restoring AQP4 polarization through A2A receptor antagonism, could provide multifaceted protection. Such polypharmacological approaches hold potential for breaking the vicious cycles that perpetuate excitotoxicity, neuroinflammation, and impaired interstitial solute clearance.
Advancing this integrated therapeutic paradigm will require a deeper understanding of the temporal dynamics and feedback loops governing these astrocytic networks. Recent progress in transcriptomics, proteomics, and single-cell technologies is beginning to illuminate the heterogeneity and context-specific plasticity of astrocyte subtypes across disease states, offering an unprecedented opportunity to develop targeted interventions with cell-type and disease-stage selectivity.
While astrocytes represent one crucial glial population involved in neurodegeneration, microglia embody the other major non-neuronal actors. The lessons learned from astrocyte-targeted strategies provide a natural transition to examining microglia, whose dual role as immune defenders and potential drivers of pathology makes them equally compelling therapeutic targets (Figure 5).

From inflammation to neuroprotection. The evolving role of microglia in neurodegenerative diseases. Classical (left) and modern (right) views of microglial phenotypes. Cell illustrations were generated with the assistance of Sora, an AI-based image generation platform.
The traditional view of microglia as simple agents of inflammation has been superseded by an understanding of their complex, dual-purpose functions in brain homeostasis maintenance and immune defense.
Microglia, discovered by Pio del Rio-Hortega [439–441], are the resident immune cells of the CNS and play a critical role in neuroinflammation, host defense against pathogens, and injury response [442]. Microglia detect harmful stimuli through pattern PRRs, particularly TLRs, which recognize pathogen-associated molecular pattern (PAMP) and DAMP molecules released by injured neurons and glia [443]. TLR4, for example, activates intracellular signaling via the MyD88 adaptor protein, leading to NF-κB activation and subsequent transcription of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), chemokines (CCL2, CXCL10), and other mediators [444]. In AD, NF-κB activation is linked to β-amyloid plaque-induced microglial stimulation [445], while in PD, α-synuclein aggregates act as DAMPs that activate TLR2 [446].
Microglia also use alternative receptors such as TREM2 (Triggering Receptor Expressed on Myeloid cells 2), which regulates the transition to the DAM phenotype [126, 447]. TREM2 interacts with phospholipids, apolipoproteins, and LPS, initiating intracellular signaling via DAP12 and DAP10. These pathways activate SYK and PI3K, which in turn promote phagocytosis, proliferation, and microglial survival [448, 449]. Loss of TREM2 disrupts mTOR signaling, increases autophagy, and impairs microglial clustering around plaques, thereby worsening pathology [29, 450].
Another key player is the NLRP3 inflammasome, a multiprotein complex that activates caspase-1 to process and release IL-1β and IL-18. In both microglia and astrocytes, this mechanism amplifies inflammation when activated by protein aggregates such as Aβ and α-synuclein that build up in neurodegenerative diseases [451, 452].
Building on the classical perspective, modern evidence reveals that microglia are dynamic participants in disease progression. Their activation patterns across disorders illustrate both harmful and protective influences. For decades, these cells were viewed primarily through the lens of neuroinflammation and neuronal damage, particularly due to their well-documented release of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6 in conditions like AD and PD [124, 453–455]. This perspective stemmed from consistent observations of microglial activation around pathological hallmarks such as Aβ plaques in AD and α-synuclein aggregates in PD. However, the emerging paradigm recognizes microglia as exquisitely plastic cells capable of adopting diverse functional states, including neuroprotective phenotypes that may actually counteract disease progression under certain conditions [120, 123, 456].
From a therapeutic standpoint, the near-ubiquitous presence of activated microglia across neurodegenerative diseases makes them a compelling target. Histopathological studies have demonstrated microglia exhibiting morphological features of activation—including enlarged cell bodies and retracted processes—not only in numerous animal models [457–459], but also in post-mortem human tissue samples [460, 461]. The development of advanced neuroimaging techniques, particularly positron emission tomography (PET) using radioligands for the translocator protein (TSPO), has enabled the visualization and confirmation of microglial activation in living PD patients [462, 463]. This technological advancement represents a significant step forward, as it may eventually allow researchers to monitor treatment-induced changes in neuroinflammation regardless of whether the therapeutic target is neuronal, astrocytic, or microglial in nature.
The reassessment of microglia’s role in neurodegeneration has been driven substantially by carefully designed animal studies. One particularly instructive example comes from research using 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced models of PD, where chronic administration of monoacylglycerol lipase inhibitors was found to not only preserve dopaminergic neurons in the substantia nigra but also induce a distinctive microglial activation profile characterized by increased release of neurotrophic factors and decreased expression of classical inflammatory markers [459]. Similarly revealing findings have emerged from studies using the APPSw/Ind transgenic mouse model of AD. This transgenic mouse overexpresses the human amyloid precursor protein (APP) carrying two familial AD mutations: The Swedish mutation (KM670/671NL), which increases total Aβ production by enhancing β-secretase cleavage, and the Indiana mutation (V717F), which alters γ-secretase cleavage to favor production of the more aggregation-prone amyloid-beta 1–42 peptide (Aβ1–42) [464–467]. Intriguingly, microglia in these mice display an activated phenotype from birth, yet cognitive impairments only become apparent several months later. This temporal dissociation suggests that early microglial activation, acting in a neuroprotective fashion, may serve a compensatory or protective role, potentially delaying the onset of clinical symptoms [468].
The traditional classification of microglial phenotypes into binary M1 (pro-inflammatory) and M2 (anti-inflammatory/neuroprotective) categories has provided a useful conceptual framework, though it is increasingly recognized as an oversimplification of their functional diversity (Figure 5). While M1-activated microglia are indeed associated with cytotoxic responses through mechanisms such as nitric oxide release, M2 cells promote tissue repair via secretion of trophic factors like IGF-1, GDNF, and transforming growth factor-β [469–471]. The concept of “disease-associated microglia” (DAM) demonstrates how transcriptomic insights reshape our understanding of microglial states [126]. Recognizing this plasticity has opened the door to therapeutic reprogramming. If microglia can be shifted toward beneficial states, they may transform from contributors to disease into mediators of neuroprotection.
In human nervous tissue, the critical role of microglia in neurodegenerative disease pathogenesis has become increasingly evident. Activated microglia have been consistently identified in post-mortem examinations of patients with AD, PD, and ALS. AD cases in particular show striking microglial changes, with cells exhibiting marked hypertrophy and forming dense clusters around Aβ plaques [472, 473]. These observations strongly implicate microglia in disease progression, though whether their activation primarily drives pathology or represents a protective response remains debated.
A particularly fascinating aspect of reactive microglia in neurodegeneration is their dynamic receptor expression profile—while these receptors are nearly undetectable in resting microglia, they become markedly upregulated upon activation. This disease-associated receptor signature, combined with growing evidence that microglial polarization can be shifted toward neuroprotective (M2-like) phenotypes, has opened exciting new avenues for targeted drug discovery. One promising therapeutic strategy involves targeting proteins that become upregulated in the DAM, such as TREM2, chemokine receptor CX3CR1, or adenosine A2A receptors, with the goal of modulating their activation state. The fundamental challenge lies in developing pharmacological agents capable of reliably shifting microglial polarization from pro-inflammatory (M1-like) to anti-inflammatory, phagocytic (M2-like) phenotypes, thereby promoting neuroprotection while mitigating disease progression.
The adenosine A2A receptor serves as a particularly instructive example of a target with relevance to human neurodegenerative diseases. This receptor is expressed not only in neurons and astrocytes but also in microglia, and preclinical studies have demonstrated that A2A receptor antagonists exert robust neuroprotective effects across multiple animal models of neurodegeneration, including both AD and PD [474–478]. In PD models specifically, these compounds appear to modulate dopaminergic transmission in the striatum, leading to improvements in motor deficits [479]. Preclinical evidence further suggests that A2A receptor antagonists can synergize with existing therapies like levodopa while potentially mitigating troublesome side effects such as dyskinesias. The clinical potential of this approach is underscored by the approval of istradefylline in Japan and the United States as an adjunct therapy for PD patients experiencing motor fluctuations, representing the most advanced A2A receptor antagonists in clinical development [480–482].
Translating this idea into practice requires identifying accessible molecular handles. Receptors upregulated during microglial activation offer precisely such opportunities, making them attractive candidates for drug development. In AD models, A2A receptor blockade has been shown to enhance synaptic plasticity and cognitive function while protecting against both Aβ and tau-induced toxicity. The ability of these compounds to counteract oxidative stress and apoptosis further supports their potential utility across multiple neurodegenerative conditions, including HD and ALS. While early research primarily focused on neuronal A2A receptors, more recent studies have highlighted the importance of microglial A2A receptors. The receptor is now recognized as a promising target for modulating neuroinflammation and microglia-mediated neuronal survival, particularly following the discovery of A2A receptor upregulation in microglia from the prefrontal cortex of AD patients [483]. The potential therapeutic benefits of blocking A2A receptor signaling in microglia appear broadly applicable across neurodegenerative diseases. In fact, adenosine acting through microglial A2A receptors potentiates nitric oxide release [484], and in PD models, A2A receptor antagonists reduce neuroinflammation by suppressing microglial activation and pro-inflammatory cytokine production, contributing to neuronal survival. Unfortunately, the neuroprotective potential of these drugs remains unproven in human clinical trials, as most studies to date have not been designed to evaluate disease-modifying effects. Several factors converge to make the assessment of neuroprotection in humans particularly challenging, including the inability to obtain brain biopsies, the lack of reliable biomarkers, species-specific differences in microglial responses, and suboptimal patient stratification. While directly measuring neuroprotection in living patients is technically unfeasible, alternative strategies, such as monitoring pro-inflammatory markers or neuroprotective cytokines in the plasma of patients receiving istradefylline, could offer valuable mechanistic insights and serve as surrogate indicators of therapeutic activity [485].
