 Review
Review

Affiliation:
Department of Experimental and Clinical Medicine, University of Florence, I50134 Florence, Italy
Affiliation:
Department of Experimental and Clinical Medicine, University of Florence, I50134 Florence, Italy
ORCID: https://orcid.org/0000-0001-8629-0878
Affiliation:
Department of Experimental and Clinical Medicine, University of Florence, I50134 Florence, Italy
Email: chiara.raggi@unifi.it
ORCID: https://orcid.org/0000-0003-2473-3535
Explor Dig Dis. 2025;4:1005100 DOI: https://doi.org/10.37349/edd.2025.1005100
Received: August 22, 2025 Accepted: October 21, 2025 Published: November 11, 2025
Academic Editor: Derek A. Mann, University of Newcastle, United Kingdom
The article belongs to the special issue Fibrosis and Hepatobiliary Cancer
Recent studies demonstrate that peripheral innervation and Schwann cells (SCs) activity within the tumor microenvironment (TME) have a significant influence on liver cancer, specifically hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA). Even though the role of SCs in HCC behavior is only hypothetical, neurotrophic factors like brain-derived neurotrophic factor (BDNF) and artemin (ARTN) enhance tumor growth, angiogenesis, and metastasis. Thus, targeting these pathways has a promising therapeutic potential. Conversely, receptors such as glial cell line-derived neurotrophic factor family receptor α-1 (GFRα1) and factors like neurotrophin-3 (NTF3) exhibit tumor-suppressive roles, indicating a context-dependent, dual impact of neurotrophic signaling in liver cancer. In iCCA, perineural invasion (PNI) correlates with poor prognosis, with SCs promoting tumor progression through the secretion of neurotrophic factors such as nerve growth factor (NGF) and BDNF, which activate signaling pathways downstream. These pathways facilitate epithelial-mesenchymal transition (EMT), invasion, and neural infiltration. This review emphasizes the complex roles of neurotrophic factors in hepatic tumors and their future potential as biomarkers and therapeutic targets via disruption of tumor-nerve interactions.
In recent years, accumulating evidence indicates that the tumor microenvironment (TME) of many types of cancer is influenced by peripheral innervation, and cancer cells can exploit biological actions related to the peripheral nervous system (PNS) to increase their malignant characteristics. Perineural invasion (PNI) is the process by which cancer cells infiltrate nerve sheaths, using nerves as pathways for spread and contributing to disease progression, as shown by the correlation between PNI and poorer survival [1, 2]. In parallel, recent advances in artificial intelligence are setting a new paradigm in hepatic pathology, as demonstrated by generative adversarial networks (GANs)-based virtual staining of hematoxylin and eosin slides to generate Masson trichrome images [3] and by transformer architectures such as the Deep Enhanced Swin Transformer (DEST) for refined histological scoring [4]. These pioneering approaches hold great promise for paving the way toward more accurate and standardized characterization of the hepatic TME, with significant potential to enhance the investigation of neurobiological interactions and PNI in the context of liver cancers. Schwann cells (SCs), the most abundant glial cells in the PNS, play key roles in immune regulation and tissue repair, and interact with tumor cells to shape a cancer-supporting TME. The interaction between SCs and cancer cells occurs through multiple signaling pathways and may create a microenvironment that facilitates tumor cell migration along nerves [5]. Understanding the interactions between SCs and cancer cells within the TME can uncover potential therapeutic targets to disrupt this invasive pathway and improve patient outcomes, limiting cancer spread and tumor-related nerve damage.
Following peripheral nerve injury, both myelinating and non-myelinating SCs dedifferentiate into “repair SCs” (rSCs). Transition to a repair phenotype represents a form of adaptive reprogramming, characterized by significant gene expression and phenotypic changes, most notably, the downregulation of myelin basic protein (MBP), myelin protein zero (MPZ), myelin-associated glycoprotein (MAG) and early growth response protein 2 (EGR2) genes, along with the upregulation of specific genes like brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), nerve growth factor (NGF), artemin (ARTN), NGF receptor (NGFR), neural cell adhesion molecule (NCAM1), and glial fibrillary acidic protein (GFAP), both primarily driven by activation of the c-Jun pathway, that act as the master regulator of SCs’ repair phenotype [6]. To provide a concise overview of the molecular profile associated with rSCs, Table 1 summarizes the main markers reported to be downregulated or upregulated during the repair phenotype transition.
