 Perspective
Perspective

Affiliation:
Institute of Molecular Pathobiochemistry, Experimental Gene Therapy and Clinical Chemistry (IFMPEGKC), RWTH University Hospital Aachen, D-52074 Aachen, Germany
Email: rweiskirchen@ukaachen.de
ORCID: https://orcid.org/0000-0003-3888-0931
Explor Dig Dis. 2025;4:100599 DOI: https://doi.org/10.37349/edd.2025.100599
Received: September 11, 2025 Accepted: October 14, 2025 Published: October 28, 2025
Academic Editor: Jose Carlos Fernandez-Checa, Institute of Biomedical Research August Pi i Sunyer (IDIBAPS), Spain
The article belongs to the special issue Advances in Hepato-gastroenterology: Diagnosis, Prognostication, and Disease Stratification
Metabolic dysfunction-associated steatotic liver disease (MASLD) is increasingly recognized as a multisystem disorder in which iron acts as both a metabolic “spark” and an accelerant of liver injury. This integrates emerging evidence that iron-driven oxidative stress and low-grade inflammation are mutually reinforcing processes in metabolic liver disease. In this perspective article, epidemiological evidence, molecular insights, and emerging clinical data are integrated to clarify how hyperferritinemia, often dismissed as a mere inflammatory marker, maps onto genuine iron redistribution and overload in the metabolic liver. Physiological iron homeostasis and its disruption by adiposity-related inflammation, hyperinsulinemia, sex hormones, and common HFE variants, creating a labile catalytic iron pool that fuels Fenton chemistry in lipid-laden hepatocytes. Population studies and expert-panel criteria are summarized that define “metabolic hyperferritinemia” and stratify dysmetabolic iron accumulation into three magnetic resonance imaging (MRI)-based grades, each linked to stepwise increases in steatosis, fibrosis, and clinical events. Mechanistically, excess Fe2+ triggers lipid peroxidation, mitochondrial dysfunction, ferroptosis, Kupffer cell activation, endoplasmic reticulum stress, and hepatic stellate cell sensitization to TGF-β, thereby accelerating the transition from steatosis to steatohepatitis and fibrosis. Finally, the diagnostic algorithms, iron-modulating therapies (phlebotomy, hepcidin agonists, diet), and prospective data supporting ferritin-based triage in clinics are discussed. Collectively, the outlined evidence positions iron not only as a biomarker but also as a modifiable driver of MASLD progression, underscoring the need for randomized trials that test whether targeted iron reduction improves hard hepatic outcomes.
The biological importance of iron lies in its ability to switch between ferrous (Fe2+) and ferric (Fe3+) catalytic centers in over 200 human oxidative enzymes (cytochromes, catalases, peroxidases) and is an integral part of different blood proteins (hemoglobin), muscles (myoglobin). Most of the 3 to 4 g of elemental iron in adults is found in hemoglobin, stored in the form of ferritin or hemosiderin in the liver, spleen, and bone marrow, or is part of myoglobin in muscle tissue [1]. Approximately 25 mg of systemic iron circulates daily, representing 0.66% of the total iron content in the body [2]. Mostly, it is recycled from aging red blood cells by splenic macrophages before being returned to developing erythroblasts through transferrin (Figure 1). Dietary iron contributes a modest but crucial 1–2 mg per day via the divalent metal transporter 1 (DMT1), also known as the solute carrier family 11 member 2 (SLC11A2), in duodenal enterocytes, a process influenced by luminal pH, ascorbate, polyphenols, calcium, and increasingly, the siderophore landscape of the gut microbiome [3]. Hepatocytes, responsible for producing the systemic “iron hormone” hepcidin (HAMP), act as central traffic controllers: when iron stores increase or inflammation spikes, hepcidin binds and internalizes ferroportin in enterocytes and macrophages, limiting further iron release [4]. Adiposity disrupts this balance: adipose-derived IL-6 and leptin increase HAMP transcription, while hyperinsulinemia enhances iron uptake by up-regulating transferrin-receptor-1 (TfR1) expression on hepatocytes. Sex hormones also play a role [5, 6]. Estrogen suppresses hepcidin, partly explaining lower ferritin levels in pre-menopausal women [7]. In men, testosterone suppresses HAMP transcription and promotes iron incorporation into red blood cells, shifting iron from storage to hemoglobin [8]. If these finely tuned mechanisms fail due to chronic caloric overload, low-grade inflammation, or mutations in the homeostatic iron regulator (HFE), HAMP, ferroportin (FPN1), or many other genes, the “labile” catalytic iron pool accumulates [9].