Beyond the A2A receptor, other microglial receptors have emerged as promising therapeutic targets. Colony-stimulating factor 1 receptor (CSF1R) inhibition, which leads to microglial depletion, has shown potential to modify disease progression in multiple neurodegenerative models. TREM2, which plays critical roles in phagocytosis and lipid metabolism, represents another compelling target, particularly as certain TREM2 variants significantly increase AD risk [29, 486, 487]. The purinergic receptor family, including P2Y12 (involved in microglial chemotaxis) and P2X7 (which activates the inflammasome), is also being actively investigated as a potential pharmacological target [488, 489].
Despite these promising developments, translating microglial modulation into effective clinical therapies presents formidable challenges. The context-dependent nature of microglial responses, which vary by disease stage, brain region, and systemic environment, complicates the identification of universal therapeutic targets [41]. Moreover, interventions that broadly suppress microglial activity risk impairing their essential physiological functions, including synaptic pruning, neurogenesis regulation, and clearance of cellular debris [490–492].
Adding further complexity, regional heterogeneity in microglial phenotypes, such as differences between striatal and cortical microglia in PD, can influence treatment responses and must be carefully considered in the design of targeted therapies. Yet, this heterogeneity may also offer a therapeutic advantage, enabling interventions to selectively modulate the most disease-relevant microglial populations in specific brain regions.
New precision-targeted strategies are emerging to address these limitations. Advances in single-cell RNA sequencing and spatial transcriptomics are revealing microglial subtypes with distinct molecular signatures that could be selectively modulated. Innovative drug delivery systems such as lipid nanoparticles and BBB–penetrant vectors are enhancing the feasibility of glia-specific interventions [493, 494].
In conclusion, our evolving understanding of microglial biology, from simplistic inflammatory mediators to dynamic regulators of CNS homeostasis, represents a paradigm shift in neurodegenerative disease research. Therapeutic strategies aimed at reprogramming microglia toward neuroprotective phenotypes offer a novel and potentially transformative approach, particularly in early disease stages when neuronal loss may still be preventable. While significant challenges remain, the convergence of advanced molecular tools, improved animal models, and innovative clinical trial designs holds considerable promise for developing effective microglia-targeted therapies in the coming years.
Limitations in biomarkers, imaging, and trial design illustrate the gap between mechanistic insight and clinical application. One of the most pressing limitations is the lack of PET tracers capable of reliably distinguishing between neurotoxic (pro-inflammatory) and neuroprotective microglial phenotypes in vivo. Current imaging approaches predominantly detect general microglial activation, without resolving the functional diversity that underpins their dualistic roles. The development of tracers that target phenotype-specific markers, such as those associated with trophic factor release, phagocytic function, or anti-inflammatory signaling, would represent a major step forward in evaluating therapeutic efficacy and disease progression.
Equally crucial is the identification of robust, translatable biomarkers of neuroprotective microglia in the human brain. While preclinical models have yielded valuable insights into microglial heterogeneity, many markers defined in rodent systems [e.g., arginase 1 (Arg1) or rodent-specific chitinase-like protein (Ym1)] have limited expression or different regulation in human microglia. The lack of conserved markers hampers clinical assessment and underscores the need for human-centric validation pipelines that integrate single-cell transcriptomic and imaging datasets with body fluid biomarkers.
Moreover, the plasticity and context-dependency of microglial states pose an inherent challenge. A phenotype beneficial in one disease stage or anatomical region may prove deleterious in another. This complexity necessitates precision targeting strategies that consider spatial and temporal factors, rather than generalized immunomodulation. Clinical trial design must account for this heterogeneity, possibly incorporating patient stratification based on imaging, genetic risk factors (e.g., APOE or TREM2 variants), or peripheral cytokine profiles.
Finally, the dynamic crosstalk between microglia and other CNS cell types, including astrocytes, neurons, and endothelial cells, complicates the isolation of microglia-specific therapeutic effects. It remains difficult to discern whether observed benefits stem from direct modulation of microglia or secondary interactions within the neuroimmune milieu. Hence, combining microglial targeting with supportive interventions on neuronal or astrocytic function may ultimately be necessary to achieve lasting neuroprotection.
Together, these challenges highlight the need for refined tools, integrative methodologies, and cautious interpretation of data when advancing microglia-targeted therapies into clinical use.
Ultimately, the success of any neuron-, microglia-, or astrocyte-based intervention hinges on reliable outcome measurement tools. Biomarkers serve as the bridge between mechanistic discovery and clinical translation, enabling researchers to assess safety, efficacy, and disease modification. Building upon the preceding analysis of cellular targets such as astrocytes and microglia, this section shifts the perspective toward biomarkers of neuroprotection. While therapeutic strategies outline what to target, biomarkers determine how to detect, monitor, and qualify disease processes and treatment effects. Conditions such as systemic lupus erythematosus (SLE) are not included, despite their potential to cause pre-senile dementia through mechanisms like autoimmune antibodies targeting sodium channels on glutamatergic neurons [495]. It is excluded because it is primarily a systemic autoimmune disease with a known etiology and a typically episodic clinical course. In contrast, we review biomarkers of several synucleinopathies, despite their multisystem involvement, since their underlying causes remain largely unknown, and their clinical progression is characteristically insidious.
Dementias of primarily non-cerebral origin are not covered in detail, as their biomarker profiles are distinct, well-characterized, and seldom controversial. Among these are infectious etiologies such as neurosyphilis [496], chronic human immunodeficiency virus-induced encephalitis (HIVE) [497], and potentially emerging links to herpes simplex virus [498]. These infections, while historically significant contributors to global dementia burden, remain clinically relevant even in developed countries. Nonetheless, with the exception of certain viral encephalitis and prion disorders [499], such conditions are not fundamentally enigmatic and are less central to current neurodegeneration-focused drug development efforts compared to diseases like AD or frontotemporal lobar degeneration.
Likewise, vascular contributions to cognitive decline account for approximately 25% of all dementia cases [500], yet are supported by robust diagnostic frameworks, including neuroimaging (e.g., MRI), and standardized treatment protocols. Other secondary causes of cognitive impairment, including genetic disorders (e.g., Wilson’s disease), autoimmune conditions (e.g., SLE), chronic epileptic syndromes, repetitive head trauma (e.g., chronic traumatic encephalopathy) [501], and neoplastic processes or their treatments (e.g., chemotherapy-related cognitive impairment or “brain fog”) [502], are similarly well defined. In each of these, diagnostic biomarkers are well established and typically integrated into differential diagnosis when clinically suspected.
Biomarkers are measurable biological characteristics that reflect physiological states or pathological processes. In the context of common senile dementias, where underlying etiologies are often elusive, a wide array of potential biomarkers has emerged, leading to multiple frameworks for their classification. They may be organized by source (e.g., fluid-based from blood or CSF, or neuroimaging-based) or by intended use, such as for diagnosis, prognosis, prediction, or patient stratification. Each classification framework captures diverse biomarker types relevant to the spectrum of neurodegenerative disorders.
This section will adopt a hierarchical approach, beginning with molecular-level indicators, e.g., genomics and proteomics, and progressing toward higher-order systems, including cognitive and behavioral assessments and emerging modalities such as radioligand-based imaging of synaptic density [503–505]. The section will conclude by underscoring the considerable and often underrecognized potential of recent metabolomics advances to transform biomarker discovery.
Having established the general scope of biomarkers, we next consider their functional classification. This framework links directly to the therapeutic challenges described earlier: selecting the right patients, proper stratification, ensuring safety during intervention, and monitoring treatment efficacy.
Biomarkers can be classified according to the function they serve in clinical practice and therapeutic development. Among these, selection biomarkers represent a critical objective in the field of neurodegenerative disease drug development. This is especially true given that many emerging therapeutics are costly, may entail significant logistical burdens for patients and caregivers, and, importantly, carry substantial risks of adverse effects. Identifying which patients are most likely to benefit from a given treatment, while avoiding harm in others, is central to optimizing both clinical outcomes and resource allocation.
Janet Woodcock, during her tenure as Chief Medical Officer at the U.S. FDA, helped formalize the categorization of biomarkers for regulatory and clinical purposes. These categories include safety biomarkers, selection biomarkers, and response biomarkers [506]. However, the main challenges in diseases related to neurodegeneration are defining optimal biomarkers for diagnosis and identifying biomarkers for assessing neuroprotection in humans.