rSCs genetic markers overview.
| Downregulation | Upregulation |
|---|---|
| Myelinating markers’ genes: MBP, MPZ, MAG, EGR2 | Neurotrophic factors’ genes: GDNF, ARTN, BDNF, NTF3, NGFTransmembrane proteins’ genes: NGFR, CDH2Other molecular markers’ genes: JUN, GFAP |
The table summarizes the downregulation of myelinating markers (MBP, MPZ, MAG, EGR2) and the upregulation of neurotrophic factors’ genes (GDNF, ARTN, BDNF, NTF3, NGF); transmembrane proteins’ genes (NGFR, CDH2); and other molecular markers’ genes (the master transcriptional regulator JUN, GFAP) during rSC reprogramming. rSCs: repair Schwann cells; MBP: myelin basic protein; MPZ: myelin protein zero; MAG: myelin-associated glycoprotein; EGR2: early growth response protein 2; GDNF: glial cell line-derived neurotrophic factor; ARTN: artemin; BDNF: brain-derived neurotrophic factor; NTF3: neurotrophin-3; NGF: nerve growth factor; NGFR: NGF receptor; GFAP: glial fibrillary acidic protein.
In various types of cancer, nerve damage or invasion by tumor cells leads to an increase in GFAP-positive SCs, reflecting a high abundance of rSCs that can severely affect tumor progression. In the TME, rSCs show a high paracrine activity, releasing a high number of soluble factors involved in these pro-tumoral events [7].
Hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA) are the two major forms of liver cancer. HCC is the most common form of primary liver malignancy and a leading cause of cancer-related mortality worldwide. Chronic liver disease and cirrhosis are the primary risk factors for its development, making surveillance essential [8]. On the other hand, CCA is a rare but highly aggressive cancer that develops from the epithelial cells within the bile ducts. About 10% of CCAs are classified as intrahepatic CCA (iCCA), i.e., developing from bile ducts located beyond the second-order branches. CCA currently accounts for 15% of primary liver cancers, and surgery offers the best potential for cure for these patients, with five-year survival rates reaching up to 40% after resection. However, only a small number of patients are candidates for surgery, and many experience a recurrence of the disease. This highlights the critical need for more effective therapeutic approaches to be used alongside surgery to improve patients’ survival [9, 10]. Ongoing research is vital to enhancing therapies and improving the survival rates of patients with these forms of liver cancer.
This review emphasizes the role of the neural microenvironment in the regulation of the malignant features of HCC and iCCA. It integrates findings from in vitro cellular models, in vivo preclinical studies, and clinical investigations based on human tumor specimens, providing a comprehensive overview of the role of neurotrophic signaling and nerve-tumor interactions in these two types of liver cancer. Since in liver cancer research, the evidence for the neuronal involvement is still at an early stage, no clinical trials specifically addressing neurotrophic signaling have yet been reported, and the exploration of novel therapeutic targets will be required in the upcoming years, since no inhibitors of neurotrophic factors or their receptors have yet been properly tested in this context. In particular, the biological effects of the neurotrophic factors released within HCC TME, the key role of SCs in orchestrating iCCA TME dynamics, as well as the involvement of PNI in tumor progression and dissemination, are highlighted.
It is important to underline that, unlike the iCCA, in HCC the available evidence predominantly concerns neurotrophic factors expressed by tumor cells themselves or more generally attributed to the TME. To date, no experimental evidence is available on the co-localization of SC markers and neurotrophic factors in human HCC specimens, and there are no functional paracrine studies demonstrating a direct contribution of SCs. Therefore, the involvement of SCs in HCC and, in particular, the contribution of their repair phenotype currently remains only hypothetical. Future studies employing single-cell/spatial transcriptomics, SCs-HCC co-culture systems, and clinical-prognostic correlations will be essential to clarify this biological aspect.