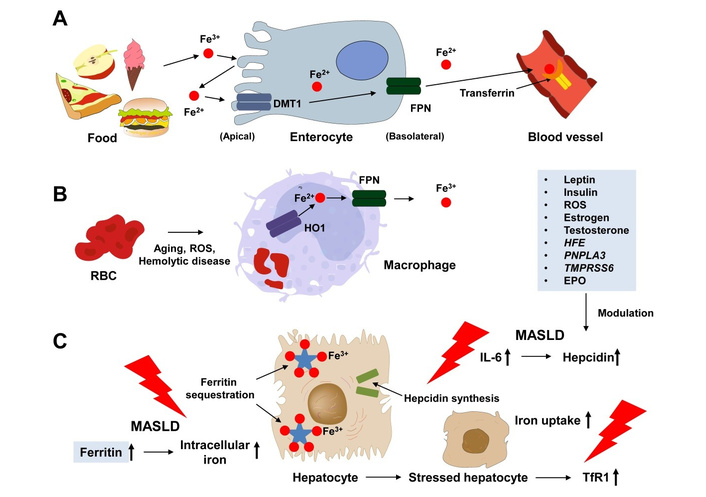
Systemic “iron traffic control” and sites of metabolic dysregulation. The schematic illustrates the major compartments and molecular players responsible for maintaining whole-body iron balance. (A) Polarized duodenal enterocytes import dietary Fe2+ through divalent metal transporter 1 (DMT1) and export it to plasma transferrin via ferroportin (FPN). (B) Macrophages recycle senescent erythrocytes, with heme oxygenase-1 (HO1) releasing iron that is then released through FPN. (C) Hepatocytes act as iron sensors and stores, emphasizing hepcidin synthesis and ferritin sequestration. Arrows indicate normal iron flow, while red lightning bolts highlight the three main disturbances in metabolic syndrome/MASLD: (i) IL-6-driven hepcidin overproduction leading to internalization of FPN, (ii) up-regulation of ferritin and intracellular iron trapping, and (iii) increased expression of transferrin-receptor-1 (TfR1) on metabolically stressed hepatocytes. Inset boxes provide a summary of hormonal (leptin, insulin, estrogen, testosterone, EPO), inflammatory (ROS), and genetic modulators (HFE, PNPLA3, TMPRSS6). EPO: erythropoietin; MASLD: metabolic dysfunction-associated steatotic liver disease; RBC: red blood cell; ROS: reactive oxygen species.
Besides genetic alterations, several infectious diseases impact hepatic iron homeostasis. In particular, the Malaria parasite Plasmodium can enter red blood cells of the host and degrade hemoglobin, converting iron into a redox-inactive Fe3+ polymer called hemozoin. This substance accumulates in large quantities and is deposited in many organs, including the liver.
In hepatocytes that are already busy oxidizing free fatty acids, even small amounts of unshielded Fe2+ can initiate Fenton chemistry. This process converts H2O2 into the highly destructive hydroxyl radical (•OH), which rapidly oxidizes organic compounds. This can lead to the degradation of lipids, proteins, and nucleic acids [10]. When uncontrolled, excessive iron can induce inflammation, fibrosis, and systemic insulin resistance by impacting numerous biological processes (Figure 2).
