 Review
Review

Affiliation:
1Department of Translational Research, College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
Affiliation:
1Department of Translational Research, College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
Affiliation:
2School of Medicine, University of California, Irvine, CA 92617, USA
Affiliation:
1Department of Translational Research, College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
Email: vrai@westernu.edu
ORCID: https://orcid.org/0000-0001-6286-2341
Explor Dig Dis. 2026;5:1005126 DOI: https://doi.org/10.37349/edd.2026.1005126
Received: March 13, 2026 Accepted: May 06, 2026 Published: June 16, 2026
Academic Editor: Nahum Méndez-Sánchez, National Autonomous University of Mexico, Mexico
The brain–gut axis, first described in the 19th century, refers to the complex bidirectional communication network between the central nervous system and the gastrointestinal tract. This dynamic system operates through neuronal, endocrine, and immune pathways. It has since expanded to include the influence of gut microbiota, given its significant role in gut motility disorders and neurological diseases. The intricate relationship involves multiple signaling mechanisms, including toll-like receptors, nuclear factor-kappa B, α-synuclein, the hypothalamic–pituitary–adrenal axis, and vagal signaling. Dysregulation of the brain–gut axis has been implicated in numerous neurological conditions, including Parkinson’s disease, stroke, multiple sclerosis, autism spectrum disorder, spinal cord injury, and peripheral neuropathies, many of which present with well-recognized gastrointestinal manifestations. Conversely, neurological sequelae are frequently associated with primary gastrointestinal disorders such as inflammatory bowel disease, celiac disease, and hepatic failure. This narrative literature review aims to examine the epidemiology, clinical presentation, and pathogenesis of common neurological and gastrointestinal diseases through the lens of the brain–gut axis. By highlighting the interconnected metabolic, immune, and physiological mechanisms underlying these conditions, this review seeks to promote a more integrated understanding of disease processes and to support improved diagnostic strategies, therapeutic approaches, and long-term patient outcomes.
The concept of the brain–gut axis has evolved since the 19th century and is rooted in the shared embryologic development and functional interdependence of the central nervous system (CNS) and gastrointestinal tract [1]. More recently, this framework has expanded to include the gut microbiota, demonstrating how bacterial organisms activate immune cells and influence the bidirectional communication that exists between the CNS and gastrointestinal tract [2]. This communication is highly complex and dependent on both cellular and molecular signals, but is fundamentally divided into three pathways: neuronal, systemic, and immune [3]. The neuronal pathway describes how the autonomic nervous system regulates gastrointestinal motility, secretion, circulation, and sensation through the balanced actions of the sympathetic and parasympathetic systems, primarily mediated by the vagus nerve. The immune pathway explains how CNS stress or injury alters gastrointestinal structure and the gut microbiome, activating immune cells by migrating back to the CNS. In fact, autoimmune disease states of the CNS have been shown to cause migration of IgA plasma cells from the intestine back to the brain and spinal cord. The systemic pathway encompasses dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis, causing the release of various downstream substrates, including glucocorticoids, altering gastrointestinal function. These mechanisms play a key role in the gastrointestinal manifestations of neurologic diseases and vice versa, which are discussed throughout this article. By highlighting the clinical perspectives and complexities of the brain–gut axis, this narrative review aims to provide a deeper understanding of CNS–gastrointestinal interactions and future therapeutic strategies that move beyond symptom-focused care toward approaches addressing the underlying mechanisms of disease [3]. Understanding the gut–brain axis is crucial because it reveals that gut microbiota dysbiosis significantly drives neurological disorders via immune, neural, and metabolic pathways. To understand this, it is important to understand etiology, mechanisms, and risk factors.
This narrative review aims to synthesize current literature on the brain–gut axis and provide a concise resource for researchers and clinicians interested in this field. A literature search was conducted in January 2025 using the database PubMed and supplemented by reference lists of relevant articles. Search terms included combinations of “Parkinson’s disease,” “gut microbiota,” “neuroinflammation,” “neurodegeneration,” “dysmotility,” “leaky gut,” “enteric neurodegeneration,” “inflammatory bowel disease,” “celiac disease,” “nonalcoholic steatohepatitis,” “malabsorption,” and “cognitive impairment,” among others. Sources were selected based on relevance to the topic and scientific rigor, with an emphasis on studies that contributed to understanding underlying mechanisms and clinical associations. Both review articles and primary research studies, including experimental and observational studies, were included.
Gut paresis arising in the context of neurological disease reflects a complex interplay between neuroinflammation, epithelial barrier dysfunction, microbial dysbiosis, and disrupted neuroenteric signaling [4–7] (Figure 1). Although clinical manifestations often converge on delayed gastric emptying, impaired motility, and altered visceral sensation, the underlying mechanisms vary across disease states such as Parkinson’s disease (PD), multiple sclerosis (MS), stroke, and autonomic neuropathies [8–11]. Growing evidence suggests that the gut is not merely a downstream target of neurological dysfunction but an active participant in disease propagation through bidirectional communication along the brain–gut axis. Gut paresis in neurological disorders arises from complex interactions involving enteric nervous system (ENS) degeneration, misfolded protein accumulation (α-synuclein), chronic immune activation, and dysbiosis. Key drivers include vagus nerve dysfunction, neuroinflammation, and loss of Interstitial Cells of Cajal (ICC), with major risk factors being Parkinson’s, MS, stroke, and gut-microbiome dysbiosis [12–14]. The following subsections outline the major mechanistic contributors and risk factors implicated in the development of gut paresis in neurological disorders [15, 16].
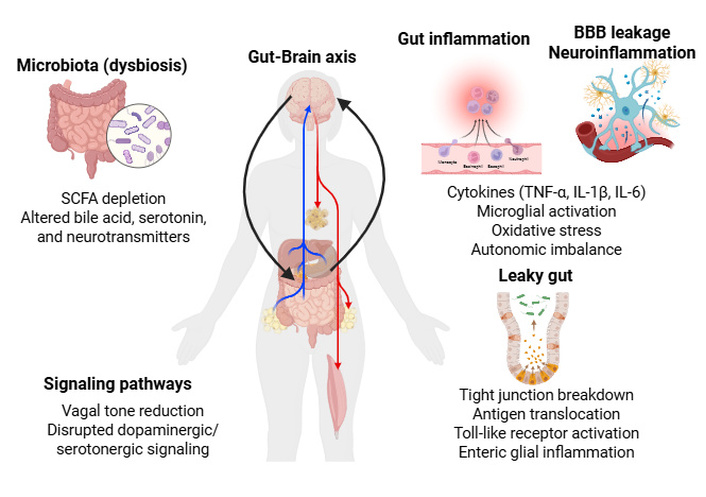
Representation of the brain–gut axis illustrating mechanisms by which neurological disease leads to gut paresis. Neuroinflammation, autonomic dysfunction, and microglial activation impair enteric neuronal signaling and motility. Leaky gut and microbial dysbiosis exacerbate inflammation via antigen translocation and altered metabolite profiles. Key signaling pathways include vagal tone reduction, disrupted neurotransmission, NF-κB activation, and HPA axis stress. Bidirectional communication between the brain and gut sustains a pathogenic loop driving gastrointestinal dysfunction. HPA: hypothalamic–pituitary–adrenal; IL: interleukin; NF-κB: nuclear factor-kappa B; SCFA: short-chain fatty acid; TNF-α: tumor necrosis factor alpha. Created in BioRender. Rai, V. (2026) https://BioRender.com/gafzhi2.
Neuroinflammation is a central driver of gut dysmotility in many neurological diseases [17] (Figure 1). In PD and MS, microglial activation and peripheral immune cell infiltration generate a systemic inflammatory milieu characterized by elevated cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 [18, 19]. These mediators can directly impair enteric neuronal function by altering neurotransmitter release, reducing neuronal excitability, and promoting oxidative stress within the myenteric plexus [20–22]. Chronic inflammation also disrupts smooth muscle contractility by modulating calcium signaling and impairing ICC, the pacemakers of gastrointestinal motility [23, 24].
Peripheral inflammation further amplifies central neuroinflammation through vagal afferents and humoral pathways [25, 26]. For example, systemic lipopolysaccharide (LPS) exposure increases hypothalamic and brainstem cytokine expression, which in turn alters vagal efferent output to the gut [27, 28]. Furthermore, the activation of toll-like receptors (TLRs) and the nuclear factor-kappa B (NF-κB) signaling pathway by microbial products or cytokines drives a destructive cycle of nitric oxide overproduction and impaired smooth muscle contractility [29]. This immune dysregulation also compromises the intestinal epithelial barrier, or “leaky gut,” allowing the translocation of luminal antigens and toxins into systemic circulation (Figure 1). Such translocation further activates peripheral immune cells and CNS microglia, creating a pathogenic feedback loop that sustains both gastrointestinal dysmotility and central neurological decline [29]. In stroke and traumatic brain injury, acute neuroinflammation triggers sympathetic overactivation and parasympathetic withdrawal, leading to ileus-like states [30, 31]. Similarly, autoimmune neuropathies such as Guillain-Barré syndrome involve inflammatory damage to autonomic fibers, resulting in profound dysmotility [32]. Thus, inflammation, whether central, peripheral, or autonomic, acts as a unifying mechanism linking neurological injury to gut paresis.
