Original Article
Original Article

Affiliation:
Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada
ORCID: https://orcid.org/0009-0001-1537-6540
Affiliation:
Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada
Affiliation:
Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada
Email: m.thompson@utoronto.ca
ORCID: https://orcid.org/0000-0002-1445-9918
Explor BioMat-X. 2026;3:101366 DOI: https://doi.org/10.37349/ebmx.2026.101366
Received: January 02, 2026 Accepted: April 27, 2026 Published: May 20, 2026
Academic Editor: Benjamin Thierry, University of South Australia, Australia
Aim: In biosensor technology, reliable attachment of protein-based probes requires careful control of the orientation of the probe molecule on the biosensor surface. In this regard, polyhistidine peptide became an attractive target for on surface immobilization. The present contribution details the total synthesis and the surface chemistry of a new antifouling organotrichlorosilane linker bearing a head function designed to immobilize the imidazole side chain of histidine for future immobilizations with polyhistidine peptide onto biosensor surface.
Methods: A novel organotrichlorosilane linker bearing the ethylene glycol backbone and a 2-chloroethyl sulfone head function (which can be converted to the vinyl sulfone group for subsequent attachment with imidazole) were synthesized via a multiple-step synthesis and carefully characterized. Surface modifications using the synthesized novel organotrichlorosilane linker, subsequent conversion to vinyl sulfone head function, and treatment with N-protected histidine were demonstrated on black lithium niobate substrate.
Results: Novel organotrichlorosilane linker was successfully synthesized, though it was also observed that organotrichlorosilane linker was quite moisture reactive. Surface characterizations also indicated successful modification of lithium niobate with the novel organotrichlorosilane linker as well as presence of N-protected histidine on the lithium niobate surface post-immobilization.
Conclusions: A novel organotrichlorosilane linker bearing the 2-chloroethylsulfone group was successfully synthesized and successful immobilization with N-protected histidine was demonstrated. The surface chemistry demonstrated onto lithium niobate herein is immediately applicable for future on-surface immobilization of protein-based probe molecules bearing polyhistidine moieties.
The ferroelectric and piezoelectric material lithium niobate (LN), LiNbO3, is being increasingly employed in medicine through applications in optical imaging [1], chemical and bio-sensing [2], biocompatible implantable devices [3] and, in the nanoparticle form, for tissue regeneration [4]. With regard to biosensor technology, LN represents a potentially attractive substrate for use as an acoustic wave (AW) detection device because of its superior electromechanical coupling coefficients relative to the more widely used piezoelectric material, quartz. In particular, LN has been employed in the shear-horizontal (SH) wave mode to detect and assay biological species [5].
Key issues for any material, including LN, to be used to fabricate biosensors are the interfacial concentration and ordination of attached surface probes, and appropriate surface chemistry to obviate fouling of the device by sample components [6]. These criteria are anticipated to be crucial in terms of the performance of devices with respect to sensitivity and selectivity. Reduction of surface fouling (or non-specific adsorption) has constituted a major research effort as it pertains to biosensor development [6]. A plethora of anti-fouling approaches have been reported, among them those incorporating surface modification and topographical alteration strategies [7–9].
In our own work, we have attempted to address said probe attachment and fouling concerns through surface modification with novel bifunctional organotrichlorosilane incorporating ethylene glycol (EG) moieties in the molecular backbone [10]. Our interest in the use of EG based molecular backbones is largely inspired by its well-documented capability to suppress fouling. Further studies involving molecular dynamics calculations [11] and neutron reflectometry experiments [12] have suggested that the incorporated EG backbone facilitates the formation of a distinct interfacial water barrier that suppresses the protein dehydration step (due to the inherent energy cost to perturbation) during attempted protein adsorption onto inorganic surfaces. To date, we have developed such EG-based organotrichlorosilanes bearing 2,2,2-trifluoroethyl ester (TFE) and benzenethiosulfonate (BTS) head function (Figure 1). The TFE head group is chosen for its capability to selectively form covalent bonds with amine bearing probes [13]. The BTS head function, on the other hand, is capable of anchoring sulfhydryl bearing probes [14].

