 Review
Review

Affiliation:
1Department of Biotechnology, School of Applied Sciences, REVA University, Bengaluru 560064, Karnataka, India
2Center for Biotechnology and Molecular Biology, REVA Research Center, REVA University, Bengaluru 560064, Karnataka, India
Email: kiranm0897@gmail.com
ORCID: https://orcid.org/0000-0002-2404-3254
Affiliation:
1Department of Biotechnology, School of Applied Sciences, REVA University, Bengaluru 560064, Karnataka, India
2Center for Biotechnology and Molecular Biology, REVA Research Center, REVA University, Bengaluru 560064, Karnataka, India
Email: halderagnik@gmail.com
ORCID: https://orcid.org/0000-0002-0725-1406
Affiliation:
1Department of Biotechnology, School of Applied Sciences, REVA University, Bengaluru 560064, Karnataka, India
2Center for Biotechnology and Molecular Biology, REVA Research Center, REVA University, Bengaluru 560064, Karnataka, India
Email: ankitachatterjee16@gmail.com
ORCID: https://orcid.org/0000-0002-0659-5469
Explor Dig Dis. 2026;5:1005116 DOI: https://doi.org/10.37349/edd.2026.1005116
Received: December 21, 2025 Accepted: March 02, 2026 Published: March 26, 2026
Academic Editor: Jose C. Fernandez-Checa, Institute of Biomedical Research of Barcelona (IIBB), CSIC, Spain
The article belongs to the special issue Inflammatory Diseases of the Gastrointestinal Tract
Microbial metabolites are now recognized as central mediators of host–microbe communication that shape intestinal immune homeostasis and influence the development of inflammatory gastrointestinal diseases. The objective of this review is to synthesize current mechanistic evidence on how microbiota-derived metabolites regulate epithelial and immune functions in the gut, with a focus on metabolite-driven inflammatory pathways. In the healthy intestine, short-chain fatty acids (SCFAs), indole derivatives, secondary bile acids, and polyamines support epithelial integrity, regulate mucosal immunity, and maintain metabolic balance. SCFAs, particularly butyrate, attenuate inflammation by serving as an energy source for colonocytes, inhibiting histone deacetylases, activating G protein-coupled receptors (GPCRs; GPR41, GPR43, GPR109A), and reinforcing epithelial barrier function. In parallel, microbial tryptophan metabolites such as indole-3-propionic acid and indole-3-aldehyde activate aryl hydrocarbon receptor signaling, promoting IL-22 production, antimicrobial peptide expression, and Th17–Treg balance. In inflammatory bowel disease, dysbiosis disrupts these protective pathways, leading to depletion of SCFA- and indole-producing taxa and accumulation of pro-inflammatory metabolites such as succinate. These metabolic shifts impair epithelial-immune crosstalk, amplify NF-κB-dependent inflammation, and compromise mucosal repair. Therapeutic strategies targeting microbial metabolites, including precision prebiotics, next-generation probiotics, engineered microbial consortia, and postbiotics, show translational promise. However, their clinical application remains constrained by interindividual variability, incomplete causal resolution, and challenges in targeted delivery. Integrative multi-omics approaches and mechanistically informed models are therefore essential to advance metabolite-based diagnostics and therapies for gut inflammation.
The gastrointestinal tract is an immunologically active organ where host and microbial processes are integrated to maintain mucosal homeostasis [1]. Microbial metabolites, small molecules produced or modified by gut microbes from dietary and host substrates, constitute a principal axis of host–microbe communication and exert pleiotropic effects on epithelial integrity, innate and adaptive immunity, and systemic metabolic regulation [2]. Short-chain fatty acids (SCFAs; acetate, propionate, butyrate) and microbial tryptophan catabolites (indole derivatives, kynurenines, and related compounds) have emerged as prototype mediators that translate microbial ecological states into specific signal cascades in host tissues [3]. The biological importance of these metabolites is underscored by convergent evidence from metabolomics, gnotobiotic models, and clinical cohorts showing that metabolite fluxes tightly correlate with inflammatory phenotypes in inflammatory bowel disease (IBD) and other chronic intestinal disorders [4].
Mechanistically, SCFAs modulate intestinal inflammation through at least three interdependent modalities: receptor-mediated signaling, epigenetic regulation, and metabolic support of the epithelium. SCFAs engage G protein-coupled receptors (GPCRs; e.g., GPR41/FFAR3, GPR43/FFAR2, GPR109A) on immune and epithelial cells to influence T-cell differentiation, neutrophil chemotaxis, and cytokine secretion, while butyrate potently inhibits class I/II histone deacetylases (HDACs) to reprogram inflammatory gene expression and promote regulatory T-cell (Treg) induction [3]. In parallel, butyrate and other SCFAs serve as primary energy substrates for colonocytes, thereby reinforcing barrier integrity and mucosal repair; the loss of these functions is mechanistically linked to barrier dysfunction and sustained nuclear factor kappa-B (NF-κB) activation in disease [5].
Tryptophan metabolism provides a complementary immunomodulatory axis. Microbial conversion of tryptophan yields indole derivatives [e.g., indole-3-propionic acid (IPA), indole-3-aldehyde (IAld)] that act as endogenous ligands for the aryl hydrocarbon receptor (AhR), driving IL-22 production, antimicrobial peptide expression, and epithelial regeneration processes central to mucosal resilience [6]. Concurrently, host-driven catabolism via indoleamine-2,3-dioxygenase-1 (IDO1) funnels tryptophan into the kynurenine pathway; kynurenines exert context-dependent effects that can either promote immune tolerance or, under persistent activation, contribute to epithelial dysfunction and pro-inflammatory signaling [4, 7]. Dysregulated tryptophan pathway balance, reflected by altered kynurenine/tryptophan (Kyn/Trp) ratios and depleted indole signals, is increasingly implicated in IBD pathophysiology and in extra-intestinal sequelae mediated by the gut-brain axis [8].
A defining feature of IBD is microbial dysbiosis accompanied by systematic shifts in metabolite landscapes: depletion of canonical SCFA- and indole-producing taxa (e.g., Faecalibacterium prausnitzii, certain Clostridia, and Akkermansia) and relative enrichment of pathobionts that generate pro-inflammatory intermediates such as succinate. These metabolomic signatures correlate with disease activity, mucosal ulceration, and impaired regenerative signaling in multiple cohort studies and mechanistic models, supporting a causal contribution of metabolite perturbations to chronic mucosal inflammation (Figure 1) [9]. However, substantial interindividual variability, context dependence of metabolite effects, and limited longitudinal causal data in humans complicate translation. Addressing these gaps will require integrated multi-omics, longitudinal cohort studies, and refined gnotobiotic and synthetic-ecology approaches to resolve causality and enable metabolite-targeted therapeutics [10].
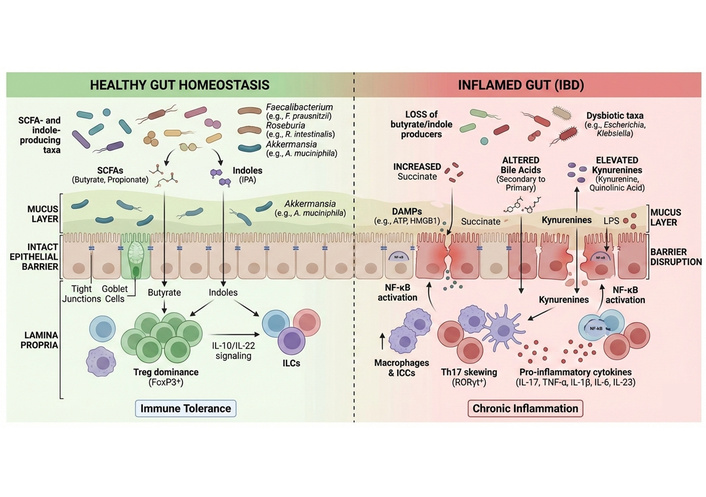
Comparative metabolite-immune landscapes in healthy gut homeostasis and IBD. This figure contrasts homeostatic vs. inflamed intestinal ecosystems. Healthy gut: SCFA- and indole-producing taxa (Faecalibacterium prausnitzii, Roseburia intestinalis, Akkermansia muciniphila) dominate, leading to high butyrate and indole levels. These metabolites maintain mucus integrity, reinforce tight junctions, and promote FOXP3+ Treg dominance and IL-10/IL-22 signaling, resulting in immune tolerance. Inflamed gut (IBD): Loss of butyrate- and indole-producing commensals is accompanied by expansion of dysbiotic taxa (e.g., Escherichia, Klebsiella). Accumulation of succinate, altered bile acid pools, elevated kynurenines, and LPS activate NF-κB signaling, macrophages, and Th17 cells. Barrier disruption, oxidative stress, and excessive pro-inflammatory cytokine release (IL-17, TNF-α, IL-1β, IL-6, IL-23) perpetuate chronic inflammation. FOXP3: forkhead box P3; IBD: inflammatory bowel disease; LPS: lipopolysaccharide; NF-κB: nuclear factor kappa-B; SCFA: short-chain fatty acid; Th17: T helper 17 cell; Treg: regulatory T-cell.