Microbial dysbiosis is increasingly recognized as a major determinant of gut paresis in neurological disease [33] (Figure 1). Alterations in microbial composition, such as reduced short-chain fatty acid (SCFA)-producing bacteria, increased pro-inflammatory taxa, and overgrowth of pathobionts, can profoundly influence ENS function, immune activation, and epithelial integrity [34]. SCFAs, particularly butyrate, support epithelial barrier health, modulate inflammation, and regulate enteric neuronal activity [35, 36]. Their depletion in PD, MS, and Alzheimer’s disease (AD) correlates with impaired motility and heightened systemic inflammation [37, 38]. Dysbiosis also alters bile acid metabolism, serotonin production, and microbial neurotransmitter synthesis [e.g., gamma-aminobutyric acid (GABA), dopamine precursors], all of which influence gut motility [39, 40]. Microbiota can shape central neurological disease as well. In PD, certain bacterial strains promote α-synuclein misfolding, while others modulate microglial activation [41]. In MS, gut microbial signatures influence T-cell differentiation and disease severity [42]. These findings underscore the bidirectional nature of the brain–gut axis: neurological disease alters gut microbiota, and microbial changes feed back to exacerbate neuroinflammation and motility dysfunction [33]. As discussed, accumulation of α-synuclein underlies the pathogenesis of neurological manifestations in the gut–brain axis; it is important to understand protein misfolding and the effects of its accumulation.
Protein misfolding and the subsequent formation of toxic aggregates represent a hallmark mechanism in the progression of neurodegenerative diseases, most notably PD. In PD, α-synuclein undergoes a conformational shift from a healthy helical structure into toxic, β-sheet-rich oligomers. These misfolded proteins aggregate into Lewy bodies, which are found not only in the CNS but also extensively within the ENS. Emerging evidence suggests that this pathology may actually originate in the gut, triggered by exposure to luminal antigens or microbial metabolites crossing a “leaky” epithelial barrier [43]. Once formed in the ENS, these α-synuclein aggregates can traverse the intestinal tract and spread to the brain via the vagus nerve through trans-synaptic transmission. This “bottom-up” propagation links early gastrointestinal symptoms, such as constipation and gastroparesis, to subsequent central neurodegeneration. Furthermore, mutations in other proteins like leucine-rich repeat kinase 2 (LRRK2) can exacerbate this process by clogging lysosomal machinery, thereby promoting further protein accumulation. This cycle of misfolding and accumulation causes structural damage to the myenteric plexus, impairing the neural regulation of motility and contributing to the chronic gut paresis observed in these patients [43]. Accumulation of misfolded α-synuclein in the ENS is a key pathogenic process in neurological diseases as it causes enteric neurodegeneration by inducing inflammation, mitochondrial damage, and synaptic dysfunction, often spreading from the gut to the brain.
Enteric neurodegeneration serves as a critical structural basis for the chronic gut paresis observed in various neurological disorders. This process primarily involves progressive damage to the myenteric plexus, the neural network residing within the intestinal wall that is responsible for coordinating peristaltic contractions. A hallmark of this degeneration is the significant loss of ICC, which function as the essential electrical pacemakers of the gastrointestinal tract [44]. When ICC networks are impaired, the regular electrical “slow waves” required for organized motility are disrupted, leading to clinical manifestations such as delayed gastric emptying, impaired transit, and altered visceral sensation. In conditions like PD, this neurodegeneration may be driven by the accumulation of misfolded proteins or chronic neuroinflammation that alters neurotransmitter release and reduces neuronal excitability within the gut. Furthermore, studies in spinal cord injury models have shown that this degeneration can manifest as a physical thinning of both the mucosal and muscular layers of the gastrointestinal tract, further compromising its functional integrity. Ultimately, the depletion of these enteric neurons and pacemakers creates a profound state of dysmotility that often precedes the onset of central motor symptoms [44].
Autonomic dysfunction represents a fundamental failure of the bidirectional communication systems that regulate gastrointestinal homeostasis. The neuronal pathway of the brain–gut axis is primarily governed by the autonomic nervous system, which coordinates gastrointestinal motility, secretion, and sensation through a delicate balance between sympathetic and parasympathetic inputs. In many neurological disorders, this balance is profoundly disrupted, often manifesting as impaired parasympathetic input via the vagus nerve or pathological sympathetic overactivity (Figure 1). Reduced vagal tone leads to a loss of the cholinergic stimulation required for organized peristalsis, resulting in clinical gut paresis [45].
These disturbances are not merely functional but often involve structural damage to autonomic fibers. For instance, in autoimmune conditions like Guillain-Barré syndrome, inflammatory damage to these fibers causes severe dysmotility. Similarly, acute neurological insults such as stroke or traumatic brain injury can trigger a state of “parasympathetic withdrawal” and sympathetic overactivation, leading to ileus-like gastrointestinal paralysis. The clinical implications of this dysfunction are vast, as autonomic symptoms like constipation and bloating frequently precede motor or cognitive decline by years. Managing these conditions often requires neuromodulatory strategies, such as vagus nerve stimulation, to restore the disrupted signaling pathways linking the CNS and ENS [46]. Autonomic dysfunction disrupts the nervous system’s control over digestion, often causing gut motility issues and chronic inflammation, which can contribute to increased intestinal permeability, or “leaky gut”.
Increased intestinal permeability or “leaky gut” is both a consequence and a contributor to gut paresis in neurological disease [47]. Tight junction proteins such as occludin, claudins, and ZO‑1 are highly sensitive to inflammatory cytokines, oxidative stress, and microbial metabolites [48]. Their disruption allows translocation of luminal antigens, bacterial components, and toxins into the lamina propria and systemic circulation [49] (Figure 1).
In PD, α‑synuclein aggregation in enteric neurons is thought to be triggered by exposure to luminal antigens crossing a compromised epithelial barrier [50]. This process may precede CNS pathology by years, suggesting that leaky gut is an early etiological factor rather than a secondary effect [51]. Similarly, in MS, increased gut permeability correlates with disease activity and may facilitate peripheral immune activation that exacerbates neuroinflammation [52].
Leaky gut also worsens motility by promoting low‑grade inflammation within the ENS, impairing ICC networks, and altering smooth muscle responsiveness [53]. Bacterial translocation can activate TLRs on enteric neurons and glia, leading to nitric oxide overproduction and inhibitory motor reflexes [54]. Thus, epithelial barrier dysfunction serves as a mechanistic bridge between luminal factors and neuroenteric pathology [55].
Multiple signaling pathways mediate communication between the brain, ENS, immune system, and microbiota [56] (Figure 1). Accumulation of α-synuclein over decades before the onset of disease plays a key role in the pathology involving the vagus nerve, as discussed below. Key pathways implicated in gut paresis include: (a) Vagal signaling: Reduced vagal tone, common in PD and autonomic neuropathies, impairs cholinergic stimulation of gut motility. Vagal afferents also transmit inflammatory and microbial signals to the brainstem, influencing central autonomic output [57]. (b) Enteric neurotransmission: Dopaminergic, serotonergic, and cholinergic pathways are frequently disrupted. Loss of dopaminergic signaling in PD slows gastric emptying, while altered serotonin availability affects peristalsis and visceral sensation [58]. (c) TLR and NF‑κB pathways: Activation by microbial products or inflammatory cytokines drives ENS inflammation, nitric oxide overproduction, and impaired contractility [59]. Accumulated phosphorylated α-synuclein in Schwann cells, acting as an endogenous agonist, is recognized by TLRs (particularly TLR2), which contributes to the activation of NF-κB and neuroinflammation. This immune response causes peripheral autonomic neuropathy, contributing to symptoms like constipation, bladder issues, and low blood pressure. (d) α‑Synuclein propagation pathways: Misfolded α‑synuclein may spread from gut to brain via trans-synaptic transmission along the vagus nerve, linking early gut dysfunction to central pathology [41]. (e) HPA axis dysregulation: Stress‑induced cortisol release alters gut permeability, immune activation, and motility, compounding neurological disease-related dysfunction (Figure 1) [60]. Dysregulation of the HPA axis is often co-associated with alpha-synuclein accumulation.
Patients with PD exhibit significant bilateral atrophy of the vagus nerve (both left and right sides) compared to healthy controls, with smaller nerve calibers directly linked to increased autonomic dysfunction. Atrophy of the vagus nerve, often seen as smaller cross-sectional areas in ultrasound imaging, correlates with severe gastrointestinal dysfunction, cardiovascular issues, and orthostatic hypotension. The Braak hypothesis suggests that misfolded α-synuclein may originate in the ENS and travel via the vagus nerve to the brainstem. Thus, the vagus nerve acts as a conduit for α-synuclein pathology from the gut to the brain, exacerbating autonomic neuropathies. Reduced vagal function causes various autonomic symptoms, including gastroparesis, chronic constipation, heart rate variability issues, and orthostatic hypotension [61, 62].
Lewy bodies (aggregated α-synuclein) are found in the enteric neurons and nerve fibers of the gut, from the esophagus to the anus, often appearing up to 20 years before PD diagnosis. This disruption of enteric neurotransmission leads to loss of neurons and changes in neurochemical signaling, disrupting the ENS’s ability to control gastrointestinal motility, secretion, and absorption. This results in autonomic neuropathies, most notably chronic constipation, gastrointestinal motility dysfunction, and cardiovascular dysfunction. The vagus nerve as a conduit plays a significant role in these neuropathies [63, 64].
In vitro and experimental models have provided important mechanistic insight into brain–gut interactions, although these findings should be interpreted as preclinical evidence rather than direct proof of human disease. Cellular studies have examined the accumulation of α-synuclein within enteric neurons and its potential effects on neuronal signaling, neurotransmission, and cell survival [65].
In addition, studies have shown that microbial products such as LPS can activate TLRs and downstream NF-κB signaling in enteric glial cells, promoting nitric oxide production and contributing to impaired smooth muscle contractility [66] (Figure 1).
Animal models have further clarified how neurologic injury and neurodegenerative disease may disrupt gastrointestinal structure and function. In rat models of spinal cord injury, CNS trauma has been associated with progressive gut remodeling characterized by thinning of the intestinal mucosa and muscular layers after injury, along with delayed gastric emptying and reduced intestinal motility, likely related to impaired neurotransmitter release [67].