Example of bifunctional organotrichlorosilane with incorporated EG moieties and replaceable TFE head groups (TFE-EG2-SiCl3) and BTS head groups (BTS-EG2-SiCl3) [10].
Nonetheless, the aforementioned silane linkers are still rather limited in scope with respect to immobilization of protein-based probes. The TFE variant, which was designed to target the amine moiety of the probe molecule, will inherently suffer from selectivity issues between the amino group of a lysine side chain and the N-terminals. The BTS counterpart required the availability of free thiols, hence necessitating the chemical reduction of disulfide bonds naturally formed between cysteine residues found in the protein-based probe pre-immobilization, which potentially leads to a compromise of the structure of the protein-based probe post-assembly. Furthermore, this chemistry does not allow the probability for the correct surface orientation of probes.
An attractive but challenging alternative consists in developing analogous EG-based organotrichlorosilane surface chemistry to selectively target polyhistidine tag (His-tag) species, a common polypeptide sequence of 6 to 10 histidine residues typically grafted into recombinant proteins (at the N or C terminal) during protein expression [15]. To date, the most well-known and documented approach for the immobilization of His-tagged proteins incorporates the nickel (II)-nitrilotriacetic acid (Ni2+-NTA) coordination complex, whose concept was inspired by the nickel affinity column chromatography used to purify said His-tagged protein. In essence, the inorganic material of interest is functionalized with the NTA head function via several approaches, which, in turn, is subsequently used in tandem with the Ni2+ ion, to coordinate to the His-tagged species [16, 17]. Importantly, there are indications that chemistry can result in specifically oriented protein on surfaces [18]. Although there are literally hundreds of articles published on use of such chemistry for biosensor development, there exists the distinct reality that the complex can be removed for the device surface by components of biological fluids. One very interesting alternative approach to circumvent these shortcomings, which appeared in the literature, consisted in substituting Ni2+ with Cobalt (III) (Co3+), which has been reported to form a more stable complex that is thermodynamically stable to commonly used chelators, reducing agents, and kinetically inert to ligand exchanges [19, 20].
Nonetheless, the Co3+-NTA system does require the use of an oxidizing agent in the form of H2O2 to oxidize Co2+ to Co3+ in situ during surface modification to attain the stable complex with the His-tagged species. Whilst this is indeed superior to the more well-known Ni2+-NTA complex with respect to a more stable His-tag coordination, the Co3+-NTA system is still limited to immobilizing protein and biological species which are not oxidation sensitive. In this light, there still remains significant room for alternative approaches to both metal coordination-based chemistries.
One solution to obviate these negative issues is the chemical functionalization of a material of interest with chemical head groups that are regioselective for the imidazole side chain of the histidine residue in the absence of activating/coupling agents. Such surface chemistry offers potential advantages of: 1) a permanent attachment of the His-tagged biomolecule and 2) better control of the orientation of the protein-based probe on the sensor area of the device. One such promising chemical head group is the vinyl sulfone (VS) moiety, which had been employed successfully for His-tag immobilization in a number of studies [21, 22]. Nonetheless, the synthesis of organotrichlorosilanes surface modifiers typically involves a final hydrosilylation step, wherein the synthesized precursor molecule, terminating with an alkene tail function, is treated with trichlorosilane in tandem with chloroplatinic acid hexahydrate (H2PtCl6 • 6H2O) to convert the terminal alkene moiety to the trichlorosilyl anchoring function [23, 24], making the prospective synthesis of an organotrichlorosilane bearing the VS head function far from trivial. In this regard, in the present contribution, we describe the development of a novel EG-based organotrichlorosilane surface modifier bearing the 2-chloroethylsulfone (CES) moiety (renamed CES-EG2-SiCl3 herein) [25], which is readily converted to the VS moiety post-immobilization via base-facilitated elimination reaction (Figure 2) [26, 27].