Gut microbial metabolism is the biochemical bridge that converts dietary macromolecules and host secretions into a chemically diverse repertoire of small molecules with potent biological activity [11]. Complex carbohydrates resistant to host digestion (e.g., resistant starches, non-digestible oligosaccharides, and plant cell wall polysaccharides) are fermented by anaerobic commensals to generate SCFAs (acetate, propionate, butyrate) and gases; concurrently, bacteria convert amino acids (notably tryptophan) into indoles and related derivatives, and transform primary bile acids into a pool of secondary bile acids with distinct receptor specificities [12]. Production rates and metabolite stoichiometry are highly context dependent, shaped by substrate availability (dietary fiber amount and composition), community structure (presence/absence of canonical producers), and ecological interactions such as cross-feeding and competition (Figure 2). Functionally relevant examples include Clostridia and members of Faecalibacterium, Roseburia, and Eubacterium taxa that possess butyryl-CoA:acetate CoA-transferase or butyrate kinase pathways and thereby sustain colonic butyrate flux, whereas specific Lactobacillus and Bacteroides spp. are prominent producers of acetate and propionate under different substrate regimes [13]. This metabolic specialization creates compartmentalized gradients of metabolite concentrations along the lumen mucosa axis and underpins microbe to host chemical signaling [14].
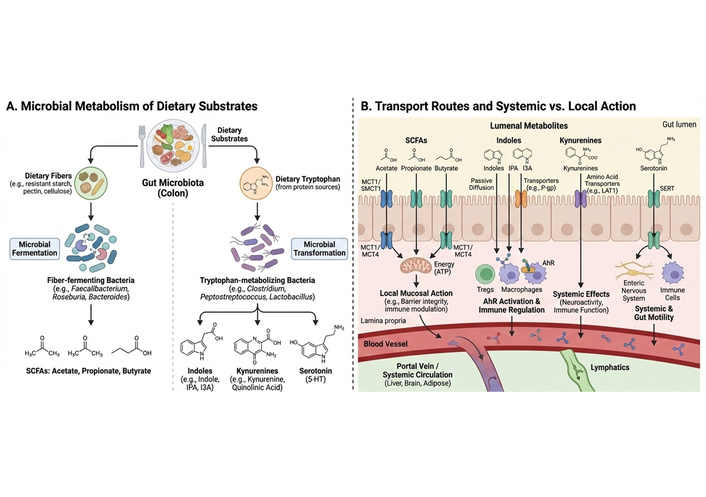
Microbial metabolism of dietary substrates and transport-dependent local vs. systemic actions of gut metabolites. (A) Microbial metabolism: Dietary fibers are fermented by gut microbiota (e.g., Faecalibacterium, Roseburia, Bacteroides) to generate SCFAs (acetate, propionate, butyrate). Dietary tryptophan is metabolized by gut microbes into indole and indole-derived compounds that signal through the AhR, or diverted toward host kynurenine pathway metabolism under inflammatory conditions. Serotonin (5-HT) is synthesized primarily by host enterochromaffin cells from tryptophan, with gut microbiota indirectly regulating its production through microbial metabolites and signaling cues, rather than serving as a direct microbial metabolite. (B) Transport and bioavailability: SCFAs are absorbed through epithelial transporters (MCT1, SMCT1) and act locally to support epithelial ATP production and immune modulation. Indoles cross the epithelium via passive diffusion or transporter-mediated mechanisms and activate AhR in immune cells. Kynurenines enter systemic circulation through amino acid transporters (e.g., LAT1), exerting effects on immune and neural tissues. Serotonin is transported via the SERT and influences gut motility and immune signaling. Portal vein and lymphatic routes distribute metabolites to peripheral organs, including the liver and brain. 5-HT: 5-hydroxytryptamine; AhR: aryl hydrocarbon receptor; ATP: adenosine triphosphate; LAT1: large neutral amino acid transporter-1; MCT1: monocarboxylate transporter-1; SCFAs: short-chain fatty acids; SERT: serotonin transporter; SMCT1: sodium-coupled monocarboxylate transporter-1.
The dominant and best-characterized microbial metabolites fall into several mechanistic classes with distinct cellular targets. SCFAs (C2–C4) function both as energy substrates for colonocytes, particularly butyrate, and as signaling ligands that engage GPCRs (GPR41/FFAR3, GPR43/FFAR2, GPR109A) and inhibit HDACs, thereby coupling cellular metabolism to epigenetic control of inflammatory gene programs. Indole and indole-derivative metabolites (e.g., IPA, IAld, indole-3-acetic acid) arise from bacterial tryptophan catabolism and act as endogenous ligands for the AhR, which orchestrates IL-22 production, antimicrobial peptide expression, and epithelial repair responses [15]. The kynurenine axis, principally a host-mediated pathway initiated by IDO1 but modulated by microbial activity, yields metabolites (kynurenine, 3-hydroxykynurenine, quinolinic acid) with context-dependent immunomodulatory properties that can promote tolerance or, upon chronic activation, contribute to oxidative stress and epithelial dysfunction [16]. Secondary bile acids and other small molecules (succinate, lactate, polyamines) further diversify the signaling landscape: for example, succinate accumulation has been linked to pro-inflammatory macrophage activation and inflammasome priming in experimental models. The distinct receptor repertoire and intracellular mechanisms engaged by each class explain why individual metabolites can have convergent or opposing effects on mucosal immunity [12].
Primary bile acids synthesized by the host (cholic acid, chenodeoxycholic acid) are extensively modified by the intestinal microbiota via deconjugation, 7α-dehydroxylation, and epimerization to yield secondary bile acids [for example, deoxycholic acid (DCA), lithocholic acid (LCA), and their derivatives]. These bacterially derived bile acids are potent signaling ligands that engage nuclear and membrane receptors, notably farnesoid X receptor (FXR) and G-protein coupled bile-acid receptor 1 (TGR5) to regulate epithelial barrier function, mucosal immune responses, and enterohepatic metabolic circuits [17]. Microbiome-driven losses in SBA-producing taxa or enzyme pathways alter the primary: secondary bile-acid ratio and shift receptor activation patterns, which in turn modulate antimicrobial peptide expression, mucosal inflammation, and epithelial regeneration. Recent clinical metabolomic studies report reduced circulating and fecal secondary bile acids and an elevated primary-to-secondary ratio in active IBD cohorts, supporting a model where dysbiosis-driven perturbation of bile acid biotransformation contributes to mucosal immune dysregulation [18].
Microbial and host pathways jointly regulate luminal and mucosal levels of polyamines—small, polycationic metabolites—as well as related amino acids such as ornithine and arginine, which support epithelial proliferation, autophagy, and barrier restitution. Polyamines act on epithelial cells and immune populations by modulating MAPK/ERK signaling, enhancing tight-junction assembly, and promoting cytoprotective transcriptional programs (including NRF2 and autophagy-related genes) [19]. Experimental models demonstrate that exogenous spermidine ameliorates chemically induced colitis and rescues epithelial barrier function via AhR-linked and antioxidant pathways, while microbiome studies indicate both loss of microbial polyamine biosynthetic capacity and altered host polyamine turnover in dysbiotic IBD samples. These observations position polyamines as metabolite effectors that can either support mucosal repair or, if dysregulated, contribute to pathological epithelial proliferation and inflammation [20].
Bioavailability of microbial metabolites depends on physicochemical properties, luminal concentration, mucosal transport mechanisms, and first-pass metabolism. SCFAs are absorbed across the colonic epithelium both by passive diffusion and via monocarboxylate transporters (MCT1/SLC16A1 and SMCT1/SLC5A8), with MCT1 prominently expressed in colonic enterocytes, facilitating butyrate uptake that fuels mitochondrial β-oxidation and supports barrier integrity [21]. Measured fecal SCFA concentrations are an imperfect proxy for mucosal exposure because substantial fractions are consumed by colonocytes or metabolized by hepatic first-pass processes; nevertheless, reductions in fecal and mucosal butyrate are consistently reported in active IBD cohorts, indicating a net loss of epithelial fuel supply in disease. Indole derivatives, smaller and more lipophilic, can diffuse across the mucosa and reach systemic compartments where they act on distant AhR-expressing cells (including immune and hepatic populations), while kynurenines are transported in plasma and have been used clinically as biomarkers, most notably the Kyn/Trp ratio, which is elevated in inflammatory conditions and correlates with disease activity in multiple studies. Interindividual variability in transporter expression, mucosal metabolism (e.g., epithelial IDO1 induction), and microbial production rates generates marked heterogeneity in circulating and tissue-level metabolite exposure, a likely contributor to the variable clinical responses observed in metabolite-targeted interventions (Figure 2) [10].