Within this framework, selection biomarkers are particularly important. Just as astrocyte and microglial modulation must be targeted with precision, clinical trials must recruit the right patients through robust diagnostic enrichment. In the realm of neurodegenerative disease, where definitive diagnostic tests are often lacking, diagnostic biomarkers play a pivotal role, not only in clinical practice but also as selection and stratification tools for clinical trials. In this context, their function often overlaps with that of selection biomarkers, as they help enrich trial populations with participants who are more likely to harbor the target pathology. This enrichment is crucial for maximizing the likelihood of detecting therapeutic effects and for avoiding the inclusion of patients unlikely to benefit from the intervention.
For instance, individuals with cognitive impairment due to unrelated conditions, such as neurosyphilis, HIV-associated encephalopathy, or neurocysticercosis, would not plausibly respond to anti-amyloid therapies. Diagnostic biomarkers thus serve to exclude such cases, preventing confounding and improving the interpretability of trial results. In this way, the traditional distinction between diagnostic and selection biomarkers becomes blurred. Although conceptually distinct, in practice, many biomarkers used in trials perform both roles simultaneously.
Given the substantial resource demands of drug development, large sample sizes, extended trial durations, and the financial and physiological burden of certain treatments (e.g., monoclonal antibodies targeting Aβ), it is ethically and scientifically imperative to identify patients for whom benefit is plausible and to avoid unnecessary exposure of those unlikely to respond.
Beyond selection, safety biomarkers address another parallel concern. Therapies acting on complex glial networks carry inherent risks, and amyloid-related imaging abnormalities (ARIAs) exemplify how biomarker-guided vigilance can prevent harm [507]. Abnormalities range from asymptomatic imaging findings to severe complications such as cerebral hemorrhages [508]. ARIA is believed to result from the clearance of Aβ plaques deposited in cerebral vasculature, which may compromise vessel integrity [509]. In the early 21st century, when no disease-modifying treatments for AD were available, diagnostic biomarkers with mechanistic specificity, such as CSF Aβ1–42/tau ratios or amyloid PET imaging, began to serve a dual role as selection biomarkers in clinical trials. They enabled enrollment of patients most likely to harbor the target pathology. However, the absence of curative therapies at that time complicated regulatory qualification of these biomarkers. Without effective treatments, it is still difficult to demonstrate that such biomarkers could reliably predict therapeutic efficacy or outcomes.
Response biomarkers complete the triad. While selection and safety establish who to treat and how to minimize risks, response biomarkers evaluate whether the intervention produces meaningful benefit, mirroring how preclinical glial modulation must be validated through functional readouts. Among the most prominent macroscopic biomarkers are neuropsychological test batteries, which aim to quantify behavioral and cognitive function. Historically, these tests have served as key diagnostic tools in neurodegenerative diseases. Alongside observable behavioral declines, such as a loss of self-care abilities, they continue to be the primary outcome measures recognized by regulatory agencies for assessing therapeutic efficacy. In cases of clinically diagnosed dementia, improvements in neuropsychological test scores are regarded with high face validity and regulatory acceptance, despite their inherent within-subject variability, even among cognitively healthy individuals.
Dementia is characterized by deficits in episodic memory followed by difficulties in problem-solving, decision-making and visuospatial deficits, such as difficulties recognizing faces, navigating familiar environments, or judging distances [510]. Mini-Mental State Examination (MMSE), developed by Folstein, Folstein, and McHugh [511], takes advantage of the impairment in memory and executive functions.
Neuropsychological evaluations are essential not only for diagnosis but also for tracking disease progression or monitoring treatment response over time. They help identify individual cognitive strengths and weaknesses, enabling clinicians to craft personalized treatment strategies aimed at mitigating cognitive deficits and promoting independence. However, there is natural variability in performance across individuals, and ceiling/floor effects that constrain the dynamic range of assessment [512].
Certain tasks, such as the Stroop Task and the Wisconsin Card Sorting Test, have demonstrated strong compatibility with neuroimaging studies, reinforcing their validity even when used outside imaging paradigms [513].
Tests evaluating visuospatial and constructional abilities assess visual perception and spatial reasoning, skills crucial for understanding the relationships between objects in space. Examples include copying complex figures or assembling block designs. The Rey-Osterrieth Complex Figure Test is widely used to assess these domains, as is the Bender Line Orientation Test, which has been employed in neuroimaging research to explore regional cerebral blood flow and metabolism (see [514, 515]). The California Verbal Learning Test (CVLT) is widely employed to assess verbal learning and memory [516], while the Boston Naming Test (BNT) evaluates confrontation naming ability [517]. These behavioral assessments can serve as diagnostic biomarkers in neurodegenerative conditions. However, due to their analog nature and limited dynamic range, their utility as response biomarkers, those that track progression or treatment response, is more constrained.
One of the most frequently cited behavioral biomarkers in PD is the University of Pennsylvania Smell Identification Test (UPSIT), developed by Richard Doty [518]. Hyposmia, or a diminished sense of smell, may precede the onset of motor symptoms by 10 to 20 years, making it a valuable early diagnostic indicator. However, the performance on this test tends to be dichotomous, and there is limited evidence that olfactory function continues to decline as the disease progresses, possibly because olfactory dysfunction appears at very early stages and rapidly approaches a floor effect, leaving little room to capture further decline.
In contrast, the Grooved Pegboard Test, a commonly used measure of fine motor coordination and dexterity, demonstrates a broader dynamic range and has been shown to decline progressively with disease worsening, making it a more reliable response biomarker in longitudinal studies [512].
The presentation of cognitive impairment in relatively young individuals necessitates a comprehensive diagnostic evaluation, as numerous potentially treatable causes must be considered. When there is a family history suggestive of early-onset AD (EOAD), genetic testing becomes particularly relevant. In such cases, screening for mutations in three well-characterized autosomal dominant genes is warranted: APP (coding for APP), PSEN1 (coding for presenilin 1), and PSEN2 (coding for presenilin 2) [519, 520]. Although mutations in these genes are often devastating for affected individuals and families, they account for only a small fraction of total dementia cases. There are other, perhaps more common, genetic variants associated with the risk of developing AD—including the late onset form of the disease (LOAD)—such as ApoE4 [521, 522] or those identified more recently (e.g., mutations in CD36 [523], see [524] for review). Some of these would likely be found at increased frequency in any population of patients with AD, thus potentially constituting “biomarkers” (perhaps even “marking” characteristic AD endophenotypes in some cases) [525]. They are not strictly specific to AD, though [526]. Genomic biomarkers are considered here as providing merely a foundation for identifying inherited risks that intersect with broader (and clinically more relevant) proteomic and fluid markers. Consequently, this review will not address them in detail, focusing instead on biomarkers with broader relevance to assess neuroprotection.
Proteomics, the study of protein expression, structure, and function, is central to biomarker discovery in dementia research. Though inherently microscopic in scale, this field is highly consequential, particularly because modern drug development largely revolves around identifying compounds that target specific proteins.
Even in the absence of clear etiologic understanding, proteomic analyses have yielded valuable insights into neurodegenerative disease processes. The depth and breadth of proteomic research far exceed the scope of this review [527]; we highlight here only proteins that are already well-integrated into diagnostic and therapeutic paradigms for common neurodegenerative diseases, while directing readers to other comprehensive sources [528, 529].
Interest in amyloid proteins has fluctuated since Dr. Alois Alzheimer’s original descriptions of extracellular plaques in the late 19th century [530]. He also noted neurofibrillary tangles, now known to consist of tau proteins, though it was Emil Kraepelin who championed the idea that amyloid plaques were the defining pathological hallmark of what came to be known as AD. This view dominated for nearly a century.
Efforts to detect these protein abnormalities in vivo have continued since the seminal work of George Glenner and Cai’ne Wong in the 1980s, who successfully isolated and sequenced the Aβ peptide from cerebral amyloid angiopathy [531]. Since then, amyloid and tau proteins have been sought in numerous biological media, including CSF, blood, urine, saliva, and skin, with varying degrees of success. More recently, noninvasive imaging techniques have enabled the detection of these proteins directly within the living brain, marking a major milestone in the field. Proteins not only embody disease pathology but also flow into CSF and plasma, linking proteomics directly with fluid-based measures.
Biomarkers derived from CSF and, more recently, blood plasma, are at the forefront of research in neurodegenerative diseases. Offering insights into brain pathology through minimally invasive methods, these markers are essential not only for early diagnosis and stratification but also for disease monitoring, crucial elements in both clinical care and trial design.
CSF remains a rich source of biomarkers, particularly in AD, where the core trio, Aβ1–42, phosphorylated tau (p-tau), and total tau (t-tau), form the basis of the ATN framework. This system categorizes biomarkers as indicators of amyloid pathology (A), tau pathology (T), and neurodegeneration (N).