In HCC, a role for neurotrophic factors is increasingly recognized, and the PNS is believed to exert significant influence over tumor behavior. PNS has been found to influence cancer cell proliferation, metastasis, immune evasion, and therapeutic resistance through the secretion of different soluble factors, including neurotrophic factors [11]. In a previous study [12], bone metastases derived from HCC exhibited significantly higher neural density compared to primary hepatic tumors. Immunohistochemical analysis of formalin-fixed, paraffin-embedded tissue samples (n = 13) with synchronous or metachronous bone metastases revealed increased perineural involvement in metastatic sites. These findings suggest a potential role for PNI in facilitating the metastatic spread of HCC to bone tissue. Previous findings [13] have shown that BDNF plays a pro-tumoral role in HCC, as indicated by the effects of lupeol, a natural compound, which significantly reduced the proliferation of the HCC cell line, HCCLM3, in a time- and dose-dependent fashion. Moreover, lupeol exerted caspase-dependent pro-apoptotic effects, mainly involving caspase-3. The compound suppressed the secretion of BDNF in the extracellular medium, and this reduction was accompanied by decreased phosphorylation of GSK-3β at serine 9, indicating its reactivation. At the transcriptional level, lupeol downregulated key genes of the PI3K/AKT and Wnt pathways, including AKT1, PIK3CA, β-catenin, c-Myc, and cyclin D1. These results suggest that lupeol promotes apoptosis in HCC cells by inhibiting BDNF secretion, reactivating GSK-3β, and attenuating signaling cascades promoting survival.
In parallel, the BDNF/tropomyosin receptor kinase B (TrkB) signaling axis has emerged as a key driver of tumor angiogenesis and growth. Murine endothelial cells engineered to overexpress BDNF displayed enhanced proliferation, migration, invasion, and resistance to apoptosis. In an in vivo co-transplantation model, these cells promoted tumor vascularization and growth when injected with transformed liver cells. Conversely, BDNF knockdown markedly reduced these effects. In clinical samples of HCC, overexpression of BDNF and TrkB was observed in 46.0% and 33.3% of cases, respectively, and high TrkB levels were associated with shorter overall survival [14]. These findings agree with a previous study [15] that analyzed human HCC specimens and found high BDNF expression in 63.1% of cases and TrkB positivity in 55.4%. These markers significantly correlated with advanced disease stage and intrahepatic multifocality. In functional studies, neutralizing antibodies against BDNF using or blockade of TrkB with the tyrosine kinase inhibitor K252a, induced apoptosis and suppressed invasion in HepG2 and HCCLM3 HCC cell lines, confirming the role of the BDNF/TrkB axis in HCC cell survival and invasiveness.
Other neurotrophic factors are involved in HCC pathobiology, especially under hypoxia, and contribute to cancer stemness. High-level expression of ARTN, a ligand for RET interacting with GFR-α3, correlates with larger tumor size, increased rate of recurrence, and poorer survival. In vitro, ARTN overexpression promotes proliferation, reduces apoptosis, enhances epithelial-mesenchymal transition (EMT), and increases migration and invasion of HCC cells. In vivo, it leads to larger tumors and more metastases. ARTN also boosts tumor sphere formation and expands the CD133⁺ cancer stem cell population, underscoring its role in tumor progression and stemness. Mechanistically, ARTN expression is induced under hypoxic conditions through HIF-1α-mediated transcriptional regulation. Its downstream oncogenic effects were largely dependent on activation of the AKT signaling pathway, because its inhibition abrogated ARTN-induced EMT and cancer stem cell expansion [16].
Based on these findings, inhibitory strategies directed to neurotrophic receptors in HCC are being developed. The dual inhibitor Indo5, which blocks the Trk and c-Met pathways, decreased cell proliferation and increased apoptosis markers like cleaved caspase-3 and PARP in vitro. In vivo, Indo5 treatment caused tumor shrinkage in mice with Huh7 xenografts, with minimal side effects, and reduced cell proliferation, as indicated by decreased Ki-67 staining. Combining Indo5 with sorafenib enhanced anti-tumor effects [17]. Based on these findings, Indo5 appears as a promising candidate to be evaluated for the treatment of HCC. Because neurotrophic signaling is involved in PNI, this approach may also help to prevent tumor spread along nerve fibers.
