 Review
Review

Affiliation:
1Department of Human Molecular Genetics and Biochemistry, Gray Faculty of Medical and Health Sciences, Tel Aviv University, Tel Aviv 6997801, Israel
Email: hwerner@tauex.tau.ac.il
ORCID: https://orcid.org/0000-0003-0154-1290
Affiliation:
2Gynecologic Oncology Research Laboratory, Department of Obstetrics and Gynecology, Hillel Yaffe Medical Center, Hadera 3820302, Israel
3The Ruth and Bruce Rappaport Faculty of Medicine, Technion - Israel Institute of Technology, Haifa 3525422, Israel
ORCID: https://orcid.org/0000-0003-4898-5462
Explor Med. 2025;6:1001342 DOI: https://doi.org/10.37349/emed.2025.1001342
Received: March 11, 2025 Accepted: June 20, 2025 Published: July 01, 2025
Academic Editor: Ferdinando Carlo Sasso, University of Campania “Luigi Vanvitelli”, Italy
Technological breakthroughs over the past quarter century have had a huge impact on the broad area of medicine. Biological processes are now amenable to integrative examination at multiple levels of analysis, ranging from molecular to organismal levels. The Human Genome Project, in particular, paved the way for a new age in medicine that is commonly referred to as Precision (or Personalized) Medicine. These changes in the health sciences world are perceived both from the patient and clinician’s perspectives. The present article focuses on the insulin-like growth factor-1 (IGF1) axis, an important endocrine network with key roles in physiological and pathological conditions. We aim to provide the reader with an overview of the basic and clinical aspects of the IGF system, with a particular emphasis on ongoing efforts to target the IGF axis for therapeutic purposes. The potential impact of precision medicine on IGF1 clinical research is discussed.
The concept of Precision (or Personalized) Medicine embodies the promise of better medical treatments for a wide range of genetic and acquired conditions based on the unique and distinctive genomic, metabolic and biochemical constitution of the patient. The identification of this characteristic makeup was initially made possible by the Human Genome Project in the early 2000s [1, 2]. Subsequently, and over the course of the next twenty years multiple translational spinoffs that emerged from this gigantic project have contributed to a better characterization of an individual’s physiological and pathological “composition”. The conceptual and technological advances that derived from this enormous project are assisting us in the identification of biomarkers, “signatures” and signaling webs that might be profoundly involved in the etiology of disease. As described in detail in this review article, these developments are expected to translate into more rational tailored therapies.
The field of oncology is one of the areas in which the new age technologies are drastically changing well-established dogmas and paradigms [3, 4]. Precision medicine employs an assortment of approaches that allow the practitioner to identify genomic signatures in tumors and metastases in real-time. The ultimate goal of these analyses is to identify clinically actionable targets. The present review article focuses on innovative state-of-the-art methodologies and tools and their potential uses in the broad field of cancer. The main emphasis of our review are the insulin-like growth factors (IGFs). We aim to provide a concise overview of basic and clinical aspects of the IGF system. Furthermore, we seek to provide the reader with an up-to-date outline of recent attempts to target the IGF system for cancer therapy. We trust that the information summarized here will be of help to basic scientists as well as clinicians.
The insulin-like growth factor-1 (IGF1) endocrine system plays a fundamental role in the regulation of growth and metabolic processes throughout life [5, 6]. IGF1, or somatomedin, was originally identified by virtue of its role as the mediator of the biological actions of pituitary-derived growth hormone (GH) [7]. Later, the liver-produced peptide was shown to be also synthesized by several extra-hepatic tissues although at levels much lower than those released by the liver. It is unclear, however, whether the local activities of IGF1 are, similar to the endocrine type of action, dependent on GH stimulus. The bioactivities of IGF1 are mediated by a cell-surface heterotetrameric receptor, the IGF1 receptor (IGF1R), which belongs to the tyrosine kinase-containing receptors superfamily and which is structurally and evolutionarily related to the insulin receptor (IR) [8–11]. It is generally agreed that, while the IR is usually involved in metabolic activities, the IGF1R is mainly responsible for growth and differentiation types of action [12, 13].