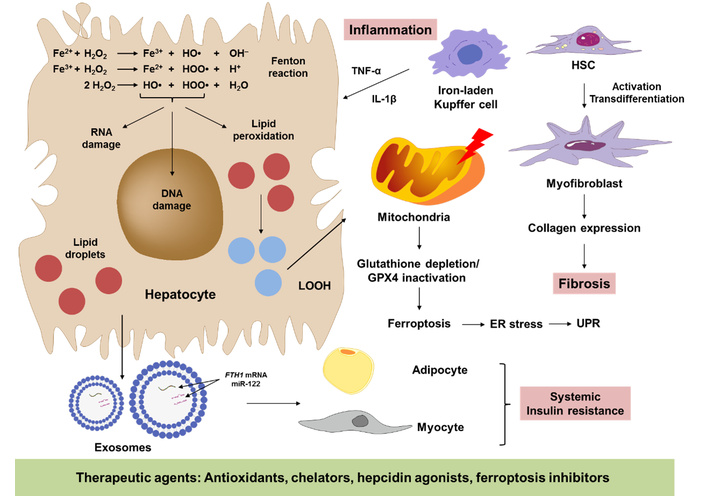
How excess iron accelerates the transition from steatosis to steatohepatitis. The pathway map integrates hepatocellular, immune, and fibrogenic events that drive the progression of metabolic dysfunction-associated steatotic liver disease (MASLD). The central hepatocyte panel depicts lipid droplet overload accompanied by Fe2+ that fuels Fenton chemistry, generating hydroxyl radicals (•OH) and lipid peroxides (LOOH). Downstream consequences shown include mitochondrial dysfunction, glutathione depletion/GPX4 inactivation leading to ferroptosis, and PERK-mediated endoplasmic reticulum (ER) stress and unfolded protein response (UPR). Surrounding panels highlight paracrine crosstalk: (i) iron-laden Kupffer cells secrete TNF-α, IL-1β, and TGF-β1, triggering inflammation, (ii) hepatic stellate cells (HSCs) activate to myofibroblasts, depositing collagen leading to fibrosis, and (iii) circulating exosomes transfer ferritin-H (FTH1) mRNA and miR-122 to distant adipocytes and myocytes, worsening systemic insulin resistance. The green block contains protective agents (antioxidants, chelators, hepcidin agonists, ferroptosis inhibitors) currently under preclinical or clinical investigation that can counteract the transition of steatosis to steatohepatitis.
Thus, iron serves as a metabolic spark that is normally controlled but becomes dangerous in the nutrient-rich, cytokine-heavy environment characteristic of metabolic dysfunction-associated steatotic liver disease (MASLD).
Moreover, iron metabolism and inflammation are intricately intertwined. On one hand, pro-inflammatory cytokines such as IL-6 transcriptionally up-regulate ferritin and hepcidin, trapping iron in macrophages. On the other hand, catalytic iron amplifies reactive oxygen species (ROS) formation and activates NF-κB, fueling the inflammatory milieu that exposes hepatocytes to lipotoxic stress. Recent work in both pre-clinical models and patients with metabolic syndrome confirms this bidirectional crosstalk: iron chelation blunts NLRP3-inflammasome activity, while hepcidin blockade exaggerates adipose tissue macrophage polarization toward a pro-inflammatory M1 state [11, 12]. Against this background, hyperferritinemia should be viewed not merely as a surrogate of inflammation but as an active participant in the inflammatory vicious circle that precipitates MASLD progression.
Serum ferritin, a 24-mer cage capable of sequestering up to 4,500 iron atoms, serves as both an indicator of iron status and an acute-phase reactant that is induced by IL-6/STAT3 and HIF-1α pathways [13]. In population studies, ferritin levels increase linearly from the first to the fifth body mass index (BMI) quintile, with notable inflections at waist circumferences greater than 102 cm (for males) and greater than 88 cm (for females) [14].