Although technical hurdles once limited blood-based detection, innovations like single-molecule array (Simoa) and mass spectrometry have enabled plasma quantification of key biomarkers such as the Aβ1–42/Aβ1–40 ratio, p-tau18, p-tau217, and NfL, which presumptively reflect central pathology with increasing accuracy. Notably, biochemical parameters in plasma are emerging as tools for assessing disease progression rate in AD [532, 533].
Yet proteins alone capture only part of the picture. To monitor dynamic shifts in cellular metabolism, closely tied to astrocytic and microglial function, metabolomics offers a complementary and more sensitive approach. The systematic profiling of small molecules reflecting dynamic metabolic states is emerging as a powerful tool in the search for biomarkers in neurodegenerative diseases. Unlike genomic or proteomic approaches that capture upstream pathology, metabolomics reveals real-time functional alterations, which may precede structural brain changes, particularly relevant in conditions such as AD.
Recent technological advances allow quantification of over 600 metabolites from minimal sample volumes (10–20 µL), using high-throughput commercial platforms like Biocrates MxP® Quant 500 (www.biocrates.com). These enable reproducible detection across a wide biochemical range (amino acids, bile acids, fatty acids, biogenic amines, indole derivatives), facilitating both discovery and translational research.
A major strength of metabolomics lies in multivariate modeling. Rather than focusing on individual biomarkers, disease-specific patterns of multiple metabolites can enhance diagnostic precision. For instance, a panel of four metabolites can distinguish between aqueous humor samples from glaucoma patients, type 2 diabetes patients, and healthy controls with 95% accuracy using a linear model. However, a nonlinear model based on only three of these metabolites can achieve 100% accuracy [534].
Similar multivariate approaches applied to plasma in AD have yielded panels of four to five metabolites, such as 5-aminovaleric acid, carnosine, cholic acid, and hypoxanthine, that classify cases with over 75% accuracy and ~80% sensitivity/specificity. Importantly, these models often integrate both up- and down-regulated compounds, emphasizing the diagnostic value of relative metabolic balance over absolute concentration shifts. Even under stringent cross-validation, such as leave-one-out testing, these panels retain robust discriminatory power [535].
Further extending its clinical potential, metabolomics can differentiate not only patients from healthy individuals but also between diseases. Biocrates platforms have the potential of to classify AD, PD, MS, and systemic conditions by applying multivariate metabolic signatures obtained from easily accessible biofluids, including plasma and tears. Neurodegenerative diseases may present overlapping clinical or imaging profiles but differ in pathways such as amino acid metabolism, bile acid homeostasis, lipid remodeling, and oxidative stress.
Validation strategies, split-sample testing, permutation analyses, and receiver operating characteristic (ROC) curves, have confirmed the discriminatory capacity of small metabolite panels (4–6 compounds), with many models reaching area under the curve (AUC) values greater than 0.85. The Conformité Européenne - In Vitro Diagnostic (CE-IVD) mark, a regulatory framework issued by the European Union, ensures that commercial kits meet standards for traceability and reproducibility, thereby enabling clinical standardization across labs.
Altogether, metabolomics offers a promising complement to existing biomarker strategies, supporting early diagnosis, differential classification of dementia subtypes, and possibly therapeutic stratification, provided robust algorithms and well-characterized cohorts are employed.
While omics highlight molecular and metabolic alterations and provide multivariate models of disease risk, imaging offer a complementary perspective by directly visualizing structural and functional brain changes, thereby bridging biochemical signals with anatomical correlates.
In the context of dementia and cognitive decline, it is crucial to recognize that many medical conditions can mimic or contribute to dementia-like symptoms. Conventional toxicology screenings and clinical imaging techniques can help rule out common causes, such as heavy metal poisoning [536] or multi-infarct dementia [537]. While many diagnostic findings lack specificity, they can still be sensitive enough to indicate that the underlying issue is not an idiopathic neurodegenerative disorder.
In the evaluation of cognitive decline, imaging remains a cornerstone for ruling out secondary causes and guiding differential diagnosis. Techniques such as MRI can detect structural abnormalities, including ARIAs, which are relevant prior to initiating anti-amyloid therapies [538]. Incidence of ARIAs in clinical trials of monoclonal antibodies has reached up to 40% in some cohorts [539], underscoring the need for appropriate imaging protocols.
Complementary efforts by consortia such as the Radiological Society of North America’s Quantitative Imaging Biomarker Alliance (QIBA) [540] and the European Imaging Biomarker Alliance (EIBALL) [541] have produced harmonized frameworks for quantifying amyloid plaque burden through functional imaging.
Proteomic imaging research has primarily focused on amyloid, tau, and α-synuclein. Isoforms of tau (3-repeat and 4-repeat) help distinguish between disorders such as progressive supranuclear palsy, corticobasal degeneration, and Pick’s disease, while α-synuclein pathology supports the diagnosis of PD and dementia with Lewy bodies (DLB) [542]. Highly sensitive detection technologies, such as Single Molecule Array (SIMOA) enable quantification of low-abundance proteins in fluids like CSF and plasma. Using antibody-coated microbeads and fluorescence-based detection, SIMOA can detect femtomolar concentrations of Aβ and tau, making it suitable for tracking disease progression or therapeutic response [543–545].
The CSF Aβ1–42/Aβ1–40 ratio remains one of the most robust biomarkers for AD. Because Aβ1–42 levels decline as the peptide aggregates into plaques, while Aβ1–40 remains stable, the ratio corrects for pre-analytical variability and enhances diagnostic accuracy [546, 547]. This metric aligns closely with amyloid PET imaging, with some studies reporting sensitivities and specificities up to 96% and 91%, respectively [548].
Blood-based assays are gaining traction as scalable and less invasive alternatives. Plasma Aβ1–42/Aβ1–40 ratios show good correlation with amyloid PET status, achieving AUC values around 0.82–0.84 [544]. A recent regulatory milestone was the FDA’s approval of the Lumipulse G test in May 2025, which measures the plasma pTau217/Aβ1–42 ratio to aid in AD diagnosis [31]. In clinical validation, this test achieved 91.7% concordance with PET-confirmed amyloid positivity and 97.3% concordance with PET negativity, with only ~20% of results deemed indeterminate. This provides a viable alternative when imaging is inaccessible or contraindicated.
Nonetheless, as with all biomarkers, real-world sensitivity and specificity remain context-dependent, and blood-based assays like Lumipulse G are best used as complements, not replacements, to comprehensive clinical assessment.
Beyond brain imaging, attention has increasingly shifted toward peripheral tissues, where accessible biopsies such as skin samples provide a minimally invasive avenue to detect hallmark proteinopathies and extend biomarker discovery beyond the CNS.
Skin biopsies are an emerging and promising tool for diagnosing dementias, particularly those classified as synucleinopathies [549]. The rationale is based on the peripheral manifestation of neurodegenerative pathology, specifically, the accumulation of misfolded proteins such as phosphorylated α-synuclein (P-SYN) in small nerve fibers of the skin. Compared to traditional brain biopsies or lumbar punctures for CSF analysis, skin biopsies offer a minimally invasive, accessible, and well-tolerated diagnostic alternative.
The synucleinopathies, including PD, DLB, multiple system atrophy (MSA), and pure autonomic failure (PAF), are unified by abnormal deposits of P-SYN within the autonomic and somatosensory fibers of the skin. These aggregates can be detected via immunohistochemistry or immunofluorescence. In large multicenter studies, such as the Synuclein-One Study, skin P-SYN detection has shown diagnostic sensitivities up to 93% for PD and nearly 100% for PAF. Specificity is also high, as healthy individuals do not appear to exhibit detectable P-SYN aggregates in cutaneous nerves [550, 551], though some clinical features alone may distinguish these patients from controls.
There is also preliminary evidence suggesting that hallmark proteins of AD, such as p-tau and Aβ, may be detectable in skin tissue. Reports have documented scattered p-tau aggregates in epidermal and dermal compartments, including nerve terminals and fibroblasts, of AD patients, as well as Aβ immunoreactivity within the dermis [552, 553]. While not yet definitive, these findings open the door to potential skin-based diagnostics for tauopathies and AD.
Commercial development has followed suit. One example is the DISCERN™ test (SYNAPS Dx, Rockville, MD), a fibroblast-based skin assay designed to identify AD pathology. DISCERN™ involves culturing fibroblasts from a skin biopsy and assessing multiple molecular signatures. Published reports suggest that it can achieve sensitivity and specificity values close to 100% for detecting AD, even in the presence of comorbid neurodegenerative conditions [551, 553, 554].
Less studied is the observation that fibroblasts from AD patients show altered expression of EAAT1 amino acid transporter, with the degree of alteration correlating with dementia severity [555]. This finding warrants further investigation to determine whether peripheral expression of this transporter could serve as a useful biomarker for neurodegenerative diseases.
Together, these findings suggest that peripheral biomarkers have the potential to become a valuable diagnostic aid across multiple forms of dementia, offering a rare blend of accessibility, biomarker specificity, and emerging commercial viability.
These peripheral approaches, when combined with molecular, imaging, and behavioral measures, underscore the need to evaluate biomarkers not in isolation but as part of an integrative framework, setting the stage for broader implications in both research and clinical practice.