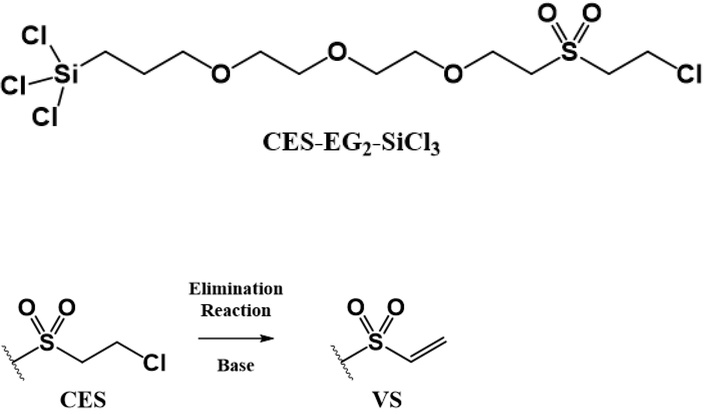
Novel EG-based trichlorosilane surface modifier bearing the CES moiety, CES-EG2-SiCl3 (Top), which is easily converted to the desired VS head function via elimination reaction (Bottom) [25].
Surface modification with the synthesized CES-EG2-SiCl3, as well as subsequent on-surface functionalization with N-protected histidine analogue, was further demonstrated on black LN, a variant of the LiNbO3 substrate which was treated with N2/H2 post growth to suppress pyroelectric charging and enhance electrical conductivity for better performance in prospective AW sensor [28].
Diethylene glycol monoallyl ether HO-EG2-ene (CAS 15075-50-0) was purchased from Gelest Inc. (Morrisville, Pennsylvania, United States). Chloroplatinic acid hexahydrate H2PtCl6 • 6H2O (CAS 18497-13-7) and divinyl sulfone were purchased from Sigma-Aldrich (Oakville, Ontario, Canada). Unless otherwise stated, all other reagents were purchased from Sigma-Aldrich (Oakville, Ontario, Canada), Alfa Aesar (Ward Hill, Massachusetts, United States), or Fisher (Ottawa, Ontario, Canada). Heavy-walled round bottom pressure tubes (d = 2.5 cm, h = 11.4 cm, 15 mL) for the synthesis of trichlorosilane surface modifiers were purchased from VWR (Mississauga, Ontario, Canada).
Black lithium niobate substrates (64 degree Y-cut) were obtained from Roditi international (London, UK) and cut into ~5 mm by 5 mm squares and used accordingly. Fisherbrand™ Versa-Clean™ Multi-Purpose Cleaner (Catalog No 18200701), supplied by Fisher (Ottawa, Ontario, Canada) was diluted in water to prepare the aqueous cleaning solutions used in the cleaning of LN surfaces. Ozone was generated from an OzoneSolutions (Hull, Iowa, United States) OZV-8 generator using a feed gas of 5 psi 90% oxygen. Histidine that was N-protected with tert-butyloxycarbonyl group (Boc-His-OH, CAS 17791-52-5) was supplied by AAPPTec LLC (Louisville, Kentucky, United States). The phosphate buffer (PB) used for Boc-His immobilization consisted of an aqueous solution of 0.75 M sodium phosphate dibasic and 0.25 M sodium phosphate monobasic adjusted to pH 7.0 using concentrated hydrochloric acid (37%).