Table S1 lists 22 heterogeneous investigations, including large National Health and Nutrition Examination Survey (NHANES) datasets, multi-centre cohort studies, Mendelian-randomization analyses, and small randomized trials, demonstrating a robust, cross-platform association between hyperferritinemia and MASLD. Elevated ferritin values (typically beginning around 200–300 µg L–1) consistently track with higher MASLD prevalence, magnetic resonance imaging (MRI)-quantified steatosis, significant fibrosis, liver-related events, and in several cohorts, increased all-cause mortality. The addition of ferritin enhanced the predictive accuracy of non-invasive scores such as the fibrosis-4 score (FIB-4) and non-alcoholic fatty liver disease fibrosis score (NFS). Genetic data indicate that hyperferritinemia in MASLD clusters with metabolic risk alleles (PNPLA3-rs738409, TM6SF2-rs58542926) rather than classic HFE variants, underscoring a dysmetabolic iron phenotype dubbed “metabolic hyperferritinemia” (MHF). Early interventional evidence shows that agents such as SGLT2 inhibitors or pioglitazone lower both ferritin and hepatic fat, whereas iron-directed therapies (phlebotomy, hepcidin agonists) await larger outcome trials. The studies summarized in Table S1 underscore the concept that ferritin is not merely an inflammatory by-product but a modifiable driver and biomarker of MASLD progression, validating expert-panel calls for ferritin-based triage algorithms in routine diabetes and hepatology care. Moreover, there are significant ethnic differences [15]. African-American men have the highest median ferritin levels yet the lowest transferrin saturation, indicating iron sequestration rather than excess absorption. On the other hand, East-Asian cohorts show smaller absolute differences in ferritin levels but stronger correlations with insulin resistance. Menopause eliminates the major physiological iron sink (i.e., menses), leading to a rise in ferritin levels by approximately 70 µg L–1 until approximately 60 years and then reaching a plateau, bringing them closer to male levels [16]. This partially explains the increase in MASLD prevalence among older women.
Having heterozygosity for HFE C282Y or H63D, which is present in up to 15% to over 20% of Europeans, adds an additional 20–30 µg L–1, slightly elevating the baseline for metabolic inflammation [17]. Extensive datasets provide more detailed prognostic information. NHANES data demonstrates a stepwise, log-linear relationship between ferritin levels and controlled attenuation parameter (CAP) scores, indicating increased liver fat content, while the UK Biobank reported a significant correlation between steatotic liver disease, elevated iron, and the pathogenesis of cirrhosis [18, 19].
The construct of “MHF” will only be clinically scalable if sex- and ethnicity-specific ferritin cut-offs are prospectively validated across continents. Equally important, MRI-based iron grading, while highly reproducible, remains constrained by cost, scanner availability, and the need for trained radiologists. Cost-effectiveness analyses and simplified point-of-care algorithms are therefore urgently needed before the proposed three-tier grading system can be adopted in routine hepatology or diabetology clinics.
Even in the presence of high C-reactive protein (CRP), a potential confounder, ferritin remains independently predictive of hepatic iron on MRI-R2* and stiffness measured by elastography. Therefore, although inflammation may contribute to “noise”, the consistent and graded association of hyperferritinemia with structural liver damage suggests that iron overload or redistribution is biologically significant, rather than just a by-product.
An international expert panel has consolidated these observations into a cohesive clinical framework known as “MHF” [20]. MHF is diagnosed when serum ferritin levels exceed 300 µg L–1 in men or 200 µg L–1 in women, in the presence of fatty liver, type 2 diabetes (T2D)/obesity, or at least two other metabolic dysfunction traits, after ruling out primary iron disorders, heavy alcohol use, overt inflammation and other secondary causes of iron accumulation [20]. The panel suggests reevaluating ferritin levels after ≥ 3 months of lifestyle optimization and then categorizing iron status into three levels: Grade 1 (ferritin < 550 µg L–1 or hepatic R2* < 70 s–1), Grade 2 “dysmetabolic iron accumulation” (ferritin 550–1,000 µg L–1 or R2* 70–140 s–1) and Grade 3 “dysmetabolic iron overload syndrome” (DIOS; ferritin > 1,000 µg L–1 or R2* > 140 s–1) [20]. Non-invasive MRI-R2* is the preferred method for confirming and staging hepatic iron levels, with DIOS requiring visible iron stores ≥ 140 s–1 or equivalent biopsy evidence [20]. These agreed-upon thresholds establish a shared terminology for research and clinical practice, allowing ferritin levels to be interpreted not just as an inflammatory marker, but also as a calibrated measure of iron accumulation in the dysmetabolic liver [20].