Interestingly, some neurotrophic receptors may act as tumor suppressors in HCC. GDNF family receptor α-1 (GFRα1), a glycosylphosphatidylinositol-linked cell surface receptor for GDNF and neurturin, that mediates activation of the RET tyrosine kinase receptor, is often downregulated in HCC tissues, with lower levels linked to worse prognosis. In vitro, knocking down GFRα1 increased cell proliferation, while overexpression inhibited growth, and in mice, GFRα1-deficient tumors grew larger. It was found that loss of GFRα1 activates Wnt/β-catenin signaling, which promotes tumor progression, and blocking this pathway reverses the effect. Of note, recombinant GDNF inhibited proliferation only in cells expressing GFRα1, and co-expression of GFRα1 and GDNF was associated with better outcomes. Moreover, GFRα1 deficiency increased the sensitivity to oxaliplatin. Therefore, GFRα1 functions as a tumor suppressor and may facilitate the response to chemotherapy in HCC [18].
Along these lines, neurotrophin-3 (NTF3) expression was significantly reduced in HCC tissues compared to normal liver tissues [19]. Low NTF3 levels correlated with shorter overall and disease-free survival and were identified as an independent prognostic factor.
Additional evidence indicates that NTF3 may function as a tumor suppressor in HCC. Gene-expression analyses using Gene Set Enrichment Analysis (GSEA) on publicly available datasets (e.g., GEO and TCGA) and qRT-PCR in 74 patient samples showed that NTF3 is consistently downregulated in tumors, and lower levels are significantly associated with poorer clinical outcomes. Functional studies confirmed that NTF3 inhibits tumor progression both in vitro and in vivo. NTF3 is transcriptionally regulated by c-Jun and promotes apoptosis via activation of the JNK and p38 MAPK pathways. These results suggest that NTF3 acts as a tumor suppressor in HCC [20]. It should be noted that, while GFRα1 and NTF3 consistently act as tumor suppressors in HCC according to current evidence, no studies have yet addressed potential context-dependent mechanisms leading to opposite effects, such as epigenetic regulation, splicing variation, or receptor co-expression. This represents a gap in the literature and a priority for future investigation.
All the aforementioned neurotrophic factors-mediated biological effects are illustrated in Figure 1 and listed in Table 2.
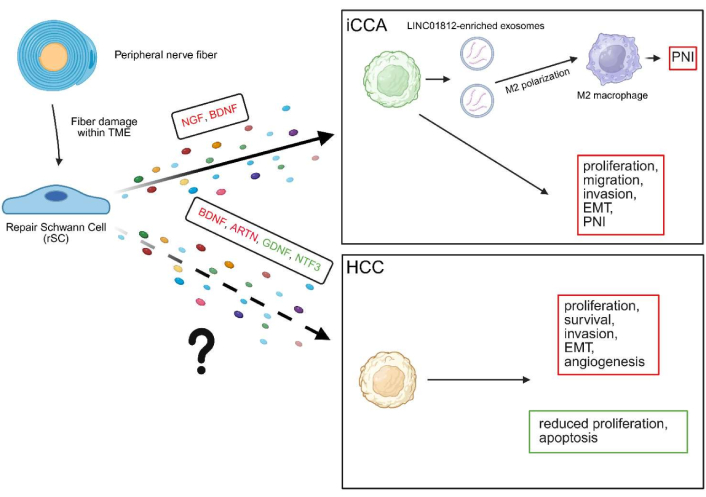
Biological effects of neurotrophic factors in iCCA and HCC. While in iCCA the impact of repair Schwann cells (rSCs)-derived neurotrophic factors is partially elucidated, in HCC there is no evidence concerning the direct influence of these factors, although various neurotrophic factors generally attributed to this tumor’s TME or directly released by cancer cells induce important biological effects. Pro-tumoral factors and effects are shown in red, while antitumoral ones are in green. The solid arrow indicates the established role of neurotrophic factors derived from rSCs, whereas the dashed arrow denotes a hypothetical role that has not yet been demonstrated. TME: tumor microenvironment; ARTN: artemin; BDNF: brain-derived neurotrophic factor; GDNF: glial cell line-derived neurotrophic factor; HCC: hepatocellular carcinoma; iCCA: intrahepatic cholangiocarcinoma; NGF: nerve growth factor; NTF3: neurotrophin-3; PNI: perineural invasion; EMT: epithelial-mesenchymal transition. Created in BioRender. Campani, C. (2025) https://www.biorender.com/ne5njqe.