However, given the large similarity between ligands (i.e., insulin, IGF1) and receptors (i.e., IR, IGF1R), a significant degree of cross-talk exists within this family [14]. As a result of this cross-talk, compensatory mechanisms exist that lead to activation of IR by insulin and/or IGF2 when blocking the IGF1R. An additional level of complexity is provided by a new family of IGF1R/IR hybrid receptors, composed of an IGF1R hemireceptor linked to an IR hemireceptor [15]. These hybrids exhibit distinct ligand-binding properties (e.g., high affinity for IGF2 and insulin) and activate unique downstream pathways, such as hyperactivation of AKT/mTOR, which drive metastasis and resistance to specific inhibitors. Finally, the bioactivities of IGF1, but not insulin, are modulated by a series of IGF-binding proteins (IGFBPs) that bind the ligand in the circulation and extracellular space and transport it to its sites of action [16, 17].
Binding of IGF1 (or IGF2) to the IGF1R activates its kinase activity, with ensuing autophosphorylation and activation of a set of cytoplasmic substrates (Figure 1) [5, 6]. These molecules include IRS1–4, Shc, and 14-3-3. Phosphorylated IRS activates two main signaling paths: the Ras-Raf-MAPK and the PI3K-AKT/PKB pathways [10]. Activated MAPK is associated with cell proliferation while activated PI3K stimulates protein synthesis and prevents apoptosis. Activated IRS1 phosphorylates the 85-kDa regulatory subunit of PI3K. Sequentially, several downstream substrates, including AKT/PKB, are activated. Phosphorylation of AKT stimulates protein synthesis via mTOR. This event triggers the classical anti-apoptotic effect of IGF1R by means of inactivation of BAD. Finally, phosphorylated IRS1 or Shc recruit Grb2/SOS, leading to enrollment of Ras and activation of the Ras-MAPK pathway. As a corollary, the huge complexity of this intracellular information system generates multiple therapeutic opportunities. Some of these aspects will be discussed in this article.
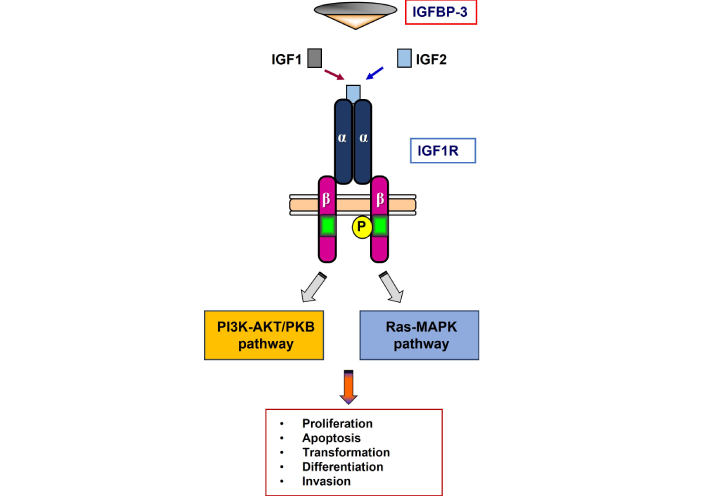
Schematic representation of the IGF1R. The biological activities of IGF1 and IGF2 are mediated by a cell-surface heterotetrameric receptor, the IGF1 receptor (IGF1R), a member of the tyrosine kinase (TK) receptors superfamily. The IGF1R is structurally and evolutionarily related to the insulin receptor. The mature receptor is composed of two extracellular a subunits that are mainly responsible for ligand binding, and two transmembrane b subunits, which include a TK domain. Ligand binding leads to structural changes with ensuing autophosphorylation of specific tyrosine residues. In turn, the activated (phosphorylated) receptor catalyzes the phosphorylation of a series of cytoplasmic substrates (PI3K-AKT/PKB and Ras-MAPK). IGFBP-3 is the main binding protein in the circulation and it modulates ligand availability. IGF1: insulin-like growth factor-1; IGFBP: insulin-like growth factor-binding protein
As described below in more detail, IGF1 functions as a progression factor that is needed by the cell in order to advance or proceed through the different phases of the cell cycle [18, 19]. In addition to its fundamental role in growth, differentiation and development, IGF1 has been also identified as an essential player in the biochemical and cellular chain of events that leads to malignant transformation and, eventually, cancer development [20]. In agreement with this pathologic function, extensive epidemiological studies have demonstrated that circulating IGF1 levels are positively correlated with cancer risk in numerous types of cancer [21–24]. In the following section, we dissect in mechanistic terms important aspects associated with IGF1 action in the context of cancer biology.