Studies in mice have also suggested that the gut microbiome may influence neurologic outcomes: germ-free mice develop larger infarct volumes after stroke, whereas fecal microbiota transplantation (FMT) has shown neuroprotective effects in experimental models, including restoration of beneficial microbial populations and reduction of pro-inflammatory NF-κB signaling in both the spinal cord and intestine [68]. In addition, mouse studies support a possible “bottom-up” pathway of disease propagation, in which misfolded α-synuclein may spread from the ENS to the brainstem through the vagus nerve [69].
Human studies support the clinical importance of brain–gut dysfunction, although much of the available evidence remains observational. In PD, gastrointestinal dysfunction is common, and dysphagia affects up to 80% of patients, increasing the risk of aspiration pneumonia. Constipation and other non-motor gastrointestinal symptoms may precede the diagnosis of PD by several years, raising interest in their potential role as early clinical markers [70]. In patients after stroke, pneumonia has been linked to translocation of gut-associated bacteria, with studies reporting that many organisms identified in pulmonary samples are commensal gastrointestinal species such as Escherichia coli, highlighting the possible clinical consequences of post-stroke dysbiosis. In MS, lesion location has also been correlated with gastrointestinal manifestations; for example, demyelinating plaques involving the area postrema have been associated with persistent hiccups, nausea, and vomiting [71, 72]. Overall, human data reinforce the clinical relevance of the gut–brain axis, but they do not yet establish the same degree of mechanistic causality demonstrated in cellular and animal models.
PD is the second most common neurodegenerative disease worldwide and is characterized by the accumulation of Lewy bodies within dopaminergic neurons in the substantia nigra [73].
The diagnosis is made clinically and is broadly defined by both motor and nonmotor symptoms, with key features including tremor, bradykinesia/akinesia, rigidity, and postural instability [74]. Nonmotor symptoms, while less visibly obvious, often precede the diagnosis of PD and are debilitating with manifestations like autonomic dysfunction, hallucinations, and depression, underscoring the clinical complexity of PD [74].
This complexity begins at the molecular level as the risk for PD development involves both genetic and environmental factors [73]. In terms of genetics, well-known mutations involve proteins such as α-synuclein, an amino acid protein that is rich within neuronal synaptic terminals. A highly pliable protein, a synuclein normally exists in the helical conformation and accomplishes its primary function of vesicular transport and neurotransmitter release; however, it is prone to convert into a highly toxic B-sheet-rich oligomer that promotes aggregation [73]. Similarly, the scaffolding protein LRRK2 aids in vesicular movement, docking, and fusion indirectly due to its effects on kinase and GTPase enzymes. When mutated, increased activity of these enzymes is detrimental to the CNS, while LRRKs become prone to clogging lysosomal machinery and against promoting protein aggregation [73]. Still, other proteins, such as Parkin and various glucocerebrosidases, can become mutated and represent the genetic factors that contribute to PD.
Studies reveal that α-synuclein is capable of traversing the intestinal tract to the brain by using the vagus nerve as a direct pathway, occurring after the misfolded protein has accumulated over the course of years within the enteric tract [73]. Gastric manifestations in PD can be subtle but include hypersalivation, dysphagia, nausea, and vomiting, amongst others [75]. Unfortunately, these are often overlooked at the expense of patients who then suffer high rates of morbidity and mortality. For instance, roughly 80% of patients will develop dysphagia at some point, regardless of whether it is oral, pharyngeal, or esophageal, which increases rates of aspiration pneumonia, a leading cause of death in PD. A fear of aspiration also contributes to weight loss, along with factors such as anosmia, loss of appetite, hormonal changes, and increased energy expenditure, such that upwards of 52% of PD patients suffer from weight loss [75]. The mechanism of gastrointestinal disorders in PD is limited, with many questioning whether it relates to protein accumulation, bacterial overgrowth, or even side effects of drugs meant to treat symptoms. For instance, levodopa-carbidopa therapies are well known to cause nausea in patients during their initiation phase [75].
Stroke is the third leading cause of death within the United States and is responsible for 134,000 deaths annually. Strokes can be broadly defined as ischemic or hemorrhagic, with manifestations dependent on the region of the brain affected; oftentimes, heavy emphasis is placed on the cerebrum, diencephalon, cerebellum, and brainstem [76]. Interestingly, stroke presentation is not limited to neuroanatomy, and growing research focuses on the impact of sex hormones on both clinical presentation and subsequent treatments for stroke [77].
Typical symptoms of stroke include hemiparesis, hemiplegia, gait and speech changes, facial asymmetry, and visual changes, all of which are considered to be focal deficits. In contrast, atypical symptoms such as altered mentation, chest pain, palpitations, generalized weakness, and even nausea and vomiting have a less succinct connection to the nervous system. These atypical symptoms are believed to be more prevalent amongst female patients and potentially play a role in health disparities, as these patients are more often misdiagnosed with stroke mimickers compared to male patients. Raising awareness of these differences in presentation can help health practitioners recognize lesser-known manifestations of stroke, such as those involving the gastrointestinal system [77].
Dysbiosis following stroke can lead to higher rates of systemic infection by altering the integrity of the epithelial barrier [78]. This manifests as higher rates of upper respiratory tract infections and pneumonia; in fact, 70% of the bacteria collected from lung samples in stroke patients were revealed to be symbiotic species from the gastrointestinal tract, including Enterococcus spp., Escherichia coli, and Morganella morganii. Residual weakness following strokes, such as impaired chewing or swallowing, can also contribute to malnutrition and dysbiosis [78]. Conversely, gut microbiota can be used to minimize the risk of stroke or potentially enhance recovery and has been applied to murine models subdivided into “germ-free” and “healthy microbiome” groups. Not only did germ-free mice suffer larger infarct sizes compared to mice with healthy microbiomes, but the infarct size was shown to significantly reduce in size following colonization by healthy bacterial species [78]. It has been postulated that these benefits are mediated by bacterial metabolites, underscoring the importance of adopting healthy diets that support a diverse gut microbiota. A growing body of research supports the use of probiotics for their cholesterol-lowering effects, particularly through the conversion of cholesterol to coprostanol and the reduction of cholesteryl esters in low-density lipoprotein (LDL) particles, thereby slowing the atherosclerotic process that contributes to stroke [79].
MS is the single most common chronic inflammatory disease of the CNS, affecting 2.5 million people worldwide with a prevalence of 33 per 100,000 [80].
MS is an inflammatory condition defined by the presence of demyelinating lesions disseminated in time and space, with a predilection for the optic nerves, brainstem, cerebellum, periventricular, and spinal cord. Its clinical manifestations vary depending on what area of the brain is involved, but generally have a gradual onset requiring a few days [81]. Common symptoms include visual loss and painful eye movements associated with optic neuritis, bilateral internuclear ophthalmoplegia, as well as loss of sensory and motor symptoms when lesions affect the spinal cord [81].
MS is a chronic autoimmune disease of the CNS that causes inflammation, demyelination, and axonal injury throughout its course [80]. MS has an onset in early adulthood and is diagnosed clinically and radiologically based on lesions disseminated in space and time [82]. Despite its prevalence, there is a lack of understanding of its pathogenesis, as some postulate that the microenvironments of the CNS harbor inflammatory cells, and others argue it’s an inflammatory process that slowly becomes neurodegenerative [80]. Regardless of the mechanism, MS is known to cause neurogenic bowel dysfunction that presents as constipation or fecal incontinence, that have major implications on quality of life [83]. Fecal incontinence is a well-known source of distress in patients with MS, who often resort to treatments such as transanal irrigation (a bowel management procedure that uses a pump or gravity system to introduce warm water into the rectum, flushing out stool to treat chronic constipation and fecal incontinence), biofeedback therapy, posterior tibial nerve stimulation, and sacral neuromodulation, amongst others [84]. Still, less obvious gastrointestinal manifestations are caused by the downstream effects of brain lesions on the gastrointestinal tract and include decreased peristalsis, abdominal straining, hiccups, and appetite loss [72]. Interestingly, these are often traced back to specific locations in the CNS; for instance, lesions within the area postrema caused by MS plaques then manifest as hiccups and vomiting [72]. Despite these studies, current treatment options for gastrointestinal symptoms in MS are limited, as most trials utilize small sample sizes and heterogeneous protocols.
According to the Center for Disease Control and Prevention, autism spectrum disorder (ASD) affects 1 in 3 children aged 8 years old, and prevails amongst all racial, ethnic, and socioeconomic groups [85]. According to a 2018 review conducted by the World Health Organization, the estimated prevalence of ASD worldwide is approximately 1.5% in developed countries [86].
ASD is a neurodevelopmental disorder marked by challenges in social interaction and communication, as well as restricted and repetitive behaviors [87]. In educational settings, ASD can present as early as preschool years, with affected children demonstrating limited language development and an increased risk of persistent nonverbalism as they mature [88]. Importantly, these deficits are responsive to intervention, highlighting the critical role of early treatment in improving long-term outcomes. For example, randomized controlled trials by Kasari et al. [88] evaluated two language-based interventions delivered in community settings through hour-long sessions over a six-month period in 164 preschool children with ASD, demonstrating overall improvement regardless of the specific treatment modality.