Neuropsychological assessments serve as foundational tools in the diagnosis and monitoring of neurodegenerative diseases. While tests such as CVLT, BNT, and UPSIT offer valuable diagnostic insight, particularly in early or prodromal stages, their limited dynamic range and susceptibility to ceiling or floor effects constrain their utility as response biomarkers. In contrast, tasks with broader performance spectra, such as the Grooved Pegboard Test, provide greater sensitivity to disease progression and therapeutic response. As the field moves toward precision medicine, refining and integrating behavioral biomarkers with imaging and molecular data will be essential for capturing the nuanced trajectories of cognitive and motor decline, ultimately improving patient stratification, treatment evaluation, and clinical outcomes.
The convergence of diverse biomarker modalities illustrates both the opportunities and the limitations of current strategies. Yet, as the field seeks to move from detection toward intervention, the central challenge becomes how these biomarkers can reliably inform the design, assessment, and validation of neuroprotective therapies. This transition introduces the conceptual and methodological hurdles addressed in the next section.
Despite decades of research and a growing repertoire of molecular targets and candidate therapeutics, the field of neuroprotection remains hindered by persistent conceptual ambiguity. The term “neuroprotection” is widely employed across preclinical and clinical contexts, but it lacks a clear, universally accepted operational definition. While some interventions, such as the use of caspase inhibitors that directly prevent apoptotic cell death [556, 557], are readily classified as neuroprotective, others, including anti-inflammatory agents or metabolic modulators, have more systemic or indirect effects, rendering their categorization less straightforward.
This definitional vagueness poses significant challenges to the development of objective and reproducible criteria for evaluating efficacy. It also contributes to heterogeneity in trial designs, encompassing a broad array of endpoints, mechanisms of action, and timescales. This variability complicates the comparison of results across studies and impairs the reliability of meta-analyses [558–564]. In the absence of a unified conceptual framework, distinctions between neuroprotection, symptomatic treatment, and disease modification often become obscured, risking the conflation of mechanistic plausibility with demonstrable clinical benefit.
In translational neuroscience, reliably demonstrating neuroprotection in humans remains particularly difficult, not only due to technical limitations but also because of this enduring conceptual imprecision. Neuroprotection is generally defined as the use of strategies to prevent, delay, or reverse neuronal injury or dysfunction caused by insults such as ischemia, trauma, or neurodegenerative processes. These strategies may involve diverse mechanisms in glial cells and neurons, including inhibition of apoptosis, reduction of excitotoxicity, suppression of neuroinflammation, enhancement of mitochondrial function, or promotion of neuronal regeneration [565, 566].
Neuroprotection in practical terms can be only assessed in preclinical studies. In fact, it remains impossible in AD to objectively determine whether an intervention provides neuroprotection in clinical settings. No validated biomarkers currently exist to confirm neuroprotective effects in patients, and thus, therapeutic efficacy is often inferred from cognitive test scores rather than biological indicators of disease modification (see [567]).
Despite the growing interest in imaging and proteomic tools, methods such as PET or CSF tau quantification do not reliably correlate with neuroprotection. Accordingly, preclinical models, mostly rodent-based, remain the main platform for assessing neuroprotective mechanisms.
Beyond conceptual ambiguities, translational neuroscience is confronted by a striking disconnect between preclinical success and clinical efficacy. A landmark analysis revealed that while approximately 86% of animal studies with positive findings proceed to human trials, fewer than 5% of those interventions achieve regulatory approval [568]. This dramatic attrition underscores not only methodological deficits, such as inadequate randomization, absence of blinding, and insufficient statistical power, but also systemic issues in model validity and disease representation.
Most preclinical studies are conducted in young, healthy, genetically homogeneous male rodents, far removed from the heterogeneity of aging human populations with comorbidities such as hypertension, diabetes, or cerebrovascular disease. These biological and environmental discrepancies limit external validity and severely constrain the generalizability of preclinical findings to real-world clinical settings. Moreover, simplified experimental paradigms often fail to capture the complex, multifactorial nature of human neurodegeneration, including chronic inflammation, metabolic dysfunction, and cumulative injury [569–571].
Together, these limitations reveal a fundamental weakness in the translational pipeline: Preclinical models, often biologically reductionist and poorly representative of human pathology, remain inadequate to support the development of interventions with reproducible and clinically relevant neuroprotective effects. It is increasingly doubtful that the expanding repertoire of animal models, regardless of their genetic or pathological sophistication, will lead to meaningful breakthroughs in this specific area. On the contrary, their proliferation may represent a conceptual burden, further distancing experimental outcomes from clinical applicability.
Beyond the bench-to-bedside gap, another critical challenge arises from the limited biological diversity represented in preclinical models. The vast majority of animal studies are conducted without considering biological variables such as age, sex, and comorbidities, including hypertension, diabetes, and metabolic syndrome. These factors are not only prevalent but often pivotal in determining disease progression and treatment response in human neurological populations. Incorporating aged animals or models with vascular and metabolic risk factors would more accurately recapitulate the clinical landscape and substantially improve the applicability of experimental findings [569, 572–574].
In stroke research, for example, models that incorporate hypertensive or diabetic phenotypes have demonstrated significantly altered responses to neuroprotective interventions compared to normotensive controls, underscoring the critical importance of patient-specific biological contexts [575]. Similarly, sex-specific differences in neuroinflammatory and neuroregenerative responses are increasingly recognized, reinforcing the need for sex-balanced study designs [576]. Contrasting sharply with the homogeneity of preclinical models, human neuropathology is inherently heterogeneous. Rodent models of stroke typically involve highly standardized focal ischemia, whereas clinical presentations vary widely in lesion size, location, and accompanying comorbidities. This biological and clinical variability reduces statistical power, introduces confounding variables, and may obscure the detection of true neuroprotective effects in trials [577, 578].
To address some of these translational gaps, biomarker identification in body fluids has emerged as a promising yet still incomplete solution. Among the most studied is NfL, a structural axonal protein that increases in CSF and plasma following neuronal injury. NfL elevations have been consistently observed in neurodegenerative conditions, stroke, traumatic brain injury, and neuroinflammatory disorders [579–581]. However, its lack of specificity remains a major limitation: NfL levels can rise following surgical procedures or systemic stress and thus cannot be used in isolation to infer neuroprotective efficacy.
Moreover, while NfL correlates with disease activity and long-term prognosis, it has not yet been validated as a surrogate endpoint in clinical trials, limiting its utility in regulatory routes. The qualification of biomarkers for drug development is not only a scientific hurdle but also a regulatory one. In the United States, the FDA Biomarker Qualification Program was designed to formalize the use of biomarkers in therapeutic development, but progress has been slow. Only a handful of biomarkers have been formally qualified due to rigorous requirements for analytical validity, clinical relevance, and demonstrated utility [582].
A major barrier lies in distinguishing between prognostic, predictive, and surrogate endpoint biomarkers. While biomarkers such as NfL can reflect ongoing neurodegeneration, they do not inherently predict treatment response. To be accepted as a surrogate endpoint, a biomarker must demonstrate that changes in its levels reliably and reproducibly predict clinical benefit across multiple interventions and study populations, an evidentiary standard that few neurodegeneration-related markers currently meet [583].
The limitations of clinical endpoints further compound the problem. Commonly used scales, such as the NIH Stroke Scale or Glasgow Coma Scale, are calibrated for acute neurological deficits but are poorly suited to detecting the gradual, molecular-level improvements that characterize many neuroprotective strategies. Functional and cognitive assessments provide valuable patient-centered data, but are heavily influenced by rehabilitation, neuroplasticity, and compensatory mechanisms, which may mask or confound the specific effects of a given intervention [584, 585].
In summary, advancing the field of human neuroprotection demands a multidimensional recalibration, spanning conceptual clarity, more representative preclinical models, validated biomarkers, and refined clinical endpoints. Regulatory frameworks must evolve in tandem, embracing flexible, data-driven approaches that account for the complexity and subtlety of neuroprotective interventions. Without these structural reforms, the field risks mistaking molecular promise for therapeutic progress and stalling the development of interventions that could meaningfully alter the trajectory of neurodegenerative disease. Given these biological and methodological constraints, the field is increasingly turning toward multimodal strategies to capture the complexity of neuroprotection.
Relying on a single biomarker, whether derived from neuroimaging, biofluids, electrophysiology, or clinical scales, has proven insufficient to capture the complexity of neuronal injury and recovery [586, 587]. A singular parameter rarely reflects the nuanced interplay between inflammation, metabolism, synaptic function, structural damage, and compensatory mechanisms. Instead, comprehensive strategies that integrate diverse streams of biological, functional, and behavioral data are now recognized as essential for advancing mechanistic understanding and therapeutic monitoring [588, 589].
This integrative shift (Figure 6) is being propelled by rapid advances in artificial intelligence (AI) and machine learning (ML), which have enabled the simultaneous analysis of large, heterogeneous datasets. By combining neuroimaging metrics (e.g., cortical thickness, functional connectivity), body fluid biomarkers (e.g., NfL, Aβ1–42/tau ratios), genetic variants (e.g., apolipoprotein E, CD36, TREM2), and cognitive/clinical profiles, AI-based models can construct multidimensional representations of disease states. These models offer powerful tools for identifying latent disease subtypes, modeling progression trajectories, and predicting therapeutic responses, capabilities that are crucial for personalizing neuroprotective strategies [523, 590–592].