As alluded to above, conceptual and technological advancements in recent years have had a major impact on our comprehension of basic aspects of the biology of the IGF1 endocrine system. These advances were made possible by the use of innovative high-throughput methods, mainly genomic and proteomic capabilities as well as powerful bioinformatic platforms [25–27]. Our analysis will adhere to a catalog of cancer-associated events, including biochemical, molecular and genetic events that are commonly referred to as Hallmarks of Cancer [28, 29]. The roles of IGF1 in cancer are summarized in Table 1.
Evidence in support of a key role for the IGF1 system in cancer biology
| Evidence |
|---|
| Epidemiological analyses identified serum IGF1 as a risk factor in various cancers |
| Cells deprived of the IGF1R do not undergo malignant transformation |
| Oncogenes adopt the IGF1 signaling pathway |
| Individuals with minimal IGF1 levels (Laron syndrome) are protected from cancer |
| Tumor cells express high IGF1R levels and exhibit a constitutively activated IGF1R pathway |
| IGF1R exhibits a very potent anti-apoptotic activity |
| The IGF1 system is an important player in angiogenesis |
IGF1: insulin-like growth factor-1; IGF1R: IGF1 receptor
While normal cells display stringently regulated growth signals that usually prevent enhanced proliferation at post-mitotic stages, cancer cells exhibit a dysregulated signaling ability [30]. The net consequence of this pathological deregulation is the ability of cancer cells to undergo unlimited proliferation. This quintessential feature of transformed cells, i.e., the capacity “not-to-die”, results in part from their ability to synthesize a large number of growth factors and their receptors [31, 32]. These molecules confer upon tumoral cells what is commonly regarded as “growth-factor independence”. The unrestricted behavior that emanates from this state allows malignantly-transformed cancer cells to traverse the cell cycle in an unopposed manner in the absence of exogenous stimuli.
In this specific context, IGF1 was shown to induce mitogenic responses in a diversity of normal as well as tumoral cells [33, 34]. In this sense, IGF1 fits the definition of a progression factor, a molecule whose expression and action are required to evade cell cycle arrest and to progress through the various phases of the cell cycle [35]. Furthermore, IGF1 induces differentiation in multiple cell types while IGF1 silencing inhibits these actions [36]. In mechanistic terms, IGF1 binding to the extracellular domain of the IGF1R leads to conformational modifications with ensuing autophosphorylation of specific tyrosine residues within the cytoplasmic domain of the receptor [37]. Next, the now-activated IGF1R is capable of phosphorylating additional “downstream” substrates, followed by stimulation of an array of intracellular cascades. It is classically accepted that IGF1-stimulated mitogenesis is linked to activation of the Ras-Raf-MAPK pathway while its anti-apoptotic effect is attributed to the PI3K-AKT pathway (see above) [38].
One of the typical features of cancer cells is their ability to evade antigrowth signals that are, under normal circumstances, aimed at keeping cells in a quiescent state or, in other words, out of the cell cycle [39]. Hence, transformed cells can regain a formerly repressed mitogenic capacity that “pushes” them through the cell cycle. This outcome, which is often regarded as a critical event in oncogenesis, is achieved by either one of two pathways: gain-of-function mutations of oncogenes or loss-of-function mutations of tumor suppressors.
Transactivation of growth factor or growth factor receptor genes, including the IGF1R, by oncogenes constitutes a common mechanistic event that is shared by different oncogenic agents. This mode of action is colloquially referred to as the “adoption” of the IGF1R pathway [40]. As an example, oncogene c-myb transactivates the IGF1R promoter, leading to an enhancement in gene transcription and IGF1R biosynthesis [41]. Likewise, oncogene pp60src accelerates the phosphorylation of the IGF1R tyrosine kinase domain in a constitutive fashion [42]. Enhanced signaling via the IGF1R, as mentioned above, is of critical importance in mitogenesis.
On the other hand, a number of tumor suppressor genes were shown to exert their antiproliferative activity via their capacity to suppress IGF1R gene transcription [43]. For example, wild-type p53 was shown to strongly inhibit IGF1R promoter activity while, on the other hand, mutant p53 stimulate this activity [44, 45]. Additional tumor suppressors shown to inhibit the IGF1R gene include the breast cancer susceptibility gene-1 (BRCA1), the Wilm’s tumor suppressor (WT1) and the von Hippel-Lindau (VHL) protein [46–50]. Thus, while anti-oncogenes differ in many aspects, including tissue-specific expression and mechanisms of action, they share the IGF1R signaling pathway as a response pathway.