A summary of neurotrophic factor-mediated biological effects in HCC and iCCA.
| Cancer | Neurotrophic factor | Biological outcomes | References |
|---|---|---|---|
| HCC | BDNF | BDNF promotes HCC progression by activating PI3K/AKT and Wnt signaling, enhancing proliferation, angiogenesis, and resistance to apoptosis. Lupeol suppresses BDNF secretion and reactivates GSK-3β, reducing tumor cell survival. Clinically, high BDNF/TrkB expression correlates with advanced stage, multifocality, and poor prognosis; its inhibition induces apoptosis and limits invasion. | [13–15] |
| ARTN | ARTN is upregulated by hypoxia via HIF-1α and promotes HCC progression through AKT-mediated EMT, stemness, and survival. Its overexpression enhances proliferation, invasion, and tumor-initiating capacity, and correlates with larger tumors, higher recurrence, and poor prognosis. | [16] | |
| INDO5 | INDO5, a dual Trk/c-Met inhibitor, suppresses HCC cell growth by reducing AKT and ERK signaling and activating apoptosis via caspase-3 and PARP cleavage. In vivo, it decreases tumor size and proliferation, with enhanced efficacy when combined with sorafenib, highlighting its therapeutic potential in targeting neurotrophic signaling in HCC. | [17] | |
| GFRα1-GDNF | GFRα1 acts as a tumor suppressor in HCC; its downregulation promotes proliferation via Wnt/β-catenin activation, while its expression enables GDNF-mediated growth inhibition. Low GFRα1 levels correlate with poor prognosis and increased sensitivity to oxaliplatin, suggesting its dual role as a prognostic and predictive marker. | [18] | |
| NTF3 | NTF3 is downregulated in HCC and correlates with poor prognosis; functionally, it acts as a tumor suppressor by promoting apoptosis and limiting tumor progression via JNK and p38 MAPK activation. Its expression is regulated by the c-Jun transcriptional factor and may serve as both a prognostic marker and therapeutic target. | [19, 20] | |
| iCCA | NGF | SCs-derived NGF promotes iCCA progression by enhancing tumor cell proliferation, migration, invasion, and PNI. These effects are mediated via TrkA signaling and are associated with EMT induction. Furthermore, high NGF/TrkA expression predicts poor prognosis and enhances proliferation and invasion via MAPK and PI3K/AKT signaling pathways. | [25, 26] |
| BDNF | BDNF expression in iCCA correlates with PNI and poor prognosis; functionally, it promotes tumor cell invasion in an autocrine/paracrine manner. | [27] | |
| LINC01812 | iCCA-derived exosomal lncRNA LINC01812 induces M2 macrophage polarization via TUBB4B/Notch/CCL2 signaling and increases the secretion of NGF and BDNF, which promote iCCA cell migration toward nerves and enhance PNI both in vitro and in vivo. | [28] |
ARTN: artemin; BDNF: brain-derived neurotrophic factor; GDNF: glial cell line-derived neurotrophic factor; HCC: hepatocellular carcinoma; iCCA: intrahepatic cholangiocarcinoma; NGF: nerve growth factor; NTF3: neurotrophin-3; EMT: epithelial-mesenchymal transition; lncRNA: long non-coding RNA; PNI: perineural invasion; TrkA: tropomyosin receptor kinase A; TrkB: tropomyosin receptor kinase B; GFRα1: GDNF family receptor α-1; TUBB4B: tubulin beta-4B chain; SCs: Schwann cells.