Butyrate is a major oxidative substrate for differentiated colonocytes, and ex vivo metabolic studies indicate that oxidation of butyrate can account for the majority of oxidative adenosine triphosphate (ATP) generation in mature colonocytes. Classic experimental work using isolated human colonocytes and tissue respiration assays estimated that bacterial SCFAs, predominantly butyrate, supply the bulk of colonic epithelial oxidative energy (early human ex vivo estimates) [22]. More recent mechanistic studies in mice (including germ-free models) using oxygen-consumption and mitochondrial respiration assays show that butyrate supplementation restores mitochondrial oxidative phosphorylation and ATP production in colonocytes that are energy-deprived in the absence of microbiota. Because these estimates derive from ex vivo colonocyte respiration measurements (human and rodent) and in vivo germ-free/rescue experiments, we therefore qualify earlier point estimates and present them as representative ranges from direct tissue/cell respiration assays rather than precise global percentages for all contexts [23].
When butyrate availability is sufficient, colonocyte β-oxidation maintains epithelial hypoxia that, in turn, preserves an anaerobic luminal niche favorable to butyrate-producing taxa, establishing a homeostatic feedback loop [24, 25]. Mechanistically, butyrate exerts anti-inflammatory effects via at least two interlinked molecular axes: (i) inhibition of class I/II HDACs, which remodels chromatin to repress pro-inflammatory transcriptional programs and promotes Treg differentiation; and (ii) engagement of GPCRs (GPR109A, GPR43/FFAR2, GPR41/FFAR3) on epithelial and immune cells to trigger downstream signaling cascades that mitigate NF-κB activation and foster IL-10/IL-22-mediated protective responses. Experimental models consistently show that butyrate supplementation or restoration of butyrate-producing taxa reduces mucosal cytokine production (e.g., TNF-α, IL-6) and preserves tight-junction protein expression, while butyrate depletion impairs epithelial energetics and predisposes to barrier breakdown [26, 27] (Figure 3).
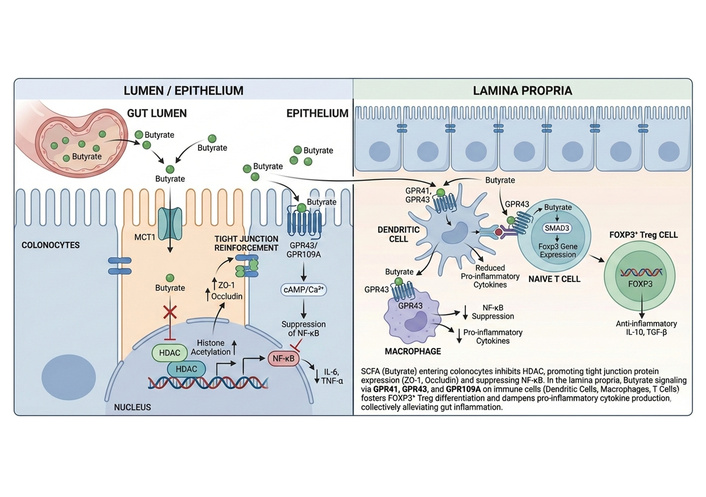
Mechanistic regulation of epithelial and immune responses by butyrate across the gut mucosa. This schematic illustrates the cell-specific actions of butyrate within the gut lumen, epithelium, and lamina propria. In the intestinal lumen, butyrate produced by microbial fermentation is transported into colonocytes via MCT1. Intracellular butyrate inhibits HDACs, increasing histone acetylation and suppressing NF-κB-driven transcription of pro-inflammatory cytokines (IL-6, TNF-α). Butyrate simultaneously enhances tight junction integrity by upregulating ZO-1 and occludin expression. In the lamina propria, butyrate activates G protein-coupled receptors (GPR41, GPR43, GPR109A) on dendritic cells and macrophages, reducing pro-inflammatory cytokine secretion and dampening innate immune activation. Butyrate signaling in naïve T-cells promotes SMAD3-dependent FOXP3 expression, driving differentiation into anti-inflammatory FOXP3+ regulatory T-cells that secrete IL-10 and TGF-β. Collectively, these epithelial and immune effects converge to restore mucosal tolerance and attenuate gut inflammation. MCT1: monocarboxylate transporter-1; HDACs: histone deacetylases; NF-κB: nuclear factor kappa-B; ZO-1: zonula occludens-1; FOXP3: forkhead box P3; TGF-β: transforming growth factor-beta.
Although often grouped with butyrate, acetate, and propionate have distinct cellular fates and immunomodulatory footprints. Acetate produced by a broad array of gut microbes is the most abundant fecal SCFA and reaches peripheral circulation at higher concentrations than butyrate; it modulates systemic immunometabolic axes (including gluconeogenesis and lipogenesis) and can indirectly influence mucosal immunity via metabolic programming of immune cells. Propionate, produced primarily by Bacteroidetes and some Firmicutes, signals through GPR41/43 to modulate dendritic cell (DC) maturation and T-cell polarization and has been shown to suppress T helper 2 cell (Th2) and Th17 responses in certain contexts [27]. Both acetate and propionate also inhibit HDACs, albeit with lower potency than butyrate, and contribute to mucin production and mucus layer integrity. Notably, the immunological effects of acetate and propionate are concentration- and context-dependent: at physiological concentrations, they are typically anti-inflammatory, but supraphysiological or ectopic exposure, depending on epithelial transporter expression and local metabolic sinks, can produce divergent outcomes in murine models [28, 29].
Multiple human studies and aggregated meta-analyses document a reproducible depletion of fecal and mucosal SCFAs, most prominently butyrate, in patients with active IBD. A recent systematic review and meta-analysis reported a statistically significant reduction in fecal butyrate concentrations in active IBD (subgroup analysis; p = 0.004) and showed consistent decreases in acetate and propionate across disease cohorts, with ulcerative colitis exhibiting particularly pronounced propionate loss. Complementary studies demonstrate reduced genomic potential for butyrate synthesis in mucosal microbiomes from inflamed regions, indicating both compositional and functional deficits in SCFA production [10]. The pathophysiological consequences are coherent: reduced butyrate availability diminishes colonocyte oxidative metabolism, increases epithelial oxygenation, and thereby favors facultative pathobionts (e.g., Enterobacteriaceae), establishing a feed-forward loop that exacerbates inflammation. However, heterogeneity exists—some cohorts report paradoxically higher fecal butyrate in subsets of patients (for example, treated or remitted individuals), underscoring that fecal concentrations reflect the net of production, mucosal uptake, and host metabolism rather than simple community output. This complexity argues for paired mucosal and luminal metabolomics, functional gene profiling, and standardized sample handling in clinical studies to resolve causal relationships and identify which patient subgroups may benefit from SCFA-targeted interventions [30, 31].
Microbial conversion of dietary tryptophan yields a chemically diverse set of indole derivatives: IPA, indole-3-acetate (IAA), IAld, indole-3-carboxaldehyde (I3C), and related compounds that function as low-molecular-weight, high-affinity ligands for the AhR. Activation of AhR in innate and adaptive mucosal immune cells is a recurring mechanistic theme linking microbial metabolism to tissue-protective programs: AhR signaling promotes IL-22 production from group 3 innate lymphoid cells (ILC3s) and certain T-cell subsets, upregulates antimicrobial peptides [e.g., regenerating islet-derived protein 3 gamma (Reg3γ)], and supports epithelial regeneration and goblet-cell function. Mechanistic and interventional studies demonstrate that supplementation or restoration of indole-producing taxa (for example, Lactobacillus species that convert Trp to IAld) increases mucosal IL-22 and reduces susceptibility to chemically induced colitis in mice, while genetic or pharmacologic blockade of AhR abrogates these protections [32]. These relationships are robust across models and have been replicated in human-relevant systems, indicating that indole–AhR–IL-22 signaling is a conserved axis of mucosal resilience. Quantitative targeted metabolomics of tryptophan-derived species indicate that indole and indole-derivative compounds are often the dominant fraction of microbial Trp catabolites detected in luminal samples, with primary data from targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses in mouse caecal contents and human feces reporting that indole species may comprise roughly ~50–75% of measured tryptophan-derived molecules in those compartments [33]. These estimates reflect stool/caecal lumen measurements obtained using targeted liquid chromatography-mass spectrometry (LC-MS) platforms and should be interpreted as compartmental (luminal) proportions that may not reflect mucosal or systemic distributions [34, 35] (Figure 4).