Steatotic hepatocytes contain a mixture of saturated fatty acids (such as palmitate and stearate) and polyunsaturated fatty acids (PUFAs) such as arachidonate and linoleate [21]. Iron triggers the peroxidation of PUFAs in cells, producing reactive aldehydes such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) [22]. Studies have found that secondary lipid peroxidation products, such as MDA and 4-HNE, are significantly higher in metabolic dysfunction-associated steatohepatitis (MASH) patients compared to those with simple hepatic steatosis [23]. These aldehydes react with lysine, histidine, and cysteine residues on mitochondrial proteins, impairing β-oxidation and respiratory chain complexes [22]. This dysfunction leads to increased leakage of superoxide, exacerbating iron-induced ROS in a harmful cycle. Additionally, iron overload shifts cystine uptake towards the mevalonate pathway, depleting glutathione and deactivating GPX4, a crucial enzyme that controls lipid peroxides and allows for ferroptosis [22]. Histological features, like condensed mitochondria with ruptured outer membranes, have been observed in liver biopsies with high ferritin levels in patients with MASLD [24, 25]. Consequently, targeting mitochondrial dysfunction has been discussed as an effective therapeutic means in mitigating the progression of MASLD [26].
Apart from hepatocytes, iron-rich Kupffer cells change to a proinflammatory and profibrotic phenotype, releasing TNF-α, IL-1β and ROS that cause damage in neighboring cells [27]. Exosomes released from these macrophages may contain ferritin-heavy-chain mRNA and microvesicles loaded with miR-122, which can activate insulin-resistant genes in distant adipocytes and myocytes, demonstrating how iron imbalance contributes to overall metabolic decline, new-onset metabolic syndrome, and T2D [28, 29]. Iron also interacts with the unfolded protein response [30], as excess Fe2+ intensifies endoplasmic reticulum (ER) stress through PERK-eIF2α activation, leading to CHOP-mediated apoptosis and increasing sensitivity of hepatic stellate cells (HSCs) to TGF-β [31]. In vitro, Fe3+ ammonium citrate increases the severity in the progression of liver fibrosis in primary mouse HSCs within 24 hours, a response that can be reversed by the iron chelator deferiprone or the antioxidant N-acetylcysteine [32]. These mechanisms show how iron plays a crucial role in every stage of MASLD development, from initial oxidative damage to the formation of a fibrotic scar.
Guideline committees are increasingly recommending ferritin in first-line MASLD work-ups for predicting long-term outcomes in patients with MASLD, but nuanced interpretation is essential [33]. A pragmatic algorithm might classify ferritin levels below 300 µg L–1 for males and below 200 µg L–1 for females as low risk (to be monitored annually), and levels equal to or greater than 300 µg L–1 for males and 200 µg L–1 for females as intermediate or high risk for developing liver-related events [34]. In addition to the FIB-4 score, data from transient elastography, screening for occult inflammation, and data on transferrin saturation, or HFE genotyping data would help estimate the risk of developing MASLD-related complications. Moreover, artificial intelligence models that integrate parameters such as ferritin with age, alanine aminotransferase (ALT), platelet count, and CAP can achieve area under the receiver operating characteristic curves (AUROCs) greater than 0.85 for fibrosis stage F3 or higher, potentially improving disease prediction beyond single-marker performance [35]. Therapeutically, serial 500 mL phlebotomies every 2–3 weeks until ferritin levels drop below 100 µg L–1 can be presented as “therapeutic blood donation in iron overload conditions”, enhancing patient acceptance and local blood-bank stocks [36]. Small RCTs report histological improvement in steatosis grade and liver damage markers [ALT, aspartate aminotransferase (AST), and γ-glutamyl transferase (GGT)] after iron depletion [37]. However, other studies have shown no reduction in hepatic fat content after phlebotomy in subjects with fatty liver diseases [38].
To date, interventions such as phlebotomy and iron-modulating treatments in MASLD have produced mixed results, primarily based on surrogate outcomes. Small open-label or non-randomized studies have shown some improvement in aminotransferases and ballooning scores, but well-controlled trials have not shown a decrease in MRI-proton density fat fraction (PDFF) or histological steatosis. The varying inclusion criteria, levels of phlebotomy, and short follow-up periods likely contributed to these conflicting results, underscoring the need for large, sham-controlled RCTs with histological endpoints lasting at least 48 weeks before guideline endorsement can be supported.