The capacity of tumor cells to survive results not only from their proliferative potential but also from their acquired ability to oppose death, in particular apoptosis. In fact, resistance toward apoptosis has been identified by Hanahan and Weinberg as a typical hallmark of transformed cells [28]. The molecular and cellular mechanisms that are responsible for the evasion of apoptosis by cancer cells have been well characterized over more than fifty years.
When compared to most other growth factor receptors, the anti-apoptotic activity of IGF1R is one of the strongest described to date [51–53]. Interestingly, there is no overlap between the IGF1R domains that are required for inhibition of apoptosis from domains required for mitosis [54]. Of importance, it seems to be a correlation between IGF1R cell-surface concentrations and cell survivability. Hence, cells (R-) deprived of IGF1R are protected from oncogene-induced transformation [55, 56]. Only upon reintroduction of an intact receptor, the cells become susceptible to the oncogenic action of transfected oncogenes. Combined, these studies support the notion that IGF1R expression and activation are fundamental requisites for tumor development.
While epidemiological studies have provided ample evidence for a positive correlation between high circulating IGF1 levels and enhanced cancer risk, the question whether low IGF1 concentrations can provide protection from tumor development was much more difficult to address. The identification of a highly atypical genetic endocrinopathy in the late 1950s by Prof. Laron [57, 58] had a huge impact on our current understanding of endocrine networks and, particularly, on the pathophysiology of the GH-IGF1 pathway. Whereas Laron syndrome (LS) patients resemble children with typical GH deficiency (i.e., dwarfism, hypoglycemia, obesity), radioimmunoassays revealed that patients have extremely high GH levels, essentially in the acromegalic range. GH receptor (GHR) gene cloning in the late 1980s led to the identification of an autosomally transmitted mutated GHR gene in LS patients [59]. Mutations include nonsense, frameshift and missense mutations as well as exon deletions [60]. GHR gene disruption leads to defective GHR biosynthesis with impaired GHR signaling and ensuing major reduction in liver IGF1 production.
An epidemiological study based on 538 patients with diminished IGF1 levels, including 230 people with LS and 308 individuals with GH deficiency, GH-releasing hormone receptor (GHRH-R) anomalies and multiple pituitary hormone deficiency, demonstrated that none of the 230 LS patients had a history of cancer (up to the age of 85) [61]. In contrast, 30 individuals with cancer were reported among 752 heterozygote family members (4%). Moreover, 31 cases of cancer were reported among 131 further relatives (23.7%). Despite the small size of this cohort, differences in cancer incidence between LS patients and relatives were regarded as statistically significant. Similarly, protection from cancer was also reported in an Ecuadorian cohort of LS patients [62]. In summary, epidemiological analyses support the assumption that congenital IGF1 deficiency confers protection against future development of tumors.
To identify the complete set of genes and signaling molecules associated with cancer protection we conducted genomic analyses on lymphoblastoid cells derived from people with LS [63]. Analyses discovered families of genes that were either under- or over-represented in people with LS. Moreover, studies identified signaling pathways that are altered in this disease (Table 2). Differential expression was detected in several gene families, including JAK-STAT signaling, cell cycle, metabolism, PI3K-AKT signaling, etc. Discrepancies between LS and controls were also seen in pathways associated with apoptosis, cell cycle distribution, and autophagy.
List of signaling pathways altered in Laron syndrome
| Signaling pathways |
|---|
| Metabolic pathways |
| Cell adhesion |
| Pathways in cancer |
| Oxidation reduction |
| Apoptosis |
| JAK-STAT signaling pathway |
| Regulation of cytokine production |
| Immune response |
| Cell migration and motility |
| G-protein coupled receptor signaling pathway |
The list highlights the broad involvement of the IGF1 system in multiple body and cellular functions. IGF1: insulin-like growth factor-1
More specifically, the genomic data summarized in Table 2 highlights specific pathways that are differentially expressed in LS, including FOXO3A-mediated apoptosis and PTEN-driven suppression of PI3K/AKT. Data indicates that LS cells exhibit AMPK hyperactivation, which inhibits mTORC1, a master regulator of anabolic metabolism. This mimics the effects of pharmacologic mTOR inhibitors (e.g., everolimus), suggesting that IGF1 deficiency induces a “natural mTOR inhibition” state. Additionally, LS models show elevated autophagy flux (via LC3B-II upregulation), which may purge damaged organelles and proteins, reducing genomic instability [63]. These findings suggest that combination of IGF1R inhibitors with AMPK activators (e.g., metformin) or autophagy inducers (e.g., hydroxychloroquine) could, probably, replicate LS-associated cancer protection. In summary, the identification of novel downstream targets of the IGF1 pathway associated with tumor protection highlights the key role of the GH-IGF1 axis in the initiation and progression of cancer [64–66].