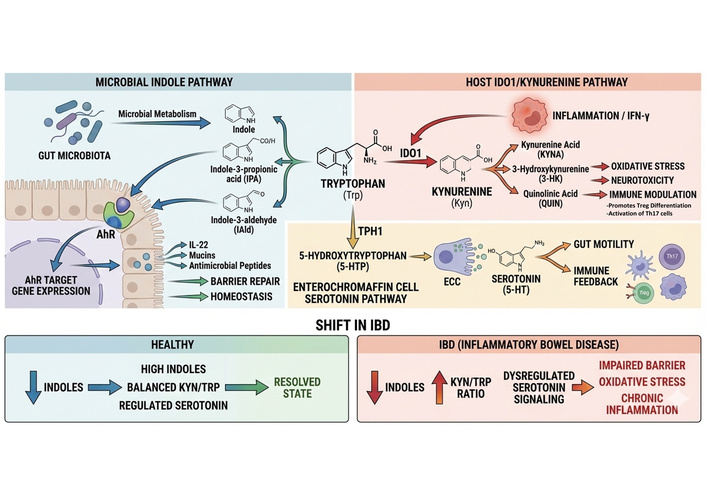
Tryptophan metabolic partitioning between microbial indole, host kynurenine, and serotonin pathways in gut inflammation. This figure depicts the three major fates of dietary tryptophan (Trp) at the host–microbe interface and their immunological consequences. Left panel (microbial indole pathway): gut microbiota convert Trp into indole derivatives, including IPA and IAld. These metabolites activate the AhR in epithelial and immune cells, inducing IL-22, mucins, and antimicrobial peptides, thereby promoting barrier repair and intestinal homeostasis. Right panel (host IDO1/kynurenine pathway): inflammatory signals (e.g., IFN-γ) induce IDO1, diverting Trp toward kynurenine (Kyn) and downstream metabolites (kynurenic acid, 3-hydroxykynurenine, quinolinic acid). These metabolites contribute to oxidative stress, neurotoxicity, and immune dysregulation, including suppression of Tregs and promotion of Th17 responses. Bottom panel (serotonin pathway): enterochromaffin cells metabolize Trp via TPH1 to produce serotonin (5-HT), which regulates gut motility and immune feedback. Disease shift: In IBD, indole production is reduced, the Kyn/Trp ratio increases, and serotonin signaling becomes dysregulated, collectively driving barrier dysfunction and chronic inflammation. 5-HT: 5-hydroxytryptamine; AhR: aryl hydrocarbon receptor; IAld: indole-3-aldehyde; IBD: inflammatory bowel disease; IDO1: indoleamine-2,3-dioxygenase-1; IFN-γ: interferon-gamma; IPA: indole-3-propionic acid; TPH1: tryptophan hydroxylase-1; Tregs: regulatory T-cells.
Despite clear protective activities, indole effects are context dependent. Different indole ligands exhibit distinct AhR binding affinities and downstream transcriptional programs; IAld and IPA preferentially bias IL-22 and barrier functions, whereas other indoles can engage additional receptors [e.g., pregnane X receptor (PXR)] to modulate epithelial metabolism and fibrosis. Furthermore, excessive or dysregulated AhR activation in chronically inflamed tissue can shift signaling toward fibrogenic or tolerogenic pathways that paradoxically impair pathogen clearance [36]. Thus, although indole supplementation or probiotic strategies that boost indole production have demonstrated efficacy in preclinical colitis models (reduced histological scores, lower mucosal TNF-α and IL-6), careful ligand selection and dosing will be essential for clinical translation [37].
Tryptophan metabolism in the intestine is bifurcated between microbial indole-producing routes and host-mediated oxidative catabolism via IDO1 and tryptophan 2,3-dioxygenase (TDO), which channel Trp into the kynurenine pathway. Kynurenines (3-hydroxykynurenine, kynurenic acid, quinolinic acid) have pleiotropic, concentration- and cell-type-dependent effects: some metabolites foster immune tolerance [e.g., kynurenic acid acting at GPR35 or N-methyl-D-aspartate (NMDA) receptors], whereas others (notably 3-HK and quinolinic acid) are pro-oxidant and neuroactive. Clinical and experimental evidence implicates up-regulation of the IDO1 Kyn axis in intestinal inflammation: IDO1 expression is induced in inflamed mucosa and correlates with histological severity, and experimental colitis models show increased mucosal and systemic Kyn and an elevated Kyn/Trp ratio, reflecting increased Trp catabolic flux. A 2023 systematic review and meta-analysis across inflammatory and rheumatologic disorders reported a standardized mean difference (SMD) of 0.88 for the Kyn/Trp ratio in disease vs. control cohorts, indicating a consistent shift toward kynurenine production in inflammatory states; similar directional changes have been observed in IBD cohorts and in dextran sodium sulfate (DSS) colitis models. Functionally, short-term activation of IDO1 can limit excessive T-cell responses and dampen inflammation, but chronic IDO1 induction may sustain a metabolite milieu that favors oxidative epithelial injury, altered mitochondrial function, and impaired mucosal restitution. These dual roles complicate the straightforward interpretation of elevated Kyn/Trp as uniformly protective or pathogenic and underline the need for temporal and compartmental resolution in clinical measurements [38, 39] (Figure 4).
From a translational perspective, the kynurenine pathway offers both biomarker and target opportunities. Kyn/Trp is increasingly used as a peripheral marker of Trp catabolism and systemic immune activation; in several cohorts, its elevation correlates with disease activity indices and extra-intestinal manifestations [33]. Modulating IDO1 enzymatic activity, or targeting downstream enzymes that control the balance between neurotoxic and neuroprotective kynurenines, has shown promise in preclinical models to reduce colitis severity and associated neuroinflammation, but clinical application remains nascent due to concerns about immune suppression, off-target CNS effects, and the pathway’s metabolic pleiotropy [40].
Beyond indoles and kynurenines, tryptophan is the precursor for peripheral serotonin [5-hydroxytryptamine (5-HT)], synthesized predominantly by enterochromaffin (EC) cells via tryptophan hydroxylase-1 (TPH1). Gut-derived 5-HT shapes motility and secretion but also exerts immune-modulatory effects by acting on enteric neurons, epithelial cells, and multiple immune receptors; notably, 5-HT influences DC phenotype, modulates T-cell polarization, and can alter epithelial antimicrobial peptide release [41]. Emerging experimental work indicates that altered mucosal 5-HT homeostasis contributes to colitis susceptibility: elevated mucosal 5-HT is associated with more severe inflammation in several animal models, and microbiota composition both influences and is influenced by mucosal 5-HT levels. 5-HT selects for a more colitogenic microbiota in a species-dependent manner and can suppress epithelial β-defensin production, thereby creating a feedback loop that perpetuates mucosal inflammation (Figure 4). Clinically, measurements of mucosal and systemic 5-HT in IBD patients have yielded variable results, reflecting differences in sampling (tissue vs. plasma), treatment status, and EC cell plasticity; nonetheless, these data collectively highlight serotonin as an important effector of Trp-derived signaling and a potential modulator of microbiome immune crosstalk [42, 43].
Tryptophan metabolism in the gut represents a competitive and dynamically regulated interface between microbial and host pathways, wherein flux toward indole derivatives or kynurenine metabolites is determined by substrate availability, inflammatory cues, and enzymatic dominance. Under homeostatic conditions, commensal bacteria expressing tryptophanase and related enzymes convert dietary tryptophan into indole and indole-derived ligands, thereby limiting the pool of free tryptophan available for host IDO1 [44]. This microbial sequestration of tryptophan indirectly constrains kynurenine pathway activation and sustains AhR-dependent epithelial and immune homeostasis. Stable-isotope tracing and targeted metabolomics in murine models have demonstrated that enhanced microbial indole production is associated with reduced kynurenine accumulation and diminished IDO1 flux in the intestinal mucosa, supporting a substrate-competition model of pathway regulation [45].
During intestinal inflammation, this balance is shifted by cytokine-driven induction of host IDO1, particularly in response to interferon-γ and other pro-inflammatory signals. Elevated IDO1 expression diverts tryptophan toward kynurenine synthesis, resulting in depletion of luminal tryptophan and a concomitant reduction in microbial indole output. Recent experimental studies combining gnotobiotic mouse models with LC-MS/MS metabolomics demonstrate that inflammatory activation of the kynurenine pathway suppresses indole AhR signaling, leading to impaired epithelial barrier function and altered Th17–Treg equilibrium. These findings indicate that kynurenine pathway dominance is not merely a downstream consequence of inflammation but actively reshapes microbial metabolic output [46].
Importantly, indole derivatives and kynurenine metabolites exert reciprocal immunoregulatory effects that further reinforce pathway bias. Indole-AhR signaling promotes epithelial regeneration, IL-22 production, and mucosal defense, whereas excessive kynurenine accumulation engages immunosuppressive signaling through AhR and other receptors, contributing to immune tolerance, metabolic reprogramming, and, in chronic contexts, epithelial dysfunction. Primary human cohort studies using paired fecal and mucosal metabolomics have shown that increased Kyn/Trp ratios correlate inversely with indole abundance and AhR target gene expression in IBD, highlighting this metabolic switch as a disease-relevant axis [47].