iCCA is considered a neurotropic cancer due to the high incidence of PNI, which is observed in approximately 75% of cases and is associated with reduced overall survival [2, 21]. KRAS mutation has been identified in 22% of a large iCCA cohort (n = 86) and showed a strong phenotypic association with PNI [22]. In this context, it has been shown that SCs play a pivotal role in modulating the TME. Indeed, previous studies indicate that SCs promote iCCA aggressiveness, as indicated by reduced expression of cytokeratin-7 in tumor cells invading nerves, an indication of partial EMT. Additionally, in a 3D in vitro model, iCCA cells were found to actively migrate toward nerve explants and to establish contact with SCs. These SCs secrete soluble factors that enhance tumor cells’ migration, invasion, proliferation, and survival. Such effects were observed using indirect co-culture methods and were associated with a cadherin switch, where expression of E-cadherin was replaced by N-cadherin. Interestingly, SB-431542, an antagonist of TGFβ receptor 1, reduced migration and invasion, indicating that TGFβ is a key driver in inducing EMT in this context. Proteomic analysis showed that SCs-derived factors induced upregulation of oncogenes and downregulation of tumor suppressor genes, further supporting the role of SCs in driving iCCA progression. In particular, proteins associated with EMT, migration, and invasion [i.e., collagen α-1 (VII) chain, nexin 1, neurogenic locus notch homolog protein 3], proliferation and survival [i.e., cytoplasmic dynein 1 intermediate chain 1, protein 4.1, NADH dehydrogenase (ubiquinone) 1 alpha subcomplex subunit 1], or with both processes (i.e., β-enolase 3, serine/threonine-protein phosphatase 2A catalytic subunit α isoform) were identified [23].
In tumor sections from CCA patients stained with S100, a SC specific marker, SCs were present with weak to moderate signal intensity in 5 out of 14 cases. Elevated levels of S100 correlated with shorter recurrence-free survival, implying a possible role for SCs in promoting disease progression. In a complementary in vitro study, wound healing assays using co-cultures of SCs and iCCA cells showed that SCs were primarily responsible for closing the gap and migrating toward the tumor cells. Therefore, the presence of iCCA cells significantly increases SCs’ motility [24]. This mutual interaction, in which iCCA cells attract SCs and then benefit from the signals they produce, reinforces the notion that SCs’ infiltration is not simply a consequence of tumor spread, but may actively promote PNI.
Additional studies have focused on the molecular mechanisms related to neurotrophic signaling, such as the contribution of SCs-derived NGF in iCCA. Several lines of evidence [25] indicate that SCs accumulate in PNI-positive iCCA tissues and that this accumulation is associated with reduced patient survival. In vitro, conditioned medium of iCCA cells promoted proliferation and migration of SCs, and transcriptomic data showed upregulation of NGF expression after co-culture. NGF secreted by SCs enhanced iCCA cell proliferation, migration, and invasion, and these effects were replicated by recombinant NGF and suppressed by specific inhibitors. Mechanistically, the effects of NGF were mediated by tropomyosin receptor kinase A (TrkA) and involved in EMT. In vivo, NGF-treated iCCA cells formed larger and heavier tumors and showed more PNI when injected into NOD-SCID mice. Although compelling, these results require further validation using human SCs.
Furthermore, NGF and TrkA expression was assessed by immunohistochemistry in tumor samples (n = 83), revealing that their high co-expression represents an independent prognostic factor significantly associated with poor differentiation, intraneural invasion, and reduced survival, while functional assays demonstrated that NGF/TrkA signaling promotes cancer cells’ proliferation and invasion through activation of MAPK and PI3K/AKT pathways [26].
SCs are known to release a range of neurotrophic molecules [6]. Among these, BDNF has drawn attention for its relevance in the context of iCCA. This association was explored in a previous study, which aimed to clarify the relationship between BDNF levels and both invasiveness and PNI. In a cohort of patients with iCCA, BDNF was mainly localized within the cytoplasm of cancer cells, and its expression was significantly correlated with PNI. Patients with higher BDNF levels tended to have worse clinical outcomes. iCCA cells silenced for endogenous BDNF showed impaired ability to invade the matrix in Matrigel transwell assays. Furthermore, adding recombinant human BDNF to the system enhanced invasiveness [27]. Given its secretory nature, BDNF may allow tumor cells to promote their own invasiveness through autocrine or paracrine activation of its receptor. These data support the role of BDNF as a marker of aggressive disease and also as a contributing driver of neural infiltration in iCCA.