Orchestrated efforts that derive from both the preclinical and clinical spheres led to the identification of the IGF1 axis as a potential therapeutic target in oncology. We describe here some of the discoveries that constitute the basis for this translational development.
In a prospective study conducted in the late 1990s known as the Nurses’ Health Study, the relative risk (RR) of breast cancer in premenopausal women with IGF1 levels in the upper tertile of the spectrum was 4.6 times higher than that in women in the lower tertile [22]. The RR increased to 7.3 when the levels of IGFBP-3 were taken into consideration. Similar analyses conducted within the framework of the Physicians’ Health Study showed a positive correlation between IGF1 levels and risk of developing prostate cancer. Thus, men with the highest IGF1 levels had an RR of 4.3 compared to men with low IGF1 [21]. In addition, IGF1 was also identified as a risk factor in a number of non-hormone dependent tumors, including bladder, colon and lung [23]. Combined, the epidemiological association between endocrine IGF1 levels and cancer risk had a profound impact in the IGF1 therapy field and helped to shape some of the current paradigms in this area.
Malignant transformation is usually associated with a reversal to a more “primitive” pattern of gene expression, which includes enhanced IGF1R gene expression and, in a significant portion of the cases, also IGF2 gene expression. IGF2 is synthesized in situ by most tumor cells and serves as a paracrine ligand for the IGF1R. These early findings led to the, now broadly accepted, idea that IGF1R expression is a crucial pre-requisite for the establishment of most types of neoplasia [67]. Moreover, constitutive phosphorylation of the cell-surface IGF1R is regarded as a universal hallmark of cancer cells.
Finally, the observation that cells deprived of the IGF1R do not undergo transformation was also of critical relevance towards the identification of the IGF1R as a potential therapeutic target. Specifically, in a series of seminal papers from the laboratory of Renato Baserga, Sell and colleagues [55] demonstrated that cells derived from embryos in which the IGF1R gene was abrogated by homologous recombination, with a small number of exceptions, do not undergo malignant transformation when exposed to viral or cellular oncogenes [56]. Hence, these experiments demonstrate that the presence of the IGF1R is of cardinal importance in the establishment of a tumor.
Based on the concepts and rationale described above, several approaches have been developed to therapeutically targeting the IGF1 axis [68–71]. The main goals of these strategies are:
Inhibition of tumor cell proliferation and survival.
Reversal of cancer growth and metastasis formation.
Sensitization to radiotherapy, chemotherapy, and biological therapies.
The main strategies employed to date to target the IGF1 pathway include:
Small molecule IGF1R tyrosine kinase inhibitors (TKIs).
Antibodies directed against the IGF1R.
Neutralizing antibodies against the IGF1 and IGF2 ligands.
TKIs have shown cancer growth inhibitory properties in experimental and preclinical models. Given that the IGF1R and IR kinase domains are structurally similar most TKIs can inhibit both receptors. However, they display a 15–30-fold higher potency against IGF1R.
IGF1R targeting with specific (usually monoclonal) antibody has been the most applied method for abolishing IGF signaling. Anti-IGF1R block signaling via two putative mechanisms: (1) hormone binding inhibition; and (2) internalization and degradation of the receptor. Consequently, antibodies targeting IGF1R inhibit IGF1/2-induced proliferation. Moreover, combined treatment with cytotoxic drugs enhanced the effect of IGF1R antibodies [72]. Of note, adaptive trial frameworks like I-SPY2 could test IGF1R inhibitors in biomarker-enriched arms (e.g., high IGF1R extracellular vesicles + low IRS-1), accelerating translation.