A consistent signal emerging from cohort studies and meta-analyses is that compositional dysbiosis in IBD is tightly coupled to predictable shifts in the gut metabolome. Across independent case control studies, taxa that are canonical producers of anti-inflammatory metabolites, most notably Faecalibacterium prausnitzii, members of Clostridium clusters IV and XIVa, Roseburia, and Eubacterium are reproducibly depleted in active disease, whereas facultative anaerobes and pathobionts such as Enterobacteriaceae expand. Meta-analytic efforts quantify this loss: early pooled analyses reported significantly reduced abundance of Faecalibacterium prausnitzii in Crohn’s disease (CD) and ulcerative colitis relative to healthy controls, a finding that has been corroborated by more recent whole-genome and metagenomic surveys showing reduced genomic potential for butyrate synthesis in inflamed mucosal sites [30, 48].
These taxonomic changes manifest as altered metabolite landscapes. Multiple systematic reviews and meta-analyses now report a statistically significant reduction in fecal SCFAs, most prominently butyrate, but also acetate and propionate in active IBD vs. controls (subgroup-level p-values often < 0.01), with ulcerative colitis cohorts showing particularly consistent deficits. One meta-analysis encompassing data from eleven studies concluded that total SCFAs and individual SCFA species were decreased in ulcerative colitis patients compared with healthy subjects; more recent aggregations through 2025 reaffirm these trends and expand them to include functional gene loss for SCFA biosynthesis. Because a substantial fraction of produced SCFA is consumed by colonocytes or hepatic first-pass metabolism, reduced fecal concentrations generally reflect both diminished production and impaired epithelial uptake, creating an energetic deficit at the mucosal interface that favours dysbiosis and inflammation [10, 49].
Concurrently, dysbiosis frequently correlates with the accumulation of putatively pro-inflammatory metabolites. Succinate exemplifies a microbial metabolite with concentration-dependent immunological effects that diverge between homeostatic and inflammatory contexts. Under physiological conditions, luminal succinate concentrations in the healthy colon are typically maintained in the low micromolar range (~0.1–0.5 μM), where succinate functions primarily as a metabolic intermediate and does not elicit overt inflammatory signaling. In contrast, dysbiosis and IBD are associated with pathological succinate accumulation, with fecal and luminal concentrations reported in the millimolar range (~1–5 mM) in both murine colitis models and human IBD cohorts, as determined by targeted gas chromatography-mass spectrometry (GC-MS) and LC-MS/MS metabolomics [50]. At these elevated concentrations, succinate activates the succinate receptor SUCNR1 (GPR91) on intestinal epithelial and immune cells, promoting pro-inflammatory signaling, enhanced IL-1β production, and amplification of Th17-skewed immune responses. Importantly, experimental studies demonstrate that SUCNR1 activation and downstream inflammatory effects are negligible below ~0.5 mM but become pronounced at ≥ 1 mM succinate, supporting a threshold-dependent switch from metabolic intermediate to inflammatory signal. These findings underscore the necessity of interpreting succinate biology within a quantitative and compartment-specific framework, rather than as a uniformly pro- or anti-inflammatory metabolite. Importantly, recent reviews emphasize context dependence, while transient succinate accumulation can support epithelial repair and type-2 immune responses under some circumstances, sustained succinate elevation during dysbiosis is associated with Treg instability, reduced forkhead box P3 (FOXP3) function, and worsened clinical indices in IBD cohorts [9].
Tryptophan-derived metabolites follow a parallel yet more complex pattern: indole derivatives (AhR ligands) are often reduced in inflamed mucosa, whereas host-driven kynurenine production (as reflected by an elevated Kyn/Trp ratio) is increased. The Kyn/Trp ratio has been reported to correlate with inflammatory burden; in recent clinical work, a Kyn/Trp cut-off was shown to associate with disease activity in approximately 82% of patients in the studied cohort, illustrating the translational potential of this metric as a biomarker of Trp partitioning toward host immunoregulatory pathways during active inflammation [51] (Table 1).
Key microbial metabolites regulating gut inflammation: sources, host targets, and pathophysiological relevance.
| Metabolite | Primary microbial producers | Host receptors/Targets | Major immunological & epithelial effects | Alterations in IBD/Inflammation | References |
|---|---|---|---|---|---|
| Butyrate | Faecalibacterium prausnitzii, Roseburia spp., Eubacterium rectale | GPR41 (FFAR3), GPR43 (FFAR2), GPR109A; HDAC inhibition | Enhances epithelial ATP metabolism; strengthens tight junctions (ZO-1, occludin); induces FOXP3+ Tregs; suppresses NF-κB, TNF-α, IL-6 | Significantly reduced in active UC and CD; correlates with barrier dysfunction and disease severity | [52–54] |
| Propionate | Bacteroides spp., Veillonella spp. | GPR41, GPR43; partial HDAC inhibition | Modulates dendritic cell maturation; suppresses Th17 polarization; supports mucin production | Reduced in IBD; systemic immunomodulatory effects are less pronounced than butyrate | [55, 56] |
| Acetate | Broad range including Bifidobacterium spp. | GPR43; central metabolic pathways | Supports epithelial metabolism; influences peripheral immune cell energy balance | Decrease in IBD stool samples; altered systemic availability | [57, 58] |
| Indole-3-propionic acid (IPA) | Clostridium spp., Peptostreptococcus spp. | AhR; PXR | Enhances IL-22 production; promotes epithelial repair; antioxidant effects | Reduced in IBD; loss associated with impaired mucosal healing | [59, 60] |
| Indole-3-aldehyde (IAld) | Lactobacillus spp. | AhR | Induces antimicrobial peptides and mucins; maintains barrier integrity | Depleted in dysbiosis; impaired AhR-IL-22 signaling in IBD | [60, 61] |
| Kynurenine (Kyn) | Host-driven (IDO1 induction), microbiota-modulated | AhR; NMDA receptors (indirect) | Immune tolerance at low levels; chronic elevation drives oxidative stress and immune dysregulation | Elevated Kyn/Trp ratio in IBD; correlates with disease activity | [62] |
| Quinolinic acid | Host Kyn pathway | NMDA receptor agonist | Neurotoxicity; oxidative stress; promotes chronic inflammation | Increased in inflammatory and neuroimmune conditions | [63] |
| Serotonin (5-HT) | Enterochromaffin cells (TPH1), microbiota-regulated | 5-HT receptors; SERT | Regulates gut motility; modulates macrophage and T-cell responses | Dysregulated in IBD; contributes to motility disorders and immune activation | [64, 65] |
| Succinate | Dysbiotic taxa (Prevotella, Enterobacteriaceae) | SUCNR1 (GPR91) | Activates macrophages; primes inflammasome; destabilizes Tregs | Elevated in IBD; promotes NF-κB signaling and Th17 skewing | [66] |
5-HT: 5-hydroxytryptamine; AhR: aryl hydrocarbon receptor; ATP: adenosine triphosphate; CD: Crohn’s disease; FOXP3: forkhead box P3; HDAC: histone deacetylase; IBD: inflammatory bowel disease; IDO1: indoleamine-2,3-dioxygenase-1; Kyn/Trp: kynurenine/tryptophan; NF-κB: nuclear factor kappa-B; NMDA: N-methyl-D-aspartate; PXR: pregnane X receptor; SERT: serotonin transporter; SUCNR1: succinate receptor 1; Th17: T helper 17 cell; TPH1: tryptophan hydroxylase-1; Tregs: regulatory T-cells; UC: ulcerative colitis; ZO-1: zonula occludens-1.
Taken together, these observations support a model in which ecological loss of metabolite-producing commensals plus expansion of metabolite-altering pathobionts produces a characteristic “metabolic fingerprint” of IBD: decreased SCFA and indole signals, elevated succinate and certain bile acid derivatives, and an upshift toward kynurenine metabolism [30]. This fingerprint correlates with clinical indices, endoscopic severity, and histological damage in multiple cohorts, but heterogeneity in magnitude and direction across studies, driven by differences in sampling site (stool vs. mucosa), treatment exposure, diet, and cohort demographics, highlights the need for standardized, paired mucosal–luminal metabolomics and longitudinal sampling to resolve causality [49].
The functional consequences of metabolite shifts are determined by the receptor landscape, cell-type specificity, and spatial distribution of metabolites relative to the mucosa. SCFAs primarily act via GPCRs (GPR41/FFAR3, GPR43/FFAR2, GPR109A) and HDAC inhibition to promote Treg differentiation, suppress NF-κB signaling in myeloid and epithelial cells, and sustain epithelial energetics [67]. Loss of these inputs reduces IL-10 and IL-22-mediated repair pathways, weakens tight junctions, and predisposes to translocation of microbial products that further stimulate innate sensors such as toll-like receptors (TLRs) and NOD-like receptors (NLRs). Parada Venegas and colleagues [54] summarized how butyrate and other SCFAs converge on epigenetic and receptor mechanisms to lower pro-inflammatory cytokines, which is functionally relevant given the documented butyrate deficits in active disease.