Recent studies underscore genetic interplay in the pathogenesis of autism, with 234 loci currently identified by exome sequencing [89]. However, much remains unknown about the pathogenesis of this disorder with profound psychiatric, neurological, and even gastrointestinal manifestations. Feeding problems remain a huge issue in the physical and psychological development of autistic children and include pica, avoidance disorders, ruminant disorders, and countless eating disorders according to the DSM-5 [90]. Data shows that autistic children are more selective with food choices, opting for low consistency foods and overall, less likely to consume fruits, dairy products, vegetables, proteins, and starches. Autistic children are also more likely to display challenging behaviors during mealtimes, such as choking, aggression, and anxiety, all of which have been shown to be correlated with the Repetitive Behavior Scales [90]. However, educated parents and caregivers can aid in overcoming barriers to feeding and subsequent nutritional deficiencies. Behavioral interventions have gained attention as of late within the scientific community and include techniques such as fading stimulus, repeated taste exposure, and positive reinforcement. These trials have shown promise; in fact, the Managing Eating Aversions and Limited Variety (MEAL) plan proved successful for 47.3% of families that implemented it [91].
There are roughly 18,000 new cases of spinal cord injury in the United States annually, with about 54 cases occurring per 1 million [92]. Globally, the incidence of spinal cord injury is estimated to range from 8 to 246 cases per million persons; however, the true incidence remains uncertain due to limited reporting in developing countries [93].
Spinal cord injuries present various upper and lower motor neuron deficits; each is associated with its own unique prognosis. Broadly speaking, these manifest as alterations in reflexes, muscle bulk and tone, and onset of pathological reflexes, including the Babinski reflex alongside fasciculations [94]. Clinical presentation varies with the level of spinal cord injury and is further classified using the American Spinal Injury Association (ASIA) scale. Injuries at or above the T6 level are particularly associated with neurogenic shock due to disruption of sympathetic outflow, in addition to other forms of shock such as hemorrhagic, cardiogenic, and obstructive. In contrast, cervical injuries, particularly at the C5 level, are associated with respiratory compromise and often require airway support or intubation [92].
Spinal cord injury is divided into a primary phase characterized by the trauma-induced shearing forces placed on spinal cord fibers, as well as a secondary phase that encompasses the physiological responses. The secondary phase can be better described as a propagation of the initial insult due to free radical production, immune cell invasion, and glutamate toxicity [95]. In the process, the body is placed under physiological stresses, making it unable to support a stable microbiome, such that significant reductions in the quantity of commensal organisms are observed, sometimes as much as a 1,000-fold decline [95]. This relationship, studied in rat models, demonstrated decreased gastric emptying and motility one month following spinal cord injury. The underlying mechanism was revealed to be impaired neurotransmitter release as well as direct effects on the gastrointestinal system, such as thinning of the mucosa and muscular layers [67]. Conversely, a healthy microbiome has been shown to play a neuroprotective role in clinical trials involving injured mice [96]. This was achieved through FMT, which restored bacterial populations within the Firmicutes phylum, including members of the Christensenellaceae family and the Butyricimonas genus. Additionally, these microorganisms significantly reduced NF-κB signaling activity in both the spinal cord and gastrointestinal tract. Overall, the study demonstrated improvements in neuronal axon regeneration, animal weight gain, intestinal barrier integrity, and gastrointestinal motility [96].
Peripheral and autonomic neuropathies are associated with many etiologies, including autoimmune, toxic, hereditary, amyloid, and diabetic, but remain idiopathic for 20–25% of patients [97]. More specifically, approximately 50% of patients with diabetes will develop diabetic peripheral neuropathy over the course of their disease [98].
Peripheral and autonomic neuropathies involve dysfunction of peripheral nerves, including autonomic fibers, and can result in significant gastrointestinal dysmotility due to impaired neural regulation. These neuropathies are often divided into disease processes that destroy either myelin or axons, with unique symptoms associated with each [97]. A well-known form of peripheral neuropathy is diabetic peripheral neuropathy, which significantly impacts quality of life and is characterized by symptoms such as electric shock-like sensations, deep aching pain that may ascend from the lower to upper extremities, and burning. Furthermore, patients often experience moderate to severe pain that contributes to insomnia and mood disorders, along with significantly increased healthcare costs compared to diabetic patients without neuropathy [98].
Diabetic neuropathy has a multitude of effects on the gastrointestinal system; for instance, diabetic gastroparesis is known to cause bloating, belching, postprandial vomiting, and early satiety [99]. Other esophageal symptoms include reflux, regurgitation, and dysphagia that contribute to higher rates of Barrett esophagus in diabetic patients [99]. It has been postulated that the detrimental effects of diabetes, namely hyperglycemia and poor oxygen reduction in the vasa nervosa, generate an inflammatory state that damages structures throughout the nervous system [100]. Numerous research efforts have focused on improving outcomes in patients with diabetic gastroparesis. However, many proposed theories have shown limited translation into clinical practice, as glycemic control appears to have only a modest effect on gastroparesis severity. Patients are often counseled to follow diets consisting of smaller particle sizes and reduced fiber content, but adherence remains low. For patients who do not respond to medical therapy, a final therapeutic option is enteral nutrition via feeding jejunostomy in conjunction with a venting gastrostomy [101].
Gastrointestinal diseases exert profound effects on the nervous system through immune, metabolic, and neuroendocrine pathways that mirror the bidirectional communication described in neurological disorders with gut involvement [102]. While neurological disease can precipitate gut dysfunction, the reverse is equally true, so chronic gastrointestinal inflammation, malabsorption, and hepatic metabolic failure can propagate neuroinflammation, alter neurotransmitter synthesis, and impair neuronal function. These interactions underscore the integrative nature of the gut–brain axis and highlight the importance of recognizing gastrointestinal pathology as a potential driver of neurological symptoms [103]. Across these conditions, a common mechanistic framework emerges in which intestinal inflammation, barrier dysfunction, malabsorption, or hepatic metabolic failure initiate peripheral immune and metabolic disturbances that converge on the CNS through cytokine signaling, microbial metabolites, neuroendocrine pathways, and toxic metabolite accumulation.
Gastrointestinal diseases influence the nervous system through three major mechanisms: systemic inflammation, altered microbial and neuroendocrine signaling, and nutrient deficiencies [104]. Although the relative contribution of each pathway differs by disease, these mechanisms are not independent and often interact to amplify neuroinflammation, alter neurotransmitter balance, and impair neuronal function. Chronic intestinal inflammation, as seen in inflammatory bowel disease (IBD) or celiac disease, leads to elevated circulating cytokines such as TNF-α, IL-6, and IL-1β [105]. These mediators cross the blood-brain barrier (BBB) or signal through vagal afferents, promoting microglial activation and neuroinflammation. This inflammatory crosstalk contributes to mood disorders, cognitive impairment, and heightened pain sensitivity [106].
Disruption of the epithelial barrier, “leaky gut”, further amplifies systemic inflammation by allowing microbial products such as LPS to enter the circulation. LPS activates TLR4 signaling in both peripheral immune cells and CNS microglia, reinforcing a proinflammatory state that affects neuronal excitability and neurotransmitter metabolism [107, 108].
Nutritional deficiencies represent a second major pathway linking gastrointestinal disease to neurological dysfunction [109]. Malabsorption syndromes impair the uptake of essential vitamins and micronutrients, particularly B vitamins, vitamin D, iron, and copper, each of which plays a critical role in neuronal metabolism, myelination, and neurotransmitter synthesis. Deficiencies can manifest as neuropathy, ataxia, cognitive decline, or encephalopathy [110].
Finally, alterations in gut microbiota composition influence CNS function through microbial metabolites (e.g., SCFAs), tryptophan metabolism, and immune modulation [109]. Dysbiosis in chronic gastrointestinal disease can shift the balance toward proinflammatory taxa, reduce neuroprotective metabolites, and alter serotonergic signaling, collectively contributing to neuropsychiatric symptoms [111]. Taken together, gastrointestinal disease-related neurological dysfunction can be understood as the downstream result of converging inflammatory, metabolic, microbial, and nutritional insults rather than isolated processes.
IBD, comprising Crohn’s disease (CD) and ulcerative colitis (UC), affects over 3.8 million individuals globally, with rising incidence in North America, Europe, and increasingly in newly industrialized regions [112]. Peak onset occurs in young adulthood, though a second incidence peak is observed in older adults [113]. The chronic, relapsing nature of IBD and its systemic inflammatory burden make neurological manifestations increasingly recognized components of disease morbidity [114].
IBD primarily presents abdominal pain, diarrhea, rectal bleeding, weight loss, and fatigue [115]. Extraintestinal manifestations occur in up to 40% of patients and include arthritis, uveitis, skin disorders, and hepatobiliary disease [116]. Neurological symptoms once considered rare are now recognized as clinically significant and may include peripheral neuropathy, headaches, cerebrovascular events, and cognitive or mood disturbances. These manifestations may arise from systemic inflammation, medication effects, or nutritional deficiencies secondary to malabsorption or chronic diarrhea [114].
The neuropsychiatric burden of IBD is substantial. Rates of anxiety and depression are significantly higher in IBD patients compared with the general population, with mood disorders often correlating with disease activity [117].
In IBD, these manifestations are best understood as the result of persistent intestinal inflammation that disrupts the epithelial barrier, sustains systemic cytokine signaling, alters HPA axis activity, and reshapes microbial metabolite production, with these pathways converging on CNS inflammation and altered neurotransmission.