Many factors influence iron intake, absorption, and individual requirements. Some dietary components either enhance or inhibit iron absorption [39]. Iron-modulating diets emphasize poultry, fish, and legumes while pairing plant-based iron with polyphenol-rich coffee or tea, and limiting vitamin C-rich beverages at main meals, to curb non-heme iron absorption [40]. Rusfertide, an injectable mini-hepcidin, induces normalization of iron parameters in patients with hemochromatosis in the absence of phlebotomies [41]. Possibly, combination therapies that pair iron reduction with GLP-1 receptor agonists, FGF-21 analogs, or PPAR-α/δ agonists aim to extinguish both caloric and oxidative sparks, a holistic approach suitable for the multifactorial pathology of MASLD.
A newly published prospective cross-sectional analysis of 523 adults aged 50–80 years with T2D offers crucial insights into how ferritin can be used in everyday MASLD care [42]. The investigators found that only one-fifth of participants met the study’s hyperferritinemia cut-off (≥ 300 ng mL-1 in men, ≥ 200 ng mL-1 in women), yet had a significant 78.5% prevalence of MASLD compared to 62.1% in normoferritinemic peers [42]. More clinically alarming, 35.5% of hyperferritinemic patients had significant fibrosis as defined by magnetic resonance elastography (MRE) compared to 22.1% in those with normal ferritin [42]. Multivariable modeling confirmed hyperferritinemia as an independent predictor of both MASLD (OR: 2.01) and significant fibrosis (OR: 2.33) after adjusting for age, sex, obesity, and insulin use [42]. Although the AUROC of ferritin for diagnosing MASLD was modest at 0.59, a ferritin threshold of < 117 ng mL-1 achieved a negative-predictive value of 82% for ruling out significant fibrosis [42]. These real-world data support expert-panel ideas of “MHF” and propose a practical two-step algorithm: (i) use serum ferritin as a cost-effective initial screen in all diabetes clinics, and (ii) refer hyperferritinemic or ferritin-indeterminate patients for quantitative imaging (vibration-controlled transient elastography (VCTE) or MRE) to detect silent but actionable fibrosis [42]. Implementing this strategy could direct up to one-third of high-risk patients towards earlier lifestyle, pharmacologic, or iron-modulating interventions while avoiding unnecessary specialist evaluations for low-risk individuals.
The next frontier is to prove that intentional iron modulation improves hard clinical outcomes, such as fibrosis regression, cirrhosis prevention, and reducing the risk of hepatocellular carcinoma (HCC), rather than simply normalizing laboratory indices. Longitudinal cohorts that combine quarterly ferritin measurements with MRI-PDFF, MRE, and circulating lipid-peroxidation panels (F2-isoprostanes, oxidized-LDL, 4-HNE) could help elucidate whether ferritin kinetics parallel histologic changes [42]. Multi-omics consortia like Liver Investigation: Testing Marker Utility in Steatohepatitis (LITMUS) and Non-invasive Biomarkers of Metabolic Liver Disease (NIMBLE) have already or will incorporate iron modulators (HAMP, FTL, FTH1), proteomics (ceruloplasmin, hemopexin) and metabolomics (cis-aconitate, 2-oxoglutarate) to stratify “iron-addicted” endotypes belonging to different diseases or conditions induced by distinct pathophysiological mechanisms [43, 44]. Refining polygenic risk scores, proposed for MASLD [45], may reveal common reference single-nucleotide polymorphisms (SNPs) cluster identifications (rsIDs) in SLC40A1 (ferroportin) or TMPRSS6 (matriptase-2) that identify lean MASLD patients with disproportionate iron burden.
Pediatric MASLD, often originating in utero via maternal over-nutrition, raises the intriguing question of whether prenatal iron status programs later hepatic susceptibility [46], a hypothesis testable in birth cohorts with neonatal dried-blood-spot ferritin data. Economically, ferritin measurement is inexpensive and can be easily done in human serum or plasma [47]. It is already collected for blood-donor screening, facilitating population-wide surveillance without new infrastructure [48]. Finally, cross-disciplinary clinics that unite hepatologists, endocrinologists, cardiologists, and nephrologists can treat hyperferritinemia as a shared pathophysiologic thread running through fatty liver disease, T2D, heart failure, and chronic kidney disease, rather than as siloed “organ-specific” phenomena. Turning this integrative vision into reality could transform ferritin from a passive biomarker into an actionable fulcrum in precision hepatology, capable of extinguishing the metabolic wildfire before it consumes the liver. Nevertheless, there are still concerns that need to be taken into account when using ferritin as a biomarker for predicting the long-term prognosis of patients with MASLD. Factors to be considered are potential sex-specific differences, inflammatory status, method-dependent variations in the determination of ferritin, and the impact of medications impacting baseline serum ferritin levels [49]. Moreover, it should be kept in mind that there are also findings that advise against iron depletion as a therapeutic means in MASLD, suggesting that further studies are needed to unravel the complex interplay between iron status and MASLD development [50].