Succinate introduces a different signaling axis: through SUCNR1, it links mitochondrial and microbial metabolic states to immune activation. SUCNR1 engagement on macrophages enhances IL-1β production and NLRP3 inflammasome priming; on epithelial cells, succinate can modulate barrier function and oxygenation, thereby altering ecological niches and enabling expansion of facultative anaerobes, a feed-forward loop that sustains inflammation. Recent mechanistic studies additionally implicate succinate in destabilizing Treg transcriptional programs (e.g., FOXP3 modulation), providing a direct route by which a single metabolite can shift adaptive immunity toward pathogenic phenotypes observed in IBD [68].
Tryptophan metabolites intersect with host immunity primarily via ligand-activated transcription factors and enzymatic control points. Microbial indoles activate the AhR in ILC3s and mucosal T-cells, driving IL-22 and antimicrobial peptide expression that underpin barrier resilience. Conversely, IDO1-driven kynurenine production engages multiple receptors and neurotransmitter systems; transient IDO1 activation may be anti-inflammatory by limiting effector T-cell responses, but chronic upregulation skews the milieu toward oxidative and neuroactive kynurenines that impair epithelial restitution and can drive extra-intestinal sequelae [4]. Integration of these pathways is evident at the cellular level: DCs and macrophages exposed to altered metabolite profiles exhibit shifted cytokine outputs and antigen-presentation phenotypes, which in turn reprogram T-cell differentiation (favouring Th17 over Treg in many inflamed contexts) and perpetuate mucosal injury [10].
A systems view underscores that metabolite effects are non-additive and context dependent: the same molecule may be protective in an acute, nutrient-replete environment but deleterious if chronically elevated or when receptor expression is altered by inflammation or therapy. This complexity explains why single-agent metabolite therapies (for example, oral butyrate) have produced variable clinical outcomes and argues for precision strategies that restore an ecological and metabolic balance, for example, by combining dietary substrates, targeted probiotics that reintroduce producer taxa, and delivery systems that localize metabolites to the mucosa [69]. Realizing this precision will require integrated multi-omics (metagenomics, metatranscriptomics, metabolomics), spatially resolved profiling, and standardized clinical collection protocols to identify mechanistically defined patient subgroups who are most likely to benefit from metabolite-directed interventions [69].
Therapeutic strategies to restore or mimic salutary microbial metabolites span ecological (prebiotic/probiotic), molecular (postbiotic), and synthetic-biology approaches. Each approach has a mechanistic rationale and emerging empirical support, but translational progress is uneven: ecological manipulations are safe and scalable yet variably efficacious; direct metabolite delivery produces clear biochemical effects in preclinical models but inconsistent clinical benefit; and engineered microbes show strong mechanistic promise in animals but remain early-stage in humans (Figure 5). Below, we evaluate the evidence base, mechanistic underpinnings, and practical limits for each modality.
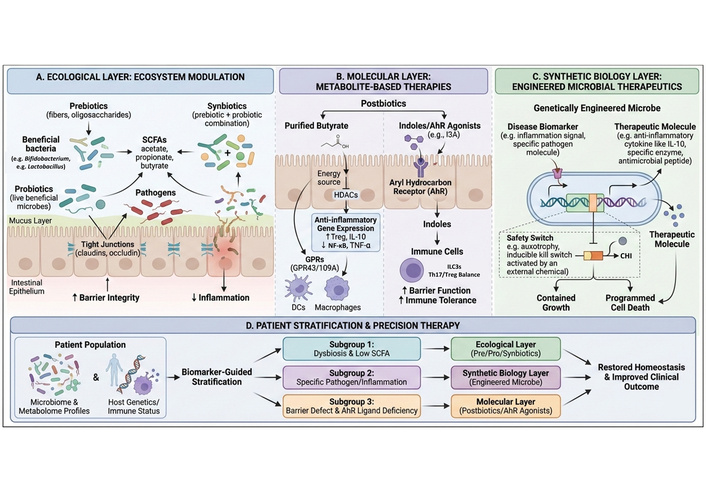
Multi-layered therapeutic targeting of the microbiota–metabolite–immune axis in gastrointestinal inflammation. This schematic illustrates a four-tier framework for therapeutic modulation of metabolite-mediated gut inflammation. (A) Ecological layer (ecosystem modulation): Prebiotics (dietary fibers and oligosaccharides) and probiotics promote the growth of beneficial commensals (e.g., Bifidobacterium, Lactobacillus), increasing SCFAs production (acetate, propionate, butyrate). Enhanced SCFA availability strengthens epithelial tight junctions (claudins, occludin), reinforces the mucus layer, suppresses pathogen expansion, and reduces inflammation. Synbiotics combine both approaches to maximize ecological stability. (B) Molecular layer (metabolite-based therapies): Postbiotics, including purified butyrate and indole-based AhR agonists (e.g., indole-3-aldehyde), act directly on epithelial and immune cells. Butyrate functions as an energy source and HDAC inhibitor, activating G protein-coupled receptors (GPR43, GPR109A) to suppress NF-κB signaling and pro-inflammatory cytokines while promoting Tregs. Indoles activate AhR signaling in immune cells, enhancing barrier function and immune tolerance. (C) Synthetic biology layer: Genetically engineered microbes detect disease-associated biomarkers (e.g., inflammatory signals) and produce therapeutic molecules (e.g., IL-10, antimicrobial peptides). Built-in safety switches (auxotrophy, inducible kill switches) ensure containment and controlled microbial survival. (D) Precision therapy: Biomarker-guided patient stratification based on microbiome, metabolome, and host immune/genetic profiles directs patients toward ecological, molecular, or synthetic biology-based interventions, ultimately restoring gut homeostasis and improving clinical outcomes. AhR: aryl hydrocarbon receptor; DCs: dendritic cells; HDAC: histone deacetylase; NF-κB: nuclear factor kappa-B; SCFAs: short-chain fatty acids; Tregs: regulatory T-cells.
Prebiotics (non-digestible fibers that stimulate beneficial taxa) target metabolite production indirectly by increasing substrate flux toward SCFAs and favorable tryptophan catabolism. Randomized human trials and systematic reviews indicate that inulin-type fructans and certain resistant starches reproducibly increase fecal SCFA concentrations and enrich canonical butyrate producers (e.g., Faecalibacterium, Roseburia), with attendant improvements in barrier biomarkers and low-grade inflammation in selected cohorts [70]. However, clinical endpoints in IBD have been mixed: systematic reviews of fiber/prebiotic interventions report improvements in symptoms or biomarkers in some small trials but lack sufficiently powered, multicenter randomized controlled trials (RCTs) demonstrating robust remission induction. Mechanistically, prebiotics rely on intact cross-feeding networks and adequate host uptake; therefore, responders are likely a biologically defined subset with retained butyrate-producer niches [71].
Precision prebiotics are structure-defined carbohydrate or oligosaccharide substrates designed to selectively enrich microbial taxa or enzymatic pathways that produce beneficial metabolites (for example, butyrate or indole-derivatives). Recent controlled human and experimental studies demonstrate that well-characterized prebiotic substrates can reproducibly increase fecal SCFA pools or enrich specific producer taxa in defined cohorts, but the magnitude and nature of the response depend strongly on the baseline microbiome and dietary context. Primary intervention studies and mechanism-focused trials emphasize that substrate specificity (chain length, branching, glycosidic linkages) determines cross-feeding networks and downstream metabolite stoichiometry, thereby enabling more predictable metabolite steering compared with non-selective fibers. However, randomized trials also document variable responder fractions across populations, underscoring that precision prebiotics are most effective when integrated with baseline microbiome stratification and dietary control [72].
Probiotics, particularly next-generation strains, seek to reintroduce metabolite producers or taxa that modulate microbial ecology. Traditional probiotic evidence in IBD is heterogeneous, yet Escherichia coli Nissle 1917 (EcN) provides a noteworthy example: meta-analyses show EcN is approximately equivalent to mesalazine for maintenance of remission in ulcerative colitis [no significant difference vs. 5-aminosalicylic acid (5-ASA); pooled odds ratio (OR) near unity], supporting its role as an adjunctive ecological therapy in defined clinical contexts. Still, many commercially available probiotics (Lactobacillus/Bifidobacterium blends) produce inconsistent SCFA- or indole-related signals in vivo, underscoring the necessity of strain-level mechanistic validation and selection for metabolite-producing capacity. Synbiotic combinations (prebiotic + probiotic) are conceptually attractive because they supply both producer strains and their substrates, but trial evidence remains preliminary and underpowered [73, 74].