Of clinical relevance, recent studies have demonstrated that co-targeting IGF1R and IR-A with bispecific antibodies reduced tumor growth in hybrid-rich models. Furthermore, hepatic feedback loops increase GH secretion upon IGF1R inhibition, elevating systemic IGF1 levels-a phenomenon requiring co-targeting GHRs (e.g., pegvisomant). Combined, the above insights might help contextualize past failures and highlight rational combinatorial approaches. Finally, clinical studies aimed at targeting the IGF1/2 ligands suggest that ligand-targeted therapies are, usually, well tolerated [68–70]. Furthermore, phase 1 clinical trials resulted in stable disease in roughly one third of the patients. On the other hand, xentuzumab (a humanized IgG1 monoclonal antibody that binds IGF-1 and IGF-2, inhibiting their growth-promoting signaling) failed in a phase 3 clinical trial [73].
Support for an enhancement effect of immunotherapy on IGF1R-targeting was recently provided by studies conducted on epithelial ovarian cancer (EOC) cells [74, 75]. EOC is the most fatal gynecologic malignancy worldwide and it accounts for 90% of all ovarian tumors. The programmed cell death protein-1 (PD-1) and its ligand (PD-L1) are critical immune checkpoints. Targeting of these molecules was shown to reverse tumor-mediated immunosuppression. Consistent with this data, we have recently demonstrated that inhibition of the IGF1R pathway elicits a very strong antitumor immunity. Furthermore, combination of IGF1R therapy with a PD-1 antagonist led to tumor recession in an animal model of EOC. In addition, we provided evidence that combination therapy led to a stronger immune response as compared to monotherapy. In mechanistic terms, IGF1R signaling in dendritic cells (DCs) upregulates PD-L1 via STAT3, while in T cells it promotes exhaustion markers (TIM-3, LAG-3). IGF1R inhibition in melanoma models was shown to reverse DC dysfunction and enhance CD8+ T-cell infiltration by suppressing IL-10 secretion from tumor-associated macrophages. This positions IGF1R as a dual modulator of tumor-intrinsic signaling and immune suppression, advocating for trials combining IGF1R inhibitors with CTLA-4 or TIM-3 blockers. In conclusion, the combined effect of PD-1 and IGF1R inhibition in EOC is of significant translational importance. Data might serve as a starting point for the development of personalized therapies for this and other malignancies.
There is wide consensus nowadays that one of the main reasons for the somehow disappointing responses reported in the IGF1 therapy field (particularly in oncology) is the fact that most early trials were conducted on unselected patients. Furthermore, initial IGF1R trials failed partly due to compensatory IRS-1/2 adaptor protein phosphorylation, which sustains PI3K/AKT signaling even after receptor blockade. For example, in Ewing sarcoma, IGF1R antibody resistance correlated with IRS-1 overexpression, which can be circumvented by adding PI3Kδ inhibitors (e.g., idelalisib). Hence, the identification of predictive biomarkers that could help in selecting patients who might benefit from this tailored type of therapy is of enormous importance [76]. In addition, biological markers are needed to monitor the response of patients to targeted therapy.
The breast cancer susceptibility gene-1, BRCA1, has key roles in many biological pathways, including transcription, control of apoptosis and DNA damage repair. In agreement with its inherent tumor suppressor role, we have shown that wild-type BRCA1 displays a potent inhibitory activity of IGF1R gene transcription [46, 77]. On the other hand, mutant BRCA1 was unable to inhibit IGF1R expression. To address the hypothesis that the mutational status of BRCA1 may serve as a biomarker capable of predicting the outcome of IGF1R-directed therapies, we used a collection of breast cancer cells that express a wild-type or a mutant BRCA1. We showed that the capacity of MK-0646, an anti-human IGF1R antibody, to inhibit IGF1-stimulated IGF1R phosphorylation was impaired in mutant BRCA1-expressing cell lines [78]. In addition, the antibody inhibited proliferation of wild-type BRCA1-expressing cells but had a diminished effect in mutant BRCA1-expressing cells. Our data strongly suggest that the mutational status of BRCA1 should be taken into consideration when selecting patients for IGF1R targeting protocols.
In a follow-up study we evaluated the hypothesis that combination therapy composed of selective IGF1R inhibitors along with chemotherapy should be more effective than individual monotherapies in the treatment of breast cancer [79]. Analyses included measurements of the synergism elicited by various therapies. Data obtained provided evidence that co-treatment of IGF1R inhibitor along with chemotherapeutic drugs markedly improved the therapeutic efficiency. Of clinical relevance, data indicated that high IGF1R baseline expression may serve as a predictive biomarker. Finally, proteomic analyses identified a ten-genes signature with potential predictive value.