Bridging the translational gap in neuroprotection: from biological diversity to multimodal innovation. Despite promising preclinical findings, clinical translation of neuroprotective strategies has been limited by persistent conceptual and methodological challenges. Conventional models often overlook biological diversity, reducing translational validity. Emerging approaches advocate for a paradigm shift, integrating multimodal biomarkers, such as neurofilament light chain (NfL), glial fibrillary acidic protein (GFAP), tau protein (Tau), with in vivo imaging (fMRI, and PET), AI-driven patient stratification and adaptive trial designs. Advancing neuroprotection requires greater conceptual clarity, inclusivity, and technological innovation to overcome current barriers. Created in BioRender. Ulrich, A. (2025) https://BioRender.com/r1pojkk.
GFAP has recently gained prominence as a sensitive biomarker of astroglial activation in AD. Elevated GFAP concentrations in plasma and CSF have been consistently associated with early amyloid accumulation, cognitive decline, and overall disease severity, even during preclinical stages. Introducing GFAP, a glial component, into biomarker panels historically focused on neuronal injury, enhances the biological resolution of neurodegenerative profiling. Recent studies demonstrate that integrating GFAP with NfL and Aβ/tau measures significantly improves diagnostic precision and phenotypic stratification, particularly when applied within ML–based frameworks for early detection and longitudinal monitoring of disease progression [593, 594].
Beyond protein-based biomarkers, metabolomics appears as a powerful tool to broaden our understanding of neurodegenerative disorders. Unlike reductionist approaches centered on single metabolites, recent studies emphasize the need to assess panels of interrelated metabolites, which may better capture the complex biochemical alterations underlying disease progression [535]. The translational imperative for multimodal strategies becomes particularly evident in complex clinical contexts such as traumatic brain injury, where the cascade of secondary insults, excitotoxicity, inflammation, oxidative stress, and delayed neurodegeneration demands comprehensive and patient-specific interventions. As reviewed by Buccilli et al. [595], neither pharmacological agents nor regenerative therapies alone can address the multifactorial nature of traumatic brain injury. Instead, therapeutic efficacy increasingly depends on integrated protocols, monitored through biomarkers that reflect dynamic changes across molecular, cellular, and network levels.
From a methodological standpoint, this shift is supported by advanced statistical and computational techniques capable of handling heterogeneous data. Bhaumik and colleagues [596] demonstrated that Bayesian modeling integrating both structural and functional MRI outperformed univariate approaches in detecting neuroprotective effects, especially in small or heterogeneous samples. This underscores the value of combining data streams, not only for diagnostics but also for increasing the sensitivity of intervention monitoring.
Similarly, multidomain preventive trials such as the FINGER study (Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability) have adopted a multimodal approach that combines dietary guidance, physical activity, cognitive training, and vascular risk management. These interventions are assessed through a range of outcome measures, including neuroimaging, blood biomarkers, and comprehensive cognitive batteries. The FINGER study demonstrated that such integrated strategies can significantly slow cognitive decline in older adults at risk [597]. Building on these findings, the World-Wide FINGERS initiative has been launched as an international extension of the original Finnish network, aiming to replicate and adapt the protocol across diverse countries while maintaining methodological harmonization [598].
Another emerging pillar of neuroprotection involves NIBS, particularly tDCS and repetitive transcranial magnetic stimulation (rTMS). These techniques offer the possibility of modulating network excitability, protein aggregation, and glial function without a pharmacological burden. A growing body of evidence suggests that NIBS may delay disease progression in Alzheimer’s and Parkinson’s diseases by targeting synaptic plasticity and neuroinflammation. Guidetti and colleagues [599] highlight preclinical findings consistent with disease modification, though clinical trials remain in early phases.
Neuroimaging, particularly combining structural MRI, functional MRI, and magnetic resonance spectroscopy, has enhanced sensitivity to early alterations in network connectivity, microstructural integrity, and metabolic function, even in AD animal models [600]. In clinical populations, approaches combining cortical thickness (structural MRI), amyloid-PET, tau-PET, and white matter lesion burden may predict domain-specific cognitive decline with precision [601, 602]. In PD, pairing novel markers, such as seed amplification assays and extracellular vesicle cargo profiling, with specialized imaging modalities (e.g., neuromelanin-sensitive MRI, susceptibility-weighted imaging for iron deposition) has yielded superior diagnostic accuracy and staging resolution [603, 604]. These fusion techniques not only improve detection but also enable mechanistic stratification of participants for targeted therapeutic interventions.
Taken together, these converging lines of evidence reinforce a central conclusion: No single modality can sufficiently capture the complexity of neurodegeneration or reliably measure neuroprotection in humans. Only by integrating molecular, cellular, systems-level, and behavioral data, analyzed with AI and interpreted within rigorous ethical frameworks, can the field realistically design robust clinical trials and develop interventions that are both mechanistically grounded and clinically effective. The imperative is clear: Neuroprotection in the 21st century must be multimodal, multidisciplinary, and multidimensional.
Building on the multimodal approaches, recent advances in AI and digital health are reshaping how disease markers are collected, analyzed, and interpreted. A multidimensional strategy would enhance both diagnostic and prognostic capabilities, offering a more integrative view of neurodegenerative pathophysiology. The real challenge ahead is to deploy multi-omic profiling in well-characterized and sufficiently powered cohorts, enabling the identification of robust biomarker combinations that guide diagnosis, prognosis, and therapeutic response in chronic neurodegenerative diseases.
In parallel, digital biomarkers, derived from continuous, real-world data captured via smartphones, wearable devices, and ambient sensors, have opened new avenues for unobtrusive, high-frequency monitoring of neurological functions. Features such as gait dynamics, speech patterns, sleep architecture, motor variability, and keystroke latency can be passively and longitudinally tracked to identify subtle deviations from individual baselines. In PD, for instance, gait parameters derived from smartphone accelerometry have shown promising diagnostic accuracy, while speech-based algorithms are under investigation for the early AD detection [605–607]. However, an important caveat remains: Many of these digital signals are more reflective of symptomatic fluctuations, often influenced by medication or contextual factors, rather than direct markers of disease progression or underlying pathology. This raises questions about their standalone utility for tracking neurodegenerative trajectories over time.
ML algorithms, such as convolutional neural networks (CNNs), random forests, and ensemble methods, are increasingly applied to neuroimaging data from large consortia like the Alzheimer’s Disease Neuroimaging Initiative (ADNI). ADNI has standardized data acquisition across more than 60 sites, covering structural MRI, PET imaging, CSF biomarkers, genomic information, and neuropsychological assessments. This initiative has enabled the longitudinal tracking of biomarker trajectories and the development of composite endpoints that are gaining acceptance among regulatory agencies for clinical trial evaluation [608]. In parallel, high-throughput and automated feature extraction techniques from structural and functional MRI are revealing complex biomarker signatures predictive of disease onset, progression, and treatment response. Notably, Vinukonda and colleagues [609] demonstrated that supervised classifiers trained on MRI data significantly outperformed traditional statistical methods in identifying early-stage AD.
Importantly, AI and ML offer powerful tools for patient stratification and for accounting for confounding factors such as overdiagnosis and polypharmacy. Within the framework of clinical trial design, predictive modeling based on imaging, body fluid biomarkers, genetics, and digital signals can enhance participant selection, identify “fast progressors” or treatment-responsive subgroups, and enable real-time adaptive modifications to trial parameters. Moreover, digital biomarkers may serve as flexible endpoints, capable of capturing meaningful physiological changes beyond the limits of traditional, static scoring systems. Collectively, these innovations hold the potential to reduce required sample sizes, shorten trial durations, and improve statistical power, thus addressing some of the most persistent limitations in the development and validation of neuroprotective therapies [610].
A critical milestone in the field would be to disentangle diagnostic accuracy from the ability to monitor disease progression, specifically, the rate of neuronal loss over time. This distinction is essential for the evaluation of neuroprotective interventions. The challenges in assessing the efficacy of drugs are compounded by the need for careful patient selection and clinical stratification. Beyond the issue of overdiagnosis in prodromal or ambiguous cases, polypharmacy remains prevalent even among correctly diagnosed patients. This introduces additional variability that can obscure the therapeutic signal of a novel compound.
The growing integration of AI and digital systems into clinical research and care also introduces significant challenges and ethical considerations. A key concern lies in the “black box” nature of many ML algorithms; despite their high predictive accuracy, often lack transparency and interpretability. This opacity hampers clinical trust, complicates regulatory approval, and limits actionable insight. Furthermore, AI models trained on non-representative or biased datasets risk perpetuating existing health disparities and may exhibit poor generalizability across diverse populations. Issues of data privacy and patient autonomy may be amplified in the context of continuous, passive monitoring through wearables and mobile devices, necessitating robust frameworks for consent, data governance, and ethical oversight [611].