Despite the consistent association between serum ferritin, MRI-derived iron indices, and hepatic outcomes, most published data remain observational. Reverse causality (i.e., inflammation leading to increased ferritin levels resulting in liver injury) and residual confounding factors such as obesity, insulin resistance, or alcohol intake cannot be fully excluded, even in propensity-matched or Mendelian-randomization analyses. Moreover, ferritin exhibits substantial biological variability (diurnal, menstrual, and assay-platform related), which may weaken risk estimates. Therefore, firm causal inference awaits adequately powered randomized trials, where intentional iron reduction is the primary exposure and histological fibrosis regression is the primary endpoint.
Iron metabolism is at the intersection of energy homeostasis, oxidative stress, and innate immunity, three axes that converge in MASLD. When there is a caloric overload, chronic inflammation, or genetic variants that tip the balance towards iron sequestration and overload, even a slight increase in ferritin levels can signal a cascade of hepatocellular lipid peroxidation, ferroptosis, and fibrogenesis that increases metabolic risk. Robust population studies and expert guidelines now recognize “MHF” as a clinically relevant condition. Proof-of-concept trials indicate that treatments like phlebotomy, hepcidin mimetics, and iron-conscious diets can help mitigate this harmful cycle. However, there is still no definitive evidence that intentional iron regulation can change the course of MASLD, slowing fibrosis, preventing cirrhosis, and reducing the risk of HCC. Future research should include regular ferritin measurements, MRI-based iron assessments, and lipid peroxidation biomarkers in longitudinal studies to identify individuals with “iron-addicted” traits who may benefit most from treatment. Integrating hepatology, endocrinology, and transfusion medicine in patient care pathways could turn ferritin into a tool for precise hepatology, stopping the metabolic damage to the liver before it becomes irreversible.
4-HNE: 4-hydroxynonenal
ALT: alanine aminotransferase
AUROCs: area under the receiver operating characteristic curves
CAP: controlled attenuation parameter
DIOS: dysmetabolic iron overload syndrome
Fe2+: ferrous
Fe3+: ferric
FIB-4: fibrosis-4 score
HCC: hepatocellular carcinoma
HSCs: hepatic stellate cells
MASLD: metabolic dysfunction-associated steatotic liver disease
MDA: malondialdehyde
MHF: metabolic hyperferritinemia
MRE: magnetic resonance elastography
MRI: magnetic resonance imaging
NHANES: National Health and Nutrition Examination Survey
PDFF: proton density fat fraction
PUFAs: polyunsaturated fatty acids
ROS: reactive oxygen species
T2D: type 2 diabetes
The supplementary table for this article is available at: https://www.explorationpub.com/uploads/Article/file/100599_sup_1.pdf.
The author is grateful to Sabine Weiskirchen (IFMPEGKC, RWTH University Hospital Aachen) for her assistance in creating the figures for this perspective.
RW: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. The author read and approved the submitted version.
Ralf Weiskirchen, who is the Guest Editor of Exploration of Digestive Diseases, had no involvement in the decision-making or the review process of this manuscript. The author declares that there are no other conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The author is supported by the German Research Foundation [WE2554/17-1], the German Cancer Aid [70115581], and the Interdisciplinary Centre for Clinical Research within the Faculty of Medicine at RWTH Aachen University [PTD 1-5]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3031
Download: 30
Times Cited: 0
Amedeo Lonardo
Amar Tebaibia ... Nadia Oumnia
Cristina Felicani ... Pietro Andreone
Rudy El Asmar ... Samer AlMasri
Vincenzo Giorgio Mirante
Giampiero Francica, Cristiano Giardiello