Circulating IGFBP-2 and IGFBP-7 have emerged as predictive biomarkers in recent studies [80–83]. IGFBP-2, when cleaved by tumor-associated proteases, releases bioactive IGF1, fostering resistance. In contrast, IGFBP-7 binds IGF1R directly, inducing receptor internalization-a mechanism exploited in phase 2 trials of recombinant IGFBP-7 [84]. Liquid biopsies detecting IGF1R extracellular vesicles or tumor-educated platelets carrying phospho-IGF1R could dynamically monitor therapeutic efficacy [85].
McCaffery et al. [86] assessed the predictive nature of baseline endocrine levels of IGF factors on the effect of monoclonal antibody ganitumab plus gemcitabine in metastatic pancreatic adenocarcinoma. A significantly stronger improvement in overall survival was seen in the patient subset with higher levels of IGF1, IGF2 or IGFBP-3, or lower levels of IGFBP-2. In another study, Yee et al. [87] examined the effect of ganitumab in combination with metformin and paclitaxel (PGM) in comparison to standard-of-care (paclitaxel alone) in the treatment of breast cancer. Authors evaluated several putative predictive biomarkers of ganitumab response (e.g., IGF1 ligand score, IGF1R signature, IGFBP-5 expression, etc.). None were specific predictors of response to PGM, although several signatures were associated with pathologic complete response in both arms. In summary, the rational use of a number of bioinformatics tools and platforms shed light on the biological pathways responsible for synergism in cancer therapy. Furthermore, these tools identify biomarkers that might predict response to treatment.
Single-cell RNA sequencing in glioblastoma revealed IGF1R+ subclones enriched in hypoxic niches, which resist radiotherapy but are vulnerable to IGF1R inhibition [88, 89]. CRISPR screens identified BRD4 as a synthetic lethal partner of IGF1R in triple-negative breast cancer [90]. BRD4 inhibitors (e.g., JQ1) synergize with IGF1R blockade by silencing pro-survival myc transcripts [91]. Finally, spatial proteomics further maps IGF1R activation to invasive tumor margins, informing localized delivery strategies (e.g., nanoparticle-conjugated inhibitors).
In view of the fact that the IGF1 endocrine axis arose as a likely therapeutic target in oncology, the discovery of networks linked to IGF1 action (“IGF1 signatures”) is of high translational relevance. It is important to emphasize that, at this point, targeting of IGF1R has failed in the vast majority of cancers and to the best of our knowledge there is no further development of these drugs with the exception of an antibody-radioisotope conjugate currently in phase 1 trial (https://clinicaltrials.gov/study/NCT03746431). We believe that lessons from the rapidly developing field of precision medicine will be implemented in the area of IGF1 targeting in the near future. In this context, the integration of spatial transcriptomics to resolve IGF1R heterogeneity within tumor microenvironments and AI-driven multi-omics platforms to predict IGF1R dependency will be of key relevance. In addition to state-of-the-art discovery platforms (genomics, proteomics, metabolomics, etc.) the development of personalized strategies requires a deep knowledge of the biochemical, molecular and cellular aspects of the IGF1 axis. Elucidation of these complex mechanisms will improve our ability to deliver IGF1-targeted therapies more effectively.
BRCA1: breast cancer susceptibility gene-1
EOC: epithelial ovarian cancer
GH: growth hormone
GHR: growth hormone receptor
IGF1: insulin-like growth factor-1
IGF1R: insulin-like growth factor-1 receptor
IGFBPs: insulin-like growth factor-binding proteins
IR: insulin receptor
LS: Laron syndrome
PD-1: programmed cell death protein-1
RR: relative risk
TKIs: tyrosine kinase inhibitors
The authors wish to thank the members of our team for their important comments and suggestions.
HW: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. IB: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. Both authors have read and agreed to the published version of the manuscript.
Haim Werner who is the Guest Editor and Editorial Board Member of Exploration of Medicine had no involvement in the decision-making or the review process of this manuscript. Ilan Bruchim who is the Guest Editor of Exploration of Medicine had also no involvement in the decision-making or the review process of this manuscript.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by a generous award from the Israel Cancer Research Fund (ICRF, Montreal) [2026011] to IB. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 8404
Download: 31
Times Cited: 0