All the concerns are mirrored in the emerging discourse on neurorights that highlights the lack of consensus around what constitutes cognitive liberty, mental privacy, and neurodata ownership. As quoted by the European Parliament, “neurorights could be defined as ethical, legal, social or natural principles of freedom or entitlement related to a person’s cerebral and mental domain” [612]. Ienca and Andorno [613] argue that the ethical foundations of brain-related protections remain underdeveloped, leaving a normative vacuum as neurotechnologies outpace legal frameworks. As emphasized by Hanslmayr [614], the rapid advances in brain-computer interfaces, non-invasive brain stimulation, and neuroimaging demand parallel innovation in governance, transparency, and public engagement.
Beyond computational tools, another frontier in neuroprotection research emerges from biology itself: the intricate interplay between the gut microbiome, immunity, and brain function. The gut-brain axis has emerged as a compelling frontier in neuroprotection, characterized by complex bidirectional communication between the CNS, the enteric nervous system, and the intestinal microbiota. Far from being confined to gastrointestinal physiology, the gut microbiome plays an active and dynamic role in modulating brain health through immune signaling, metabolic regulation, neurotransmitter synthesis, and neuroendocrine communication. Increasing evidence suggests that this axis exerts a profound influence on key neurobiological processes, including neuroinflammation, BBB permeability, neurotransmission, and synaptic plasticity, all of which are directly relevant to neurodegeneration and neuroprotection [615, 616].
Dysbiosis, or imbalance in the composition and function of the gut microbiota, has been increasingly linked to several neurodegenerative conditions. In PD, for example, patients exhibit reduced microbial diversity and an overrepresentation of pro-inflammatory taxa, patterns that correlate with systemic inflammation and greater motor symptom severity [617]. Similar microbial signatures have been observed in AD and MS, suggesting a shared microbiome-inflammation axis that contributes to disease pathophysiology [618, 619]. Additionally, there is evidence for a correlation between dysbiosis and ALS (see [620] for review); if the relationship is confirmed as causal, it could be exploited in future therapies [621].
Microbiota-targeted interventions are a novel class of neuroprotective strategies. Therapeutic approaches include the administration of probiotics and prebiotics, dietary modulation, and even fecal microbiota transplantation, aiming to restore microbial homeostasis and dampen neuroinflammatory cascades [622, 623]. Preclinical studies have shown promising results: Specific strains such as Bifidobacterium breve and Lactobacillus plantarum reduce microglial activation, increase anti-inflammatory cytokines like IL-10, promote neurogenesis, and enhance cognitive performance in rodent models of AD [624, 625].
Beyond bacterial composition, the metabolomic output of the microbiota is also central to its neuroactive potential [626]. Short-chain fatty acids are microbial fermentation products exert neuroprotective effects via epigenetic regulation, including inhibition of histone deacetylases and promotion of neurotrophic signaling [627, 628]. Short-chain fatty acids can also influence BBB integrity and immune cell polarization, adding further mechanistic depth to microbiota-mediated brain modulation.
Although clinical translation remains in its early stages, the gut-brain axis represents a promising, non-invasive, and systems-level target for neuroprotection. Its integration alongside neuroimaging, biomarkers, and digital health tools may allow for novel combinatorial approaches capable of modifying disease trajectories. Moving forward, robust longitudinal clinical studies, mechanistic elucidation, and personalized microbiome profiling will be critical to transforming this emerging science into actionable therapies for neurodegenerative disease.
Just as the microbiome underscores biological complexity, the global landscape of neuroprotection trials reminds us that diversity in participants is equally essential.
Despite decades of awareness, neuroprotection trials remain disproportionately populated by white, male, and high-income participants, limiting the generalizability and ethical validity of their findings. Women, racial and ethnic minorities, and individuals from low- and middle-income countries continue to be underrepresented, even though these populations often exhibit distinct biomarker trajectories, disease risks, and treatment responses. Individuals of African or Hispanic descent may have different patterns of AD biomarkers [629, 630], yet are frequently excluded from most large-scale trials. This exclusion stems from a combination of structural barriers, including cultural mistrust, logistical limitations, and lack of community engagement. Effective solutions include culturally sensitive recruitment strategies, decentralized or hybrid trial models, and partnerships with local health leaders. Regulatory agencies have increasingly mandated the inclusion of diverse populations in funded research, reinforcing the urgency of addressing intersectionality and social determinants of health in clinical trial design [631, 632].
The expansion of neurodegenerative disease-associated clinical trial infrastructure in low/middle-income countries is equally essential, as the burden of age-related diseases is rising sharply. Investments in research capacity-building, shared protocols, regional biobanking, and data analysis platforms are necessary to ensure that scientific innovation in neuroprotection has truly global reach and impact [633]. To match this call for inclusivity, trial design must also evolve, adopting adaptive and master protocols that reflect the complexity of neurodegeneration in the real world.
The increasing complexity of neurological and neurodegenerative disorders, coupled with the paradigm shift toward precision medicine, has exposed the limitations of traditional clinical trial designs. Conventional randomized controlled trials, long regarded as the gold standard, often fail to accommodate the biological heterogeneity, comorbidities, and evolving pathophysiological trajectories that characterize chronic brain diseases. To respond to these challenges, new frameworks are emerging that emphasize adaptability, inclusivity, and biomarker-driven personalization, aiming to increase both the efficiency and equity of neuroprotective research [634].
To address clinical and biological heterogeneity more directly, adaptive trial designs have gained prominence. These designs allow for pre-specified modifications, such as dose adjustments, reallocation of participants, or sample size recalibration, based on interim data, without compromising statistical rigor. Adaptive trials offer greater flexibility, reduce patient exposure to ineffective treatments, and align more closely with real-world clinical variability [635].
Building on this adaptability, Hammouri and colleagues [636] introduced the Neyman Weighted Multiple Testing Procedure, which incorporates stage-specific weighting, dynamic sample redistribution, and sequential testing to enhance decision-making while rigorously controlling type I error rate. This approach is particularly beneficial in trials involving multiple treatment arms or stratified subgroups, where conventional fixed designs often prove too rigid or inefficient. By prioritizing critical timepoints and enabling more flexible allocation of resources, the procedure not only improves statistical efficiency but also reinforces ethical accountability in trial conduct.
Even more transformative are master protocol designs, including umbrella, basket, and platform trials. These frameworks allow simultaneous testing of multiple therapies or targets within a shared infrastructure, reducing redundancy and accelerating discovery. Integrating computational modeling, multi-omics data, and biomarker-driven stratification within these trials are key to operationalizing precision medicine. Rather than treating trial design as static, these models allow the protocol itself to evolve alongside accumulating biological knowledge [637, 638].
Methodological innovations converge on a broader necessity: reconceptualizing neuroprotection itself in clinical contexts. Despite decades of investigation and an expanding array of molecular targets and candidate therapeutics, the field continues to struggle with the lack of a standardized and universally accepted definition of neuroprotection. Mechanisms inferred from animal models demand renewed scrutiny to establish genuine translational potential [639].
The existing conceptual uncertainty complicates trial design, data interpretation, and ultimately, clinical translation. It fosters heterogeneity in outcome measures, mechanisms of action, and temporal scales, blurring distinctions between true neuroprotection, symptomatic relief, and disease modification. Consequently, promising interventions often fail to demonstrate meaningful clinical impact, revealing the limitations of current models and endpoint [566].
Meaningful progress will require a paradigmatic shift towards integrated approaches. These include moving beyond single biomarkers to adopt multidimensional assessments that combine biomarkers, neuroimaging, digital phenotyping, and behavioral measures [580, 582, 610, 638]. AI and ML are accelerating this integration, enabling real-time modeling of disease trajectories and individualized therapeutic responses [637]. Simultaneously, advances in our understanding of the gut-brain axis and neuroimmune pathways have expanded the therapeutic landscape to include microbiota-targeted strategies and modulation of systemic inflammation.
The transformation of clinical trial infrastructure is equally critical. Adaptive designs, umbrella and platform trials, and master protocols offer a path forward, but they are not without obstacles. Their implementation requires robust computational infrastructure, interdisciplinary coordination, dynamic regulatory oversight, and enhanced training for investigators in informatics and biostatistics. Furthermore, patient communication and consent processes must evolve to accommodate the flexible and iterative nature of these trials [582, 610].
Despite these challenges, the potential benefits are substantial: increased precision, faster optimization of interventions, improved participant safety, and more nuanced insights into the biological variability of neurodegenerative conditions. Inclusive recruitment strategies and global research equity are also imperative to ensure that trials reflect the diversity of patient experiences and are generalizable across populations. Figure 6 schemes the drawbacks and advances in the progress of “measuring” neuroprotection.
Ultimately, the future of neuroprotection research hinges on embracing complexity rather than avoiding it. Progress depends on fostering collaboration across scientific domains, refining conceptual frameworks, and aligning innovation with ethical responsibility and clinical relevance. Only through this holistic and forward-thinking approach can we hope to develop therapies that go beyond modifying disease progression, preserving, and potentially restoring neural integrity in the human brain.