Next-generation probiotics (NGPs) and live biotherapeutic products (LBPs) comprise defined commensal strains selected for their metabolic capacities (for example, native butyrate-producers) or engineered strains with enhanced functional outputs. Primary preclinical work and early translational studies show that single strains or defined consortia can reintroduce missing metabolite pathways and correct dysbiotic deficits in animal models, producing durable mucosal and immunologic improvements when colonization is achieved. Nonetheless, clinical translation is constrained by strain engraftment variability, ecological competition with the resident microbiota, and regulatory considerations for live therapeutics. Recent preclinical and early translational reports underscore the need for mechanistic, strain-level validation (metabolite flux, mucosal colonization, host receptor engagement) before clinical deployment [75].
Postbiotics, the direct administration of metabolites or inactivated microbial products, aim to bypass ecological variability and deliver defined biochemical effectors. Butyrate is the canonical postbiotic: topical butyrate enemas have shown benefit in small trials of distal ulcerative colitis and diversion colitis, and multiple systematic reviews report biochemical and histological improvements in subsets of patients, though overall clinical efficacy is inconsistent across studies. Oral butyrate formulations and pro-drug approaches (e.g., tributyrin, microencapsulated butyrate) have progressed into randomized trials and early-phase studies showing improved mucosal butyrate delivery and modulation of inflammatory biomarkers, but clear clinical endpoints (sustained remission) have not yet been uniformly achieved. Indole/AhR agonists and kynurenine-targeted compounds are at an earlier stage: preclinical models demonstrate that administering specific indoles or AhR ligands increases IL-22 and reduces colitis severity, but human safety and dose-response data are limited. Importantly, postbiotics avoid risks of live biotherapeutics but introduce pharmacokinetic challenges (mucosal targeting, first-pass metabolism) and potential off-target receptor activation that require rigorous dose-finding [76].
Postbiotic strategies administer defined metabolites (for example, butyrate or tributyrin, a butyrate pro-drug) or inactivated microbial components to deliver biochemical effects without the risks of live organisms. Primary experimental studies in mice show that tributyrin and other butyrate prodrugs can restore mucosal signatures and reduce injury in models of antibiotic or chemically induced intestinal damage; early human feasibility studies and clinical trials of butyrate formulations are ongoing. Postbiotic administration avoids engraftment barriers but introduces pharmacokinetic challenges (rapid absorption, first-pass metabolism) and therefore demands delivery technologies that achieve mucosal exposure [77].
Synthetic-biology approaches use engineered commensals to produce therapeutic metabolites or immunomodulatory factors in situ, offering precision localization and sustained delivery. Preclinical work demonstrates that EcN or Lactococcus strains engineered to express anti-inflammatory cytokines (e.g., IL-10, IL-35) or to upregulate butyrate/indole biosynthetic pathways can markedly reduce experimental colitis and restore barrier metrics. Recent reviews catalog an expanding toolkit of programmable gene circuits, kill-switches, and targeted secretion systems that enhance safety and controllability [78]. Clinically, translation is nascent: early-phase human studies of engineered lactic acid bacteria delivering IL-10 or other payloads involved very small cohorts and reported proof-of-concept safety signals but limited efficacy data. Major translational hurdles include regulatory pathways for live genetically modified organisms, potential ecological perturbation of the resident microbiome, and pleiotropic effects of chronic metabolite overproduction (e.g., dysregulated epithelial proliferation or inadvertent immune suppression). Nevertheless, the capacity to combine sensor-actuator circuits (detect inflammation, produce metabolite only when needed) positions engineered microbiota as a promising precision strategy if safety and containment challenges are solved [79, 80].
Synthetic-biology approaches enable the design of microbial chassis that sense environmental cues and produce therapeutic payloads (metabolites or proteins) in situ. Recent primary experimental models demonstrate feasibility: facultative anaerobic commensals have been engineered to deliver butyrate at mucosal surfaces and to ameliorate chemically induced colitis in mice, providing direct causal evidence that targeted microbial production of a metabolite can produce therapeutic benefit. These studies also highlight critical design considerations, metabolic flux optimization, containment/kill-switches, and ecological compatibility, which must be addressed before human application [81].
The clinical translation of engineered microbial therapeutics has progressed cautiously, with early-phase trials prioritizing safety, tolerability, and biological containment. A prominent example is SYNB1020, a genetically modified EcN strain designed to metabolize ammonia, which was evaluated in Phase 1/1b clinical trials that collectively enrolled approximately 40–50 participants, including healthy volunteers and patients with gastrointestinal disease. These studies implemented dose-escalation designs and predefined safety endpoints, including treatment-emergent adverse events, gastrointestinal symptoms, systemic dissemination, and immunogenicity. Importantly, the trials demonstrated an acceptable safety profile without evidence of uncontrolled colonization or serious adverse events, establishing a clinical precedent for the use of engineered commensals in humans [82].
More recently, SYNB8802, an engineered EcN strain optimized for metabolic activity in the gut, entered early-phase clinical evaluation in IBD-relevant contexts. Ongoing Phase 1 studies (planned enrollment ~30–40 participants) are explicitly focused on safety, tolerability, and pharmacodynamic activity, with endpoints including adverse-event incidence, fecal shedding kinetics, and effects on local metabolite profiles. Although efficacy outcomes are exploratory at this stage, trial designs increasingly incorporate biomarker endpoints such as inflammatory mediators and mucosal metabolic signatures to support mechanistic validation [83].
In parallel, VE303, a defined consortium of rationally selected commensal strains developed to restore metabolic and ecological balance after dysbiosis, has advanced through Phase 1/2 clinical testing. In early trials involving approximately 80–100 participants, VE303 demonstrated favorable safety and tolerability profiles, with no serious treatment-related adverse events reported. While VE303 is not genetically engineered at the single-strain level, its consortium-based, function-oriented design provides an important translational bridge toward future engineered metabolite-producing consortia.
The clinical efficacy of metabolite-based interventions is increasingly recognized to depend on baseline metabolic and microbial phenotypes, rather than uniform application across heterogeneous patient populations. In IBD, interindividual variability in fecal and mucosal metabolite profiles necessitates phenotype-guided therapeutic stratification to optimize response and avoid overtreatment. Quantitative fecal metabolomics and targeted LC-MS/MS analyses have identified reproducible patient subsets characterized by low SCFA abundance, particularly butyrate, which correlate with impaired epithelial energy metabolism and barrier dysfunction. In such patients, butyrate or butyrate-prodrug (e.g., tributyrin) postbiotics represent a rational intervention, supported by preclinical and early translational studies demonstrating epithelial repair and anti-inflammatory effects specifically under SCFA-deficient conditions [84].
Conversely, patients exhibiting preserved SCFA levels but reduced indole and indole-derivative abundance, often associated with loss of indole-producing taxa, may benefit more from precision prebiotics or NGPs designed to restore microbial tryptophan metabolism and AhR signaling. Recent human cohort studies integrating fecal metabolomics with transcriptomic profiling indicate that diminished indole–AhR activity correlates with reduced IL-22 signaling and compromised mucosal defense, providing a mechanistic basis for indole-targeted interventions in this subgroup [85].
A distinct phenotype is characterized by elevated Kyn/Trp ratios, reflecting inflammation-driven activation of host IDO1. In these patients, strategies aimed at indirectly reducing kynurenine pathway flux—such as restoring microbial indole production or dampening inflammatory drivers of IDO1 induction may be more appropriate than direct SCFA supplementation. Primary studies using paired fecal mucosal metabolomics demonstrate that excessive kynurenine accumulation is associated with disease severity and immune dysregulation, supporting its use as a stratification biomarker rather than a universal therapeutic target [86].
Robust mechanistic inference about metabolite-mediated gut inflammation depends on rigorous analytical pipelines that link sample collection and processing to sensitive, quantitative detection and integrative data analysis. Three interdependent pillars determine study quality: (1) pre-analytical handling and metadata capture, (2) choice of targeted vs. untargeted analytical platforms with validated quantitation, and (3) quality control, normalization, and multi-omics integration to enable biological interpretation.
Pre-analytical variables are a major source of variance and must be controlled and reported. Stool collection medium, storage temperature, and time-to-freeze alter metabolite profiles: comparative evaluations indicate that 95% ethanol or immediate freezing at –80°C best preserve fecal metabolomes for downstream LC-MS/GC-MS analysis, whereas ambient delays introduce artifactual shifts in SCFAs and labile indoles [87]. Thus, standardized metadata (time-to-freeze, diet in 48–72 hours, bowel preparation, and medication exposure) and separate collection streams for metagenomics vs. metabolomics are recommended to avoid confounding. Adoption of community reporting standards [Metabolomics Standards Initiative (MSI)/Core Information for Metabolomics Reporting] and deposition of raw data in repositories substantially increases reproducibility and reuse [88].