Within this framework, the major mechanistic pathways contributing to these neurological consequences are: i) Cytokine-mediated neuroinflammation: Elevated TNF-α, IL-6, and IL-1β in active IBD can cross the BBB or activate endothelial and microglial cells (Figure 2), altering neurotransmitter metabolism (e.g., reducing serotonin availability) and promoting sickness behavior, fatigue, and depressive symptoms [118], ii) HPA axis dysregulation: Chronic inflammation activates the hypothalamic–pituitary–adrenal axis (Figure 2), leading to cortisol abnormalities that influence mood, cognition, and stress responsiveness [119], iii) tryptophan metabolism: Inflammation-induced activation of indoleamine 2,3-dioxygenase (IDO) shifts tryptophan metabolism toward, reducing serotonin synthesis and generating neuroactive metabolites implicated in depression and cognitive dysfunction [120] (Figure 2), iv) microbiota alterations: Dysbiosis in IBD reduces SCFA-producing bacteria and alters microbial neurotransmitter production, contributing to anxiety-like behavior and impaired stress resilience [121] (Figure 2), v) medication effects: Long-term corticosteroid use can exacerbate mood disorders, while anti-TNF therapies may improve depressive symptoms by reducing systemic inflammation [122, 123] (Figure 2). These pathways are interrelated rather than discrete, and together they reinforce the bidirectional inflammatory and neuroendocrine signaling that links intestinal disease activity to CNS dysfunction (Figure 2). Collectively, these mechanisms highlight the intertwined nature of intestinal inflammation and CNS dysfunction in IBD (Figure 2 and Table 1).
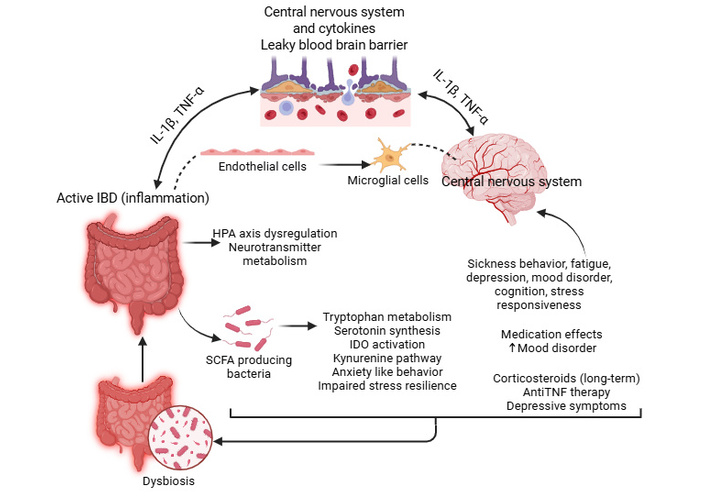
Pathophysiological mechanisms linking IBD to mood disorders. Intestinal inflammation in active IBD triggers a multi-pathway cascade affecting the CNS through the translocation of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) across the BBB. These cytokines, along with HPA axis dysregulation and microbiota-driven reductions in SCFAs, alter neurotransmitter metabolism and promote neuroinflammation. The activation of the IDO enzyme shifts tryptophan metabolism away from serotonin synthesis toward neurotoxic kynurenine metabolites. BBB: blood-brain barrier; HPA: hypothalamic–pituitary–adrenal; IBD: inflammatory bowel disease; IDO: indoleamine 2,3-dioxygenase; IL: interleukin; SCFA: short-chain fatty acid; TNF-α: tumor necrosis factor alpha. Created in BioRender. Rai, V. (2026) https://BioRender.com/8srspo6.
Pathophysiology and neurologic consequences in IBD.
| Mechanism | Pathophysiological features | Neurological consequences |
|---|---|---|
| Cytokine-mediated neuroinflammation [118] | Elevated TNF-α, IL-6, and IL-1β cross the BBB or activate microglia; altered serotonin metabolism. | Fatigue, sickness behavior, depressive symptoms. |
| HPA axis dysregulation [119] | Chronic inflammation activates the HPA axis; cortisol imbalance affects brain function. | Mood instability, cognitive impairment, stress sensitivity. |
| Tryptophan metabolism shift [120] | IDO activation diverts tryptophan to the kynurenine pathway; reduced serotonin synthesis. | Depression, cognitive dysfunction. |
| Microbiota alterations [121] | Dysbiosis reduces SCFA-producing bacteria; disrupted microbial neurotransmitter production. | Anxiety-like behavior, impaired stress resilience. |
| Medication effects [124] | Corticosteroids worsen mood; anti-TNF agents may reduce systemic inflammation. | Exacerbation or improvement of mood symptoms depending on therapy. |
BBB: blood-brain barrier; HPA: hypothalamic–pituitary–adrenal; IBD: inflammatory bowel disease; IDO: indoleamine 2,3-dioxygenase; IL: interleukin; SCFA: short-chain fatty acid; TNF: tumor necrosis factor.
Celiac disease affects approximately 1% of the global population, though many cases remain undiagnosed [125]. It is triggered by an immune-mediated reaction to gluten in genetically susceptible individuals (HLA-DQ2/DQ8) [126]. Beyond classical celiac disease, a spectrum of malabsorption syndromes, including chronic pancreatitis, small intestinal bacterial overgrowth, and short bowel syndrome, can similarly impair nutrient absorption and contribute to neurological complications [127].
Celiac disease presents with variable gastrointestinal symptoms, including diarrhea, bloating, abdominal pain, and weight loss [128]. Extraintestinal manifestations are common and may include anemia, osteoporosis, dermatitis herpetiformis, and neurological symptoms such as peripheral neuropathy, ataxia, headaches, and cognitive impairment [129]. Malabsorption syndromes present with steatorrhea, weight loss, and deficiencies in fat-soluble vitamins and micronutrients [130].
Neurological complications in celiac disease and malabsorption arise from immune-mediated mechanisms, micronutrient deficiencies, and inflammatory signaling. In contrast to IBD, where chronic cytokine excess and dysbiosis are prominent drivers, celiac disease and other malabsorptive states often exert neurological effects through a combination of autoimmune cross-reactivity and prolonged deficiency of nutrients required for myelin maintenance, mitochondrial function, and neurotransmitter synthesis.
Within this framework, the major mechanistic pathways contributing to these neurological consequences are: i) gluten ataxia: Autoantibodies against transglutaminase 6 (TG6) can target cerebellar tissue, leading to progressive ataxia independent of gastrointestinal symptoms. Early gluten-free diet initiation can halt progression [131] (Figure 3). ii) peripheral neuropathy: Both axonal and demyelinating neuropathies occur, driven by immune-mediated injury and deficiencies in vitamins B1, B6, B12, and E [132] (Figure 3). iii) cognitive impairment (“brain fog”): chronic inflammation, cytokine release, and micronutrient deficiencies contribute to impaired concentration, memory deficits, and fatigue [133] (Figure 3). iv) Wernicke’s encephalopathy: thiamine (vitamin B1) deficiency, common in severe malabsorption or prolonged vomiting, can precipitate acute confusion, ophthalmoplegia, and ataxia. This condition is a medical emergency and underscores the neurological vulnerability associated with malabsorption [134] (Figure 3). v) vitamin E deficiency: Leads to spinocerebellar degeneration, neuropathy, and myopathy [135]. vi) vitamin B12 deficiency: Causes subacute combined degeneration of the spinal cord, presenting with ataxia, paresthesia, and cognitive changes [136] (Figure 3). Together, these mechanisms show that neurological injury in malabsorption is not merely a secondary consequence of gastrointestinal symptoms, but a direct result of immune and metabolic injury affecting both the central and peripheral nervous systems (Figure 3). These manifestations highlight the importance of early recognition and correction of nutritional deficiencies in gastrointestinal disease (Figure 3 and Table 2).
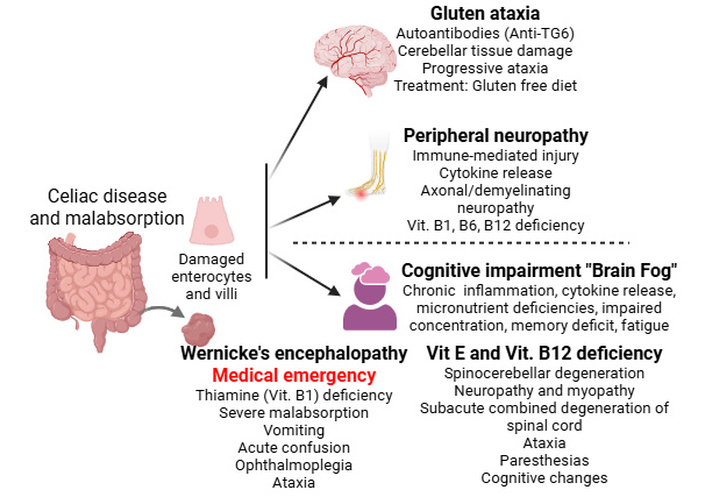
Neurological sequelae of malabsorption and micronutrient deficiencies in celiac disease. Chronic malabsorption and immune-mediated pathways in gastrointestinal disease led to diverse neurological complications, including gluten ataxia via anti-transglutaminase 6 (TG6) antibodies and “brain fog” driven by systemic inflammation. Deficiencies in vitamins (vit) B1, B6, B12, and E contribute to peripheral neuropathies and cognitive impairment, with specific vit E and B12 deficits causing spinocerebellar and subacute combined degeneration. Most critically, severe thiamine (B1) deficiency can precipitate Wernicke’s encephalopathy, a medical emergency characterized by ataxia, ophthalmoplegia, and acute confusion. Created in BioRender. Rai, V. (2026) https://BioRender.com/eme8jcf.