Taken together, these results suggest that SCs are not passive bystanders but active contributors during iCCA progression, providing a plausible cellular explanation for the high frequency of PNI observed in this disease.
Signals originating from the tumor itself represent another mechanism through which the composition of the TME and PNI may be modulated. iCCA cells may actively promote PNI through macrophage modulation. The long non-coding RNA (lncRNA) LINC01812, secreted by the tumor and enriched in exosomes, was shown to be internalized by THP-1 macrophages and to interact with tubulin beta-4B chain (TUBB4B), favoring macrophage polarization toward an immunosuppressive M2 phenotype. This M2 shift was mechanistically mediated by activation of the Notch signaling pathway and by upregulation of CCL2. M2-polarized macrophages, stimulated by LINC01812-rich exosomes, released NGF and BDNF, two neurotrophic factors known to facilitate neural invasion. Furthermore, in vitro assays showed enhanced migration of iCCA cells toward dorsal root ganglia when exposed to LINC01812-containing exosomes, and in vivo xenograft models where iCCA cells were co-injected with M2 macrophages pretreated with LINC01812-rich exosomes, demonstrated increased tumor size and significantly higher incidence of PNI [28, 29]. Together, these results reveal a new functional axis that facilitates immune-supported neural infiltration in iCCA and may serve as a future therapeutic target.
All the aforementioned neurotrophic factors-mediated biological effects are illustrated in Figure 1 and listed in Table 2.
The dynamic interplay between neural microenvironment, SCs activity, and associated signaling considerably shapes the progression and invasiveness of liver cancers such as HCC and iCCA. While certain neurotrophic factors promote malignant behavior and neural infiltration, others act as tumor suppressors, reflecting the complexity of neuro-immune-tumor interactions. Targeting these pathways offers promising therapeutic potential, either by inhibiting pro-tumoral neurotrophic signals or by restoring tumor-suppressive mechanisms. Future research should focus on elucidating the precise molecular mechanisms governing these interactions to develop more effective, targeted treatments that can hinder tumor progression and dissemination through nerve pathways, ultimately improving patient outcomes.
ARTN: artemin
BDNF: brain-derived neurotrophic factor
CCA: cholangiocarcinoma
DEST: Deep Enhanced Swin Transformer
EGR2: early growth response protein 2
EMT: epithelial-mesenchymal transition
GANs: generative adversarial networks
GDNF: glial cell line-derived neurotrophic factor
GFAP: glial fibrillary acidic protein
GFRα1: glial cell line-derived neurotrophic factor family receptor α-1
GSEA: Gene Set Enrichment Analysis
HCC: hepatocellular carcinoma
iCCA: intrahepatic cholangiocarcinoma
MAG: myelin-associated glycoprotein
MBP: myelin basic protein
MPZ: myelin protein zero
NGF: nerve growth factor
NGFR: nerve growth factor receptor
NTF3: neurotrophin-3
PNI: perineural invasion
PNS: peripheral nervous system
rSCs: repair Schwann cells
SCs: Schwann cells
TME: tumor microenvironment
TrkA: tropomyosin receptor kinase A
TrkB: tropomyosin receptor kinase B
TUBB4B: tubulin beta-4B chain
Prof. Raggi is a member of the European Network for the Study of Cholangiocarcinoma (ENSCCA) and participates in the initiatives COST Action EURO-CHOLANGIO-NET and Precision-BTC-Network granted by the COST Association (CA18122, CA22125).
LM: Conceptualization, Writing—original draft. FM: Writing—review & editing. CR: Writing—review & editing, Supervision, Funding acquisition. All authors read and approved the submitted version.
Fabio Marra and Chiara Raggi, who are the Guest Editors and Editorial Board Members of Exploration of Digestive Diseases, had no involvement in the decision-making or the review process of this manuscript. The other author declares no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was provided by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) [IG23117]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3495
Download: 26
Times Cited: 0
Patrizia Pontisso, Maurizio Parola
Siva Bala Subramaniyan, Balasubramaniyan Vairappan
Anna Fichera, Mirella Fraquelli