Analytical strategy should be chosen according to the biological question. Targeted assays [LC-MS/MS, GC-MS with derivatization, or gas chromatography-flame ionization detection (GC-FID)] provide the sensitivity, linear dynamic range, and absolute quantitation required to measure low-molecular-weight metabolites such as SCFAs and tryptophan derivatives. Recent method developments achieve limits of detection in the low-nanomolar range for SCFAs: an isotope-dilution LC strategy (SQUAD) reported LODs ≈ 40 nM and LOQs between 160–310 nM, enabling reliable quantification in plasma and feces across physiological ranges [89]. Complementary LC-MS/MS workflows with 3-nitrophenylhydrazone or aniline derivatization similarly permit sub-μM quantitation and rapid throughput. For tryptophan and its > 20 downstream metabolites, validated HPLC-MS/MS panels now permit simultaneous quantification across feces, serum, and tissue with inter-assay CVs < 15% when stable isotope standards and appropriate extraction controls are used. These targeted platforms are essential when absolute concentrations, kinetic fluxes, or biomarker thresholds (e.g., Kyn/Trp ratio) are the readout of interest [90].
Untargeted, discovery metabolomics (high-resolution LC-MS or NMR) remains indispensable for hypothesis generation but requires careful downstream annotation and validation; putative features should be confirmed by targeted methods or authentic standards to reach Level-1 identification per MSI guidelines [91]. Importantly, fecal concentrations are an imperfect proxy for mucosal exposure: a sizable fraction of produced SCFAs is consumed by colonocytes or metabolized hepatically, so paired mucosal biopsy metabolomics or direct mucosal lavage measurements provide far greater mechanistic resolution when feasible. Publicly curated paired microbiome metabolome datasets are now available and accelerate cross-cohort meta-analysis, but analysts must harmonize sample handling and analytical methods to avoid batch artifacts [92, 93].
Quality control, normalization, and statistical modeling require stringent attention. Use of isotope-labeled internal standards for each analyte class, pooled quality control samples to monitor drift, and assessment of matrix effects are minimal requirements for credible quantitation. Normalization strategies should be selected to match the hypothesis: per-gram fecal concentration is appropriate for production studies, while normalization to host protein or tissue weight suits mucosal measurements [94]. For clinical biomarker work (for example, Kyn/Trp ratio as an index of IDO1 activity), analytical precision maps directly onto clinical utility: several studies report that Kyn/Trp elevates robustly in inflammatory states (effect sizes and ROC characteristics vary by cohort), but reproducible thresholds require harmonized assays and population-specific reference ranges [87].
Finally, integrative interpretation demands multi-omics fusion (metagenomics/metatranscriptomics + metabolomics + host transcriptomics) and experimental validation in gnotobiotic or defined-consortia models. Statistical causal inference, Mendelian randomization where possible, longitudinal trajectory modeling, and perturbation experiments are required to move beyond correlation [91]. In practice, the field benefits most when analytical rigor (standardized collection, validated targeted assays with isotope standards, paired mucosal measurements) is combined with systems-level modeling and mechanistic follow-up, producing reproducible metabolic fingerprints that can be translated into mechanistic biomarkers and intervention targets [87].
A sober assessment of the field shows that mechanistic insights into metabolite-mediated gut inflammation have outpaced our ability to translate them reliably into clinical interventions. Major limitations fall into four interrelated categories: measurement and sampling heterogeneity, causal inference, clinical-translational gaps, and safety/feasibility constraints for advanced therapeutics. First, pre-analytical and analytical variability differences in stool vs. mucosal sampling, collection media, time-to-freeze, extraction protocols, and platform (GC-MS, LC-MS, NMR) produce systematic biases that confound cross-study comparisons and reduce statistical power; interlaboratory studies and method comparisons consistently show that sample handling explains a substantial fraction of observed variance in fecal metabolomes [95]. Second, most human datasets are cross-sectional: while multiple meta-analyses report reproducible trends (for example, significant reductions in fecal butyrate in active IBD cohorts), these associations do not establish temporality or causation and are blurred further by medication, diet, and disease-stage confounders [10].
Third, translation into therapeutics has been uneven. Interventions that modify metabolites, prebiotics, probiotics, and postbiotics, such as butyrate, produce promising mechanistic signals but inconsistent clinical endpoints: systematic reviews of butyrate enemas and oral formulations show biochemical or histologic benefits in subsets of patients but no uniform remission advantage across trials, highlighting the gap between biochemical modulation and durable clinical effect [96]. Finally, next-generation approaches (engineered probiotics, sensor-actuator consortia) offer precision but raise regulatory, ecological, and safety questions; early first-in-human phase-1/2a work demonstrates feasibility and short-term safety for engineered Escherichia coli in metabolic disease, but robust efficacy and long-term containment data in inflammatory indications are lacking [97].
To move forward, the field should prioritize (1) harmonized standards for sample collection and reporting (paired mucosal and luminal sampling, immediate freezing or validated stabilizers), (2) longitudinal, deeply phenotyped cohorts with dietary and medication metadata to resolve temporality and identify responder endotypes, (3) integration of targeted, isotope-labelled quantitative metabolomics with metagenomics/metatranscriptomics and host transcriptomics to define mechanistic axes, and (4) stratified early-phase trials that use metabolomic biomarkers for patient selection and as mechanistic end points [98]. Finally, the development of mucosa-targeted delivery systems (microencapsulation, pro-drugs) and programmable biological containment for engineered therapeutics will be essential to balance efficacy with safety. These steps, including technical standardization, causal experimental designs, precision clinical trials, and responsible engineering, are necessary if microbial metabolites are to transition from biomarkers and mechanistic tools into reproducible, safe therapies for inflammatory gut disease [10].
This review highlights microbial metabolites as central mechanistic mediators of gastrointestinal inflammation, rather than passive consequences of dysbiosis. Key metabolite classes, particularly SCFAs and tryptophan-derived metabolites, emerge as critical regulators of epithelial energy metabolism, barrier integrity, and immune homeostasis. In IBD, disruption of these metabolite-driven networks is characterized by reproducible signatures, including depletion of protective SCFA and indole signaling and enrichment of pro-inflammatory metabolic intermediates, which collectively drive epithelial dysfunction and persistent immune activation. By integrating microbial ecology with metabolite signaling and host immune responses, this review provides a mechanistic framework that links dysbiosis to intestinal inflammation. These insights establish microbial metabolites as biologically actionable nodes for understanding disease pathogenesis and for guiding future metabolite-informed therapeutic strategies in gastrointestinal inflammatory disorders.
5-ASA: 5-aminosalicylic acid
5-HT: 5-hydroxytryptamine
AhR: aryl hydrocarbon receptor
ATP: adenosine triphosphate
CD: Crohn’s disease
DC: dendritic cell
DCA: deoxycholic acid
DSS: dextran sodium sulfate
EC: enterochromaffin
EcN: Escherichia coli Nissle 1917
FOXP3: forkhead box P3
FXR: farnesoid X receptor
GC-FID: gas chromatography-flame ionization detection
GC-MS: gas chromatography-mass spectrometry
GPCRs: G protein-coupled receptors
HDACs: histone deacetylases
I3C: indole-3-carboxaldehyde
IAA: indole-3-acetate
IAld: indole-3-aldehyde
IBD: inflammatory bowel disease
IDO1: indoleamine-2,3-dioxygenase-1
ILC3s: group 3 innate lymphoid cells
IPA: indole-3-propionic acid
Kyn/Trp: kynurenine/tryptophan
LCA: lithocholic acid
LC-MS/MS: liquid chromatography-tandem mass spectrometry
LC-MS: liquid chromatography-mass spectrometry
MSI: Metabolomics Standards Initiative
NF-κB: nuclear factor kappa-B
NGPs: next-generation probiotics
NLRs: NOD-like receptors
NMDA: N-methyl-D-aspartate
OR: odds ratio
PXR: pregnane X receptor
RCTs: randomized controlled trials
Reg3γ: regenerating islet-derived protein 3 gamma
SCFAs: short-chain fatty acids
SMD: standardized mean difference
TDO: tryptophan 2,3-dioxygenase
TGR5: G-protein coupled bile-acid receptor 1
Th2: T helper 2 cell
TLRs: toll-like receptors
TPH1: tryptophan hydroxylase-1
Treg: regulatory T-cell
We, the authors, sincerely acknowledge REVA University for providing an opportunity and platform for research.
NSK: Conceptualization, Writing—original draft, Resources, Data curation, Writing—review & editing, Visualization. AH: Data curation, Writing—review & editing, Visualization, Supervision. AC: Conceptualization, Writing—review & editing, Visualization, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 5449
Download: 39
Times Cited: 0
Mohamed Ahmed Mohamed ... Faith Bishop