Neurological complications in celiac disease and malabsorption.
| Mechanism/Deficiency | Pathophysiological features | Neurological consequences |
|---|---|---|
| Gluten ataxia [131] | Autoantibodies against TG6 target cerebellar neurons; independent of gastrointestinal symptoms. | Progressive ataxia; halted by early gluten-free diet |
| Peripheral neuropathy [132] | Immune-mediated axonal/demyelinating injury; deficiencies in B1, B6, B12, E. | Sensory loss, paresthesia, motor weakness |
| Cognitive impairment “brain fog” [133] | Chronic inflammation and micronutrient deficits impair neurotransmission and energy metabolism. | Memory deficits, poor concentration, fatigue |
| Wernicke’s encephalopathy [134] | Severe B1 deficiency from malabsorption or vomiting; astrocyte dysfunction. | Confusion, ophthalmoplegia, ataxia; medical emergency |
| Vitamin E deficiency [135] | Oxidative stress and neuronal degeneration in spinocerebellar tracts. | Ataxia, neuropathy, myopathy |
| Vitamin B12 deficiency [136] | Demyelination of dorsal columns and corticospinal tracts. | Subacute combined degeneration: ataxia, paresthesia, cognitive decline |
Hepatic failure, whether acute or chronic, results from diverse etiologies including viral hepatitis, alcohol-associated liver disease, nonalcoholic steatohepatitis (NASH), autoimmune hepatitis, and drug-induced liver injury. Chronic liver disease affects over 1.5 billion individuals globally, with cirrhosis representing a major cause of morbidity and mortality [137].
Patients with hepatic failure present with jaundice, coagulopathy, ascites, portal hypertension, and metabolic disturbances [138]. Neurological manifestations range from subtle cognitive changes to profound encephalopathy, seizures, and coma [139]. Neuropsychiatric symptoms such as depression, anxiety, and sleep disturbances are common in chronic liver disease.
Acute liver failure is characterized by the rapid onset of hepatic dysfunction and encephalopathy in individuals without preexisting liver disease. Cerebral edema and intracranial hypertension are major causes of mortality [140]. Chronic liver failure (cirrhosis) develops gradually, with progressive fibrosis, portal hypertension, and recurrent episodes of hepatic encephalopathy [141]. Chronic neuroinflammation and metabolic derangements contribute to long-term cognitive impairment [142].
Neurological dysfunction in hepatic failure arises from metabolic toxins, neuroinflammation, and altered neurotransmission [142]. In contrast to celiac disease or IBD, in which inflammatory and nutritional pathways predominate, hepatic failure is characterized by the accumulation of neurotoxic metabolites in the setting of systemic inflammation, with astrocyte dysfunction serving as a central pathophysiological link between liver failure and brain injury.
Within this framework, the major mechanistic pathways contributing to these neurological consequences are: i) Ammonia accumulation: Impaired hepatic detoxification leads to elevated ammonia, which crosses the BBB and is metabolized by astrocytes into glutamine. Astrocyte swelling, oxidative stress, and impaired neurotransmission contribute to hepatic encephalopathy [143] (Figure 4). ii) Neuroinflammation: Systemic inflammation sensitizes the brain to ammonia toxicity. Elevated cytokines exacerbate astrocyte dysfunction and impair neuronal signaling [144] (Figure 4). iii) Altered neurotransmitter systems: Increased GABAergic tone, reduced glutamatergic signaling, and impaired monoamine metabolism contribute to cognitive slowing, lethargy, and mood disturbances [145] (Figure 4). iv) Wilson’s disease: A genetic disorder of copper metabolism causing hepatic dysfunction and neuropsychiatric symptoms, including tremor, dystonia, personality changes, and cognitive decline. Copper accumulation in the basal ganglia and liver exemplifies the metabolic neurological interface [146] (Figure 4). v) Mental health consequences: Depression, anxiety, and sleep disorders are prevalent in chronic liver disease (driven by inflammation), metabolic dysfunction, and psychosocial stressors [147] (Figure 4). These mechanisms are closely interconnected, as systemic inflammation increases susceptibility to ammonia toxicity, while neurotransmitter imbalance contributes to the spectrum of symptoms ranging from subtle mood changes to overt encephalopathy (Figure 4). Hepatic encephalopathy remains one of the most clinically significant neurological complications, with both reversible and irreversible components depending on duration and severity (Figure 4 and Table 3).
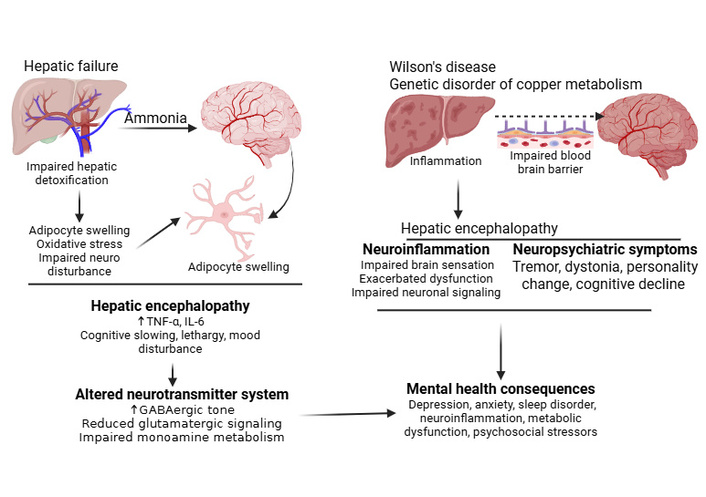
Pathophysiological mechanisms linking liver disease to neuropsychiatric manifestations. Hepatic failure leads to hepatic encephalopathy through ammonia-induced astrocyte swelling, oxidative stress, and neurotransmitter system alterations (left panel). Wilson’s disease, a genetic disorder of copper metabolism, causes hepatic encephalopathy via inflammation-mediated blood-brain barrier (BBB) disruption and subsequent neuroinflammation (right panel). Both pathways ultimately contribute to shared neuropsychiatric symptoms, including cognitive decline, mood disturbances, and personality changes, driven by convergent mechanisms of metabolic dysfunction and inflammatory processes. Created in BioRender. Rai, V. (2026) https://BioRender.com/jinoc73.
Neurological complications in hepatic failure.
| Mechanism/Condition | Pathophysiological features | Neurological consequences |
|---|---|---|
| Ammonia accumulation [143] | Impaired detoxification leads to elevated ammonia, astrocyte glutamine overload, and oxidative stress. | Hepatic encephalopathy: confusion, lethargy, impaired cognition |
| Neuroinflammation [144] | Systemic cytokines sensitize the brain to ammonia; astrocyte dysfunction. | Exacerbated hepatic encephalopathy, impaired neuronal signaling |
| Altered neurotransmitter systems [145] | Increased GABAergic tone, reduced glutamatergic signaling, and monoamine imbalance. | Cognitive slowing, mood disturbances, psychomotor impairment |
| Wilson’s disease [146] | Copper accumulation in the liver and basal ganglia, a genetic defect in copper metabolism. | Tremor, dystonia, personality changes, cognitive decline |
| Mental health consequences [147] | Chronic inflammation and metabolic stressors affect the brain and behavior. | Depression, anxiety, and sleep disturbances |
The clinical management of gut paresis in the context of neurological and gastrointestinal disease requires an integrated, systems‑level approach that acknowledges the bidirectional nature of the brain–gut axis [148]. Because motility disturbances arise from intertwined inflammatory, microbial, neuroendocrine, and autonomic mechanisms, effective care must extend beyond symptomatic treatment to address upstream drivers of dysfunction [16].
Clinical implications involve managing disorders of gut–brain interaction, such as IBD, optimizing metabolic, immune, and psychological health, and targeting microbiomes for therapeutic interventions like probiotics, prebiotics, and FMT [33, 149]. A central clinical implication is the need for early recognition of gastrointestinal symptoms in neurological disorders such as PD, MS, stroke, and autonomic neuropathies. Constipation, delayed gastric emptying, and bloating often preceded overt neurological decline, particularly in PD, where prodromal gut dysfunction may reflect early α‑synuclein pathology. Routine screening for gastrointestinal symptoms can therefore support earlier diagnosis, improve quality of life, and potentially slow disease progression by interrupting inflammatory and microbial feedback loops [8–11].
Management strategies typically combine pharmacologic, dietary, microbial, and neuromodulatory interventions. Prokinetic agents such as dopamine‑independent promotility drugs and serotonergic agonists remain foundational for symptomatic relief, though their efficacy varies across disease states [150]. Inflammatory contributors may be addressed through disease‑modifying therapies in MS or anti‑cytokine biologics in IBD, which can indirectly improve motility by reducing systemic inflammation [151, 152]. For patients with malabsorption or celiac disease, strict dietary adherence and targeted micronutrient repletion are essential to prevent irreversible neurological injury such as ataxia or neuropathy [153].
Microbiota‑directed therapies represent an expanding therapeutic frontier. Microbiota-based therapies for gut–brain axis neuropathies aim to modulate the gut microbiome to alleviate neuroinflammation and neurological disorders. Probiotics (beneficial bacteria), prebiotics (fiber for bacteria), and dietary fiber can help restore SCFA‑producing taxa and strengthen epithelial barrier integrity, while FMT is being explored for refractory dysbiosis‑associated motility disorders. Although evidence remains preliminary, microbial modulation may offer dual benefits by improving both gut function and neuroinflammatory tone [154, 155]. This is achieved by immune system modulation by targeting intestinal inflammation to prevent systemic inflammation and subsequent neuroinflammation. Neurotransmitter regulation by modulating the production of neurotransmitters like serotonin and dopamine, which are heavily influenced by gut bacteria, is another strategy. Strengthening the gut-blood barrier (tight junctions) to prevent toxic products from reaching the CNS may have therapeutic effects [156, 157]. This notion is supported by the results of gut microbiota-mediated modulation of distal symmetric polyneuropathy in patients with diabetes [158].