Together, these insights lay the groundwork for a final roadmap: a multitarget, biomarker-guided vision of neuroprotection for the decades ahead.
Neurodegenerative diseases such as AD, PD, HD, and ALS share the tragic feature of progressive neuronal loss, but they also reveal a deeper truth: Neurons do not degenerate in isolation. Rather, they fail within a complex, multicellular environment in which astrocytes, microglia, oligodendrocytes, vascular elements, and systemic immune components actively participate. This review has highlighted how neuroinflammation, oxidative stress, mitochondrial dysfunction, and impaired proteostasis converge to create a vicious cycle that overwhelms neuronal resilience. At the same time, the evidence gathered underscores that each of these mechanisms represents not only a challenge but also an opportunity for therapeutic intervention.
From this perspective, neuroprotection must be reframed as a systemic, integrated endeavor. Future therapies will need to combine neuron-directed approaches with strategies that modulate glial function, restore homeostatic communication across cell types, and exploit emerging biomarkers for early detection and patient stratification. The days of single-target interventions, such as focusing exclusively on Aβ in AD or dopaminergic neurons in PD, are coming to an end. The next era will be defined by multimodal interventions capable of addressing the layered complexity of neurodegeneration.
Another major theme is the role of biomarkers. Reliable diagnostic, prognostic, and pharmacodynamic markers remain indispensable for translating laboratory advances into clinical benefit. Blood-based and imaging biomarkers not only allow the identification of individuals at risk during prodromal stages but also provide measurable endpoints for clinical trials. Without this integration of biomarkers, it will remain difficult to distinguish symptomatic effects from true disease modification. Conversely, with them, the field can design adaptive trials, refine patient selection, and accelerate therapeutic discovery.
Importantly, the concept of neuroprotection must evolve from a defensive stance to a proactive, regenerative paradigm. Enhancing synaptic plasticity, stimulating intrinsic cytoprotective pathways, and delivering neurotrophic support are necessary, but insufficient steps. Long-term success will require regenerating functional circuits, possibly through glial reprogramming, neuromodulation, or advanced gene therapy. Though many of these strategies remain experimental, their convergence with real-world data, digital phenotyping, and precision medicine frameworks heralds a transformative decade ahead.
Equally vital is the adoption of predictive, preventive, personalized, and participatory (P4) medicine. This framework aligns with the multifactorial nature of neurodegeneration and ensures that interventions are matched not only to disease stage but also to individual patient biology and lifestyle. The integration of genomics, proteomics, metabolomics, imaging, and clinical metadata will be essential to construct patient-specific trajectories and therapeutic windows.
Finally, the social and economic burden of neurodegenerative disorders demands urgent innovation. As the global population ages, the prevalence and impact of these diseases will continue to grow, straining healthcare systems and eroding quality of life for millions of patients and families. Research must therefore maintain a dual focus: mechanistic rigor in the laboratory and translational efficiency in the clinic. Collaboration among neuroscientists, clinicians, data scientists, patients, and policymakers will be necessary to transform incremental advances into real-world impact.
In conclusion, the path forward lies in embracing complexity rather than simplifying it. By acknowledging the active role of glial cells, securing robust biomarkers, and designing multimodal, personalized strategies, the field can move closer to genuine disease modification. Neuroprotection beyond neurons is no longer an aspiration; it is a necessity. Only through an integrated, systemic, and forward-looking approach can we hope to not merely extend life but preserve its quality in the face of neurodegeneration.
AD: Alzheimer’s disease
ADHD: attention-deficit/hyperactivity disorder
ADNI: Alzheimer’s Disease Neuroimaging Initiative
Aeg-1: Astrocyte Elevated Gene-1
AI: artificial intelligence
ALS: Amyotrophic Lateral Sclerosis
AMPA: alpha-amino-3-hydroxy-5-methyl-4-isooxazole-propionate
APP: amyloid precursor protein
AQP4: aquaporin-4
ARE: antioxidant response element
Arg1: arginase 1
ARIAs: amyloid-related imaging abnormalities
AUC: area under the curve
Aβ: amyloid-β
Aβ1–42: amyloid-beta 1–42 peptide
BBB: blood-brain barrier
BDNF: brain-derived neurotrophic factor
BNT: Boston Naming Test
CDNF: cerebral dopamine neurotrophic factor
CNS: central nervous system
CSF: cerebrospinal fluid
CVLT: California Verbal Learning Test
DALYs: Disability-Adjusted Life Years
DAM: disease-associated microglia
DAMP: damage-associated molecular pattern
DLB: dementia with Lewy bodies
EAAT2: excitatory amino acid transporter 2
EOAD: early-onset AD
GABA: gamma-aminobutyric acid
GDNF: glial cell line-derived neurotrophic factor
GFAP: glial fibrillary acidic protein
GFRα: GDNF family receptor α-1
HD: Huntington’s disease
IGF-1: insulin-like growth factor 1
iNOS: inducible nitric oxide synthase
LOAD: late onset form of AD
LTP: long-term potentiation
MAO-B: monoamine oxidase B
MAPK: mitogen-activated protein kinase
mGluRs: metabotropic–G protein-coupled–glutamate receptors
mHTT: mutant huntingtin
ML: machine learning
MS: Multiple Sclerosis
mTOR: mechanistic target of rapamycin
NADPH: nicotinamide adenine dinucleotide phosphate
NfL: neurofilament light chain
NIBS: non-invasive brain stimulation
NMDA: N-methyl-D-aspartate
Nrf2: nuclear erythroid related transcription factor 2
OS: oxidative stress
P4: predictive-preventive-personalized-participatory
PAF: pure autonomic failure
PD: Parkinson’s disease
PET: positron emission tomography
PI3K: phosphatidylinositol 3-kinase
PINK1: Phosphatase and Tensin Homolog-induced kinase 1
PRRs: pattern recognition receptors
P-SYN: phosphorylated α-synuclein
p-tau: phosphorylated tau
PTEN: Phosphatase and Tensin Homolog
RNS: reactive nitrosylation species
ROC: Receiver Operating Characteristic
Shh: Sonic Hedgehog
SIMOA: Single Molecule Array
SLE: systemic lupus erythematosus
tDCS: transcranial direct current stimulation
TLRs: Toll-like receptors
TMS: transcranial magnetic stimulation
Tregs: regulatory T cells
TREM2: Triggering Receptor Expressed on Myeloid cells 2
TrkB-FL: tropomyosin receptor kinase B-full length
UPSIT: University of Pennsylvania Smell Identification Test
Ym1: rodent-specific chitinase-like protein
The research group of the University of Barcelona is considered of excellence (grup consolidat #2021 SGR 00304) by the Regional Catalonian Government. Sara Costa Pinto helped to design and produce some of the Figures.
During the preparation of this work, RF and CV utilized the Sora tool, available at the time through ChatGPT, to generate illustrative images of neural cells displaying a range of morphologies and cellular extensions, effectively highlighting their phenotypic diversity. These images were generated under Sora’s terms of use, which did not assign copyright restrictions at the time of their creation, allowing free use.
CV, GH, CAS, CV Jr., JAS, SHV, AMS, PDM, FV, VS, YT, HU: Investigation, Writing—original draft. FTV: Investigation. VJB: Writing—review & editing. RF: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision, Project administration. All authors reviewed, edited, and approved the final submitted version.
Rafael Franco is Editor-in-Chief of Exploration of Neuroprotective Therapy. Christopher Shaw, Yong Tang, and Henning Ulrich are Associate Editors of Exploration of Neuroprotective Therapy. Cinzia Volonté, Vladimir J Balcar, P. David Mozley, and Vincenzo Silani are Editorial Board Members of Exploration of Neuroprotective Therapy. Guoku Hu and Claudio Viegas Jr. are Editorial Board Members and Guest Editors of Exploration of Neuroprotective Therapy. Rafael Franco, all the Associate Editors, Editorial Board members, and Guest Editors mentioned above had no involvement in the decision-making or the review process of this manuscript. The other authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
HU’s research on neuroprotection and purinergic signaling has been supported by National Council for Scientific and Technological Development (CNPq)—the CNPq-funded National Institute of Science and Technology (INCT) for Purinergic Signaling: Challenges for 21st Century Health (CNPq, Grant No. [409156/2024-8], the CNPq project No. 406396/2021-3 and 440993/2023-7). His research was also funded by a Thematic Project granted by the São Paulo Research Foundation (FAPESP, Grant No. [2018/07366-4]). FAPESP is acknowledged for a postdoctoral fellowship awarded to FTV (project No. 2024/17387-0). YT and HU are supported by grants from NSFC-RSF [82261138557]. CV is supported by FATALSDRUG Project (Progetti di Ricerca@CNR, SAC.AD002.173.058) from the National Research Council, Italy. Ana M. Sebastião is supported by Fundação para a Ciência e Tecnologia, Portugal (PTDC/MED-FAR/4834/2021 and 2023.17919.ICDT) and COST Actions CA24130 (PSY-NET) and CA21130 (PRESTO) and HORIZON-WIDERA-2023-ACCESS-04-01 (GA 101160180 - PANERIS). The funders had no role in review design, collection of information, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