Neuromodulation strategies, including vagus nerve stimulation, biofeedback, and autonomic rehabilitation, target the disrupted signaling pathways that link central and enteric dysfunction [159]. These approaches are particularly relevant in stroke‑related ileus, autonomic neuropathies, and conditions characterized by reduced vagal tone. Stress‑reduction interventions, such as cognitive behavioral therapy (CBT) or mindfulness‑based programs, may further support motility by mitigating HPA axis overactivation [160]. Vagus nerve stimulation improves gut–brain neuropathies by reducing inflammation, enhancing gut motility, and strengthening the connection between the gastrointestinal tract and the CNS. By modulating the “cholinergic anti-inflammatory pathway,” vagus nerve stimulation lowers proinflammatory cytokines, decreasing chronic low-grade intestinal inflammation, helping treat disorders such as irritable bowel syndrome (IBS), IBD, and associated neuropsychiatric conditions. Vagus nerve stimulation strengthens the vagovagal reflex to improve gut motility, gastric emptying, and digestion. Vagus nerve stimulation shows potential to modulate gut-related neurodegenerative issues by influencing the brain’s response to gut microbiota imbalances [161, 162].
Both microbiota‑directed therapies and vagus nerve stimulation have a common denominator: suppressing inflammation. This suggests that targeted anti-inflammatory approaches may significantly improve gut–brain related neuropathies by reducing neuroinflammation, modulating the gut microbiota, and restoring intestinal barrier integrity [163]. Given the high prevalence of nutritional deficiencies in chronic gastrointestinal disease and hepatic failure, routine assessment of B vitamins, vitamin D, iron, copper, and fat‑soluble vitamins is critical. Early correction can prevent severe neurological sequelae such as Wernicke’s encephalopathy or subacute combined degeneration [164]. Nutritional supplementation improves gut–brain-related neuropathies by modulating the gut microbiome, reducing inflammation, and strengthening the gut barrier.
Mind-body therapies, particularly CBT and gut-directed hypnotherapy, represent a shift in gastrointestinal management from purely symptom-focused care toward addressing the underlying psychological and neuroendocrine drivers of disease. These therapies function by mitigating overactivation of the HPA axis, thereby reducing the stress-induced cortisol release that otherwise increases intestinal permeability and triggers immune activation [165]. By targeting bidirectional communication between the CNS and ENS, CBT can modulate visceral hypersensitivity and improve patient resilience to the chronic pain and bloating associated with IBS [166]. Similarly, gut-directed hypnotherapy has been shown to restore a more balanced autonomic tone, potentially enhancing the parasympathetic “rest and digest” signals that facilitate coordinated peristalsis [167]. These interventions are increasingly integrated into multidisciplinary care frameworks, alongside dietitians and gastroenterologists, to optimize the psychological health of patients while simultaneously improving physical gut motility and barrier function. Ultimately, these therapies acknowledge that managing the brain's perception of gut signals is as critical as managing the physiological state of the gut itself in achieving long-term clinical remission.
Ultimately, optimal management requires multidisciplinary coordination among neurologists, gastroenterologists, dietitians, and mental health professionals. As research continues to clarify mechanistic pathways, particularly those involving microbiota, epithelial permeability, and neuroimmune signaling, future therapies will likely shift toward personalized, mechanism‑based interventions that target the root causes of gut–brain dysfunction rather than its downstream manifestations [56].
The identification of biomarkers within the gut–brain axis represents a pivotal frontier in the transition toward precision medicine for neurological and gastrointestinal disorders. Microbiome-derived metabolites, particularly SCFAs such as butyrate, acetate, and propionate, have emerged as primary candidates for non-invasive disease markers due to their significant roles in maintaining the intestinal epithelial barrier and modulating systemic inflammation [168].
In neurological conditions such as PD, MS, and AD, clinical studies have consistently demonstrated a correlation between the depletion of SCFA-producing bacterial taxa and heightened levels of neuroinflammation and impaired gut motility [33, 169]. For instance, reduced butyrate levels are associated with compromised tight junction integrity, allowing for the translocation of luminal antigens and toxins that further drive central microglial activation [170]. Conversely, the presence of specific pro-inflammatory taxa and the alteration of microbial neurotransmitter synthesis (e.g., GABA and dopamine precursors) provide a metabolic “signature” that may enable clinicians to diagnose prodromal stages of neurodegeneration years before the onset of motor or cognitive symptoms [56].
Furthermore, in gastrointestinal diseases like IBD, microbiota-driven reductions in SCFAs are linked to HPA axis dysregulation and the shifting of tryptophan metabolism toward neurotoxic kynurenine metabolites, directly contributing to comorbid mood and cognitive disturbances [171]. By integrating these metabolite profiles with immune and neurophysiological data, future diagnostic frameworks could utilize a simple stool or blood sample to detect pathological feedback loops, allowing for earlier and more targeted therapeutic interventions.
At the same time, several limitations should be acknowledged. Biomarker profiles can vary substantially across cohorts due to differences in diet, medication exposure, disease stage, geography, sample collection and storage, sequencing platforms, and analytical pipelines, which may limit reproducibility between studies. In addition, many of the proposed biomarkers remain associated rather than disease-specific, and overlapping microbial or metabolite patterns across neurological and gastrointestinal disorders may reduce their diagnostic precision in routine practice. From a clinical perspective, stool and blood-based biomarker panels remain promising, but they still require standardized methodologies, validation in larger longitudinal cohorts, and integration with clinical findings before they can be applied reliably for diagnosis, risk stratification, or treatment monitoring.
Noninvasive microbiota-based biomarkers represent a transformative approach for early diagnosis and monitoring of neurological disorders through the gut–brain axis [172]. Fecal microbial profiling has emerged as a particularly promising avenue, with machine learning algorithms achieving high diagnostic accuracy for PD using just 11 key bacterial genera, including Alistipes, Anaerotruncus, Enterococcus, Porphyromonas, Scatomorpha, Limiplasma, Bifidobacterium, Christensenella, Streptococcus, Blautia, and Butyricicoccus [173]. These biomarkers offer significant advantages over traditional diagnostic methods by potentially detecting prodromal disease stages years before motor symptom onset. In advanced PD with motor complications, multiomics integration of fecal microbiome and plasma metabolome analysis has identified distinct signatures characterized by elevated Lactobacillus and Bifidobacterium alongside depleted Agathobacter, combined with plasma metabolites such as 3-deoxysappanchalcone and N-acetylisoleucine [174]. Beyond bacterial composition, microbial-derived metabolites serve as functional biomarkers. Specifically, dihydrouracil (often identified in context with related sulfur metabolites as 2,3-dihydroxypropane-1-sulfonate or DHPS) has been identified as a shared metabolic marker across AD, PD, and amyotrophic lateral sclerosis (ALS), correlating strongly with acetylated amino acids and taurine levels [175].
SCFA profiles, particularly butyrate-producing capacity, provide additional noninvasive indicators of gut–brain axis integrity, with reduced fecal butyrate correlating with intestinal barrier dysfunction and neuroinflammation. Enteric α-synuclein deposition detection in intestinal biopsies, while minimally invasive, offers pathological confirmation with phosphorylated α-synuclein detected in 72–74% of PD patients compared to healthy controls [176]. For clinical translation, quantitative PCR and 16S rRNA sequencing of fecal samples present cost-effective methodologies that could enable population-scale screening [177]. The integration of microbiome data with plasma metabolomics, such as caffeine metabolism pathways showing reduced theophylline and paraxanthine in PD patients, further enhances diagnostic specificity through multi-modal biomarker panels [178]. These noninvasive approaches hold promise for longitudinal disease monitoring and therapeutic response assessment, though standardization of analytical protocols and large-scale validation studies remain essential prerequisites for clinical implementation.
Therapeutically, current management strategies insufficiently address the interconnected nature of inflammation, dysbiosis, and autonomic dysfunction. Anti-inflammatory biologics improve systemic cytokine burden in IBD but do not consistently reverse neuropsychiatric symptoms [179]. Prokinetics offer partial relief in neurological gut paresis but do not modify disease progression. Microbiota-directed therapies remain promising but underdeveloped; probiotic formulations are heterogeneous, and FMT lacks standardized protocols and long-term safety data. Neuromodulation approaches such as vagus nerve stimulation show early potential but require rigorous trials to define optimal parameters and patient selection [159].
Based on the current literature as discussed in this review, future progress in understanding and managing gut–brain axis disorders will depend on addressing the substantial limitations of current diagnostic and therapeutic strategies. Although recognition of gut paresis and neurological complications of gastrointestinal disease has improved, clinical management remains largely symptomatic and fragmented. Most available treatments, including prokinetics, laxatives, dietary modification, immunosuppression, or micronutrient replacement, target downstream manifestations rather than the upstream neuroimmune, microbial, and epithelial mechanisms driving disease. This gap underscores the need for mechanistic, personalized approaches that integrate neurology, gastroenterology, immunology, and microbiome science. A major limitation is the absence of reliable biomarkers that capture early neuroenteric dysfunction. In conditions such as PD, α-synuclein pathology may begin in the gut years before motor symptoms, yet clinical tools to detect early ENS involvement remain rudimentary. Similarly, in IBD or celiac disease, neuropsychiatric symptoms often precede overt malabsorption or severe inflammation but are rarely incorporated into routine assessment. Future research must prioritize biomarker discovery, ranging from microbial signatures and metabolomic profiles to epithelial permeability markers and neurophysiological measures, to enable earlier diagnosis and risk stratification.
Despite growing interest in the gut–brain axis, much of the available literature remains associative rather than mechanistic. Many studies are cross-sectional, involve small sample sizes, or are derived from single-center cohorts, which limits generalizability and makes it difficult to determine whether gut alterations are causal, compensatory, or secondary to systemic disease. This is particularly relevant in disorders such as PD, MS, and TBI, where changes in diet, mobility, medication exposure, and disease stage may independently influence the microbiome and gastrointestinal function.
There is also substantial heterogeneity across studies. Reported microbial signatures are not always consistent between cohorts, and findings may vary depending on sequencing platform, geographic population, sample handling, dietary patterns, and concurrent therapies. Likewise, the clinical significance of increased intestinal permeability, inflammatory markers, or autonomic dysfunction is not uniform across disease states. In some settings, these abnormalities correlate with neurological burden or gastrointestinal symptom severity, whereas in others the relationship is weaker or remains uncertain. As a result, the field still lacks a clear distinction between shared downstream features of chronic disease and truly disease-specific gut–brain mechanisms.
Conflicting evidence is also evident at the therapeutic level. Probiotics, prebiotics, dietary interventions, and FMT have shown encouraging signals in some studies, but the magnitude and durability of benefit remain inconsistent. Differences in strain selection, treatment duration, patient population, and endpoint definition make comparison difficult. Similarly, while preclinical studies support roles for SCFAs, vagal signaling, TLR modulation, and epithelial barrier restoration, many of these findings have not yet been translated into robust human evidence. Animal models continue to provide important mechanistic insight, but they may oversimplify the chronic, heterogeneous, and bidirectional nature of human gut–brain disorders.
These limitations highlight several important gaps in knowledge. It remains unclear whether microbiome alterations and intestinal barrier dysfunction are primary drivers of neurological disease, early biomarkers of vulnerability, or downstream consequences of established pathology. It is also not yet known which gut-derived signals are most relevant to disease initiation versus progression, or which patient subgroups are most likely to benefit from targeted intervention. Addressing these gaps will require longitudinal human studies, standardized methodologies, better phenotyping, and closer integration of clinical, molecular, and functional data.
Emerging technologies may help address several of these limitations. Multi-omics approaches integrating metagenomics, metabolomics, transcriptomics, proteomics, and spatial profiling may better define disease-specific gut–brain signatures. Likewise, organoid systems, gut-on-chip platforms, and advanced neurophysiological assessment of enteric and autonomic pathways may provide more precise tools for studying host-microbe interactions and testing therapeutic strategies under controlled conditions. These methods may also help bridge the current gap between descriptive associations and actionable mechanisms.
Future clinical trials should adopt multidimensional endpoints that reflect both gastrointestinal and neurological outcomes. Trials integrating microbiome modulation with anti-inflammatory or neuromodulator therapies may be particularly impactful, given the synergistic nature of these pathways [180, 181]. For example, combining SCFA-enhancing interventions with vagal stimulation could simultaneously strengthen epithelial integrity and restore autonomic balance. Similarly, studies exploring agents that stabilize tight junctions, inhibit TLR signaling, or modulate α-synuclein propagation may offer disease-modifying potential in neurodegenerative disorders [182]. Several ongoing and completed clinical trial themes are summarized in Tables 4 and 5, including studies of prebiotics, multi-omics profiling, microbiome changes in epilepsy, TBI, delirium, obesity, and dietary interventions. Although some of these studies remain ongoing and others have reached completion, posted results are still limited for several trials, underscoring the need for more rigorous and clinically informative translational data.
Future perspectives in managing gut–brain axis disorders.
| Therapeutic area | Examples of current/emerging trials |
|---|---|
| Microbiome modulation [180, 181] | FMT for Parkinson’s-related constipation; targeted probiotics for MS-associated dysbiosis |
| Barrier restoration [182] | Agents enhancing tight junction integrity in IBD and neurodegenerative disease |
| Neuromodulation [159] | Vagus nerve stimulation for gastroparesis and autonomic dysfunction |
| Anti-inflammatory strategies [179] | Cytokine-targeted therapies assessing cognitive and mood outcomes in IBD |
| Metabolic therapies [183, 184] | Ammonia-lowering agents and astrocyte-targeted treatments for hepatic encephalopathy |
FMT: fecal microbiota transplantation; IBD: inflammatory bowel disease.
Ongoing clinical trials on the treatment of gut–brain axis disorders (based on clinicaltrials.gov).
| Trial ID/Phase/Type of study | Aim of the study | Intervention strategy | Status/Results |
|---|---|---|---|
| NCT03835468Phase: Not ApplicableType: RCT, single-center interventional study | To evaluate the effects of galacto-oligosaccharides (GOS) on the gut–brain axis, focusing on trait anxiety, neurochemistry (GABA/glutamate via 1H-MRS), gut microbiome composition, cognition, and nutritional outcomes in healthy young females. | Galacto-oligosaccharides (prebiotic) vs. placebo | Unknown status, was recruiting patients until 2021, no results posted |
| NCT06117930Type: Observational study | To explore how microorganisms in the gut can affect the growth and progression of the brain tumors using spatial transcriptome/proteomics/multi-omics coupled with microbiome data. | Bioinformatics analysis of multi-omics data from blood, tissue, saliva, urine, and stool samples | Recruiting patients |
| NCT07010445Phase: Not ApplicableMulticenter (Italy) | Microbiota–gut–brain axis in Resistant Epilepsy (CARE): To investigate the role of gut bacteria in drug-resistant epilepsy (DRE), exploring how gut microbiota changes influence seizures and how treatments (surgery, VNS, ketogenic diet) impact these changes. | Longitudinal stool/blood sampling in epilepsy patients | Recruiting patients |
| NCT02849028Phase: Not ApplicableType: Single-center observational study | The clinical research on the relationship between circadian rhythm and gut microbiota in TBI patients: to investigate gut microbiota differences between traumatic brain injury patients with and without sleep disorders. | Observational stool sampling in TBI patients | Status unclear, was recruiting patients till 2016 |
| NCT05628480Phase: Not ApplicableType: Single-center observational study | Multi-omics analyses reveal microbiota–gut–brain axis in ICU patients with post-cardiac surgery delirium: to investigate the microbiota–gut–brain axis mechanisms in postoperative delirium following cardiac surgery. | Multi-omics (stool/blood) in post-cardiac surgery patients | Completed (April 2024), no results posted |
| NCT04788836Phase: Not ApplicableType: Observational study(BrainFood Part 2) | To elucidate the changes in food behavior among obese adults undergoing obesity treatments, with the hypothesis that the altered gut–brain axis influences food choices.To observe the effect of obesity treatments on food purchase behavior using digital receipts from grocery shopping, with the hypothesis that bariatric surgery leads to changes in food purchase behavior in obese adults. | Looking at the digital receipts from grocery shopping to analyze the behavior of the type of food they are purchasing | Active, not recruiting patients |
| NCT04813003Phase: Not ApplicableType: Observational(BrainFood Part 1) | To elucidate the neurobehavioral underpinnings of food behavior among obese adults and how food behavior is altered by different obesity treatments. | Experimental setting combining neurobehavioral tasks, computational modelling, and functional brain imaging (surgery group vs. control group) | Completed, no results posted |
| NCT06020703Phase: Not ApplicableType: Interventional | Effects of diets rich in fermented food on adult mental health: Investigating the impact of fermented food consumption on mental health outcomes through the gut–brain axis. | Fermented food-rich diet | Active, not recruiting |
Looking ahead, the most transformative advances will likely arise from precision-medicine frameworks that integrate genomic risk, microbial ecology, immune profiling, and neurophysiological data. Such approaches could enable individualized treatment algorithms that not only alleviate symptoms but also interrupt the pathological feedback loops linking the gut and brain.
Looking ahead, the most transformative advances will likely arise from precision-medicine frameworks that integrate genomic risk, microbial ecology, immune profiling, and neurophysiological data. Such approaches could enable individualized treatment algorithms that not only alleviate symptoms but also interrupt the pathological feedback loops linking the gut and brain.
Advancements in scientific knowledge have expanded our understanding of the brain–gut axis as a bidirectional communication network between the CNS and the gastrointestinal tract, mediated through neural, endocrine, and immune pathways and influenced by the gut microbiome. Dysregulation of this system contributes to both gastrointestinal manifestations of neurologic diseases—including PD, MS, and spinal cord injury—and neurologic consequences of gastrointestinal disorders such as IBS, celiac disease, and hepatic failure. Importantly, these interactions may precede overt disease, offering opportunities for earlier detection. For example, subtle gastrointestinal symptoms in PD may serve as early indicators of disease onset, thereby alerting clinicians to evolving neurologic pathology. However, a major limitation remains the lack of reliable biomarkers capable of capturing early neuroenteric dysfunction. Future research should prioritize the identification of such markers—including microbial signatures, metabolomic profiles, epithelial permeability indices, and neurophysiological measures—to enable earlier diagnosis and improved risk stratification.
This narrative review highlights the brain–gut axis as a unifying mechanism across diverse disease states and aims to promote greater awareness of these often-underrecognized interactions. By emphasizing the clinical relevance of these mechanisms, this work seeks to support more informed, comprehensive care. Although emerging therapies targeting the microbiome and neuroimmune pathways show promise, further research, particularly in human populations, is needed to clarify their role in disease progression and management. Collectively, these findings underscore the importance of multidisciplinary care to improve patient outcomes.
AD: Alzheimer’s disease
ASD: autism spectrum disorder
BBB: blood-brain barrier
CBT: cognitive behavioral therapy
CNS: central nervous system
ENS: enteric nervous system
FMT: fecal microbiota transplantation
GABA: gamma-aminobutyric acid
HPA: hypothalamic–pituitary–adrenal
IBD: inflammatory bowel disease
IBS: irritable bowel syndrome
ICC: Interstitial Cells of Cajal
IL: interleukin
LPS: lipopolysaccharide
LRRK2: leucine-rich repeat kinase 2
MS: multiple sclerosis
NF-κB: nuclear factor-kappa B
PD: Parkinson’s disease
SCFA: short-chain fatty acid
TLRs: toll-like receptors
TNF: tumor necrosis factor
AB: Conceptualization, Writing—original draft. SS: Conceptualization, Writing—original draft. KH: Writing—original draft. VR: Conceptualization, Writing—original draft, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1166
Download: 44
Times Cited: 0