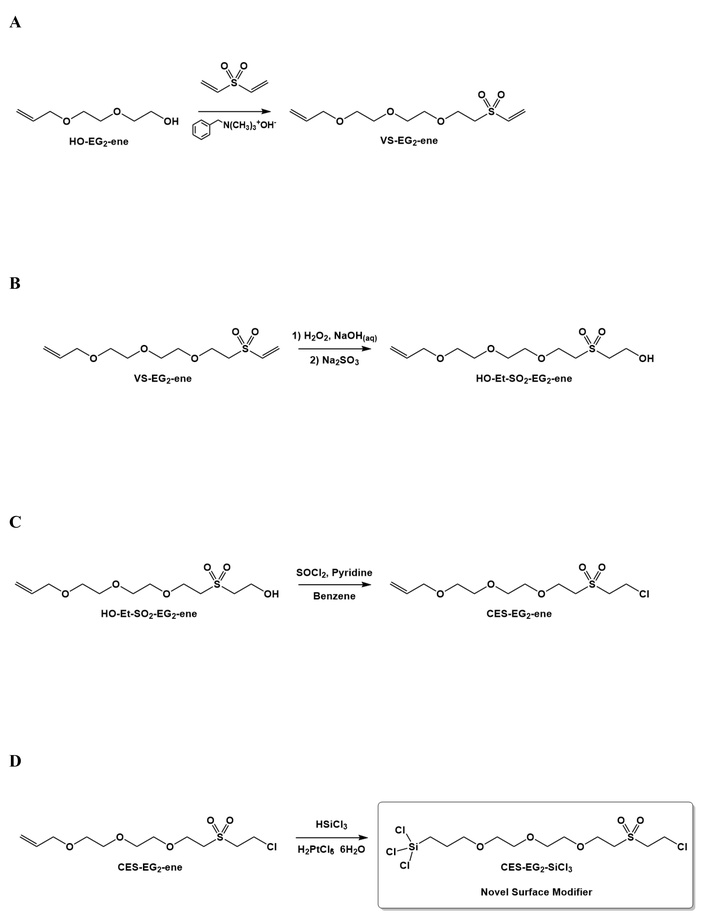
Benzyltrimethyl ammonium hydroxide (40% in methanol, 0.062 mL, 0.057 g, 0.14 mmol) was concentrated to dryness in a 20 mL scintillation vial. To the residue was added HO-EG2-ene (0.5 mL, 0.5 g, 3.4 mmol), followed by divinyl sulfone (0.4 mL, 3.98 mmol). The resulting mixture was left to stir at room temperature for 41 h, after which preliminary NMR analysis indicated presence of unreacted starting material. In an attempt to consume the remaining starting material, the reaction mixture was transferred to a 2nd scintillation vial in which additional benzyltrimethylammonium hydroxide (40% in methanol, 0.100 mL, 0.208 mmol) was concentrated to dryness prior to the addition of the reaction mixture. The reaction was stirred for an additional 4 h, after which a crude NMR indicated the absence of starting material. Purification of the crude mixture by flash silica column chromatography (100/0 to 70/30 Hexanes/acetone gradient followed by 100/0 to 50/50 Hexanes/ethyl acetate gradient) finally yielded compound VS-EG2-ene as a colourless, clear oil (0.200 g) (Figure 3A).
HO-Et-SO2-EG2-ene was synthesized using reaction conditions adapted from work of Evans et al. 2005 [29]. VS-EG2-ene (0.125 g, 0.47 mmol) is diluted in hydrogen peroxide (30% in H2O w/w, 0.239 mL, 0.266 g, 2.35 mmol). To this mixture was added NaOH(aq) (2.1 M, 0.235 mL, 0.49 mmol). The reaction was stirred at room temperature for 2.5 h, after which sodium sulfite (ca. 0.66 g) was added to the solution. The resulting solution was then stirred for an additional 0.5 h and then diluted with water (5 mL). The aqueous phase was extracted repeatedly with diethyl ether (4× 10 mL). The volatiles were evaporated from the ether phase which finally yielded crude HO-Et-SO2-EG2-ene as a clear colourless oil (0.150 g) (Figure 3B).
Thionyl chloride (0.033 mL, 0.053 g, 0.450 mmol) was added cautiously to a stirring solution of HO-Et-SO2-EG2-ene (0.042 g, 0.148 mmol) and dry pyridine (0.041 mL) in dry benzene (0.200 mL). After a further dilution with additional dry benzene (0.100 mL), the resulting mixture was stirred at room temperature for 2 days, after which the benzene solvent was evaporated. The reaction mixture was then repeatedly concentrated with heptane to azeotropically remove unreacted pyridine. The residue was then diluted with diethyl ether, which was then decanted. After the volatiles were removed, the crude residue was further concentrated with additional portions of heptane, which finally yielded CES-EG2-ene as a bright yellow clear oil (0.039 g) (Figure 3C).
CES-EG2-ene (0.015 g, 0.049 mmol) and H2PtCl6 • 6H2O (17 mg, 0.033 mmol) were combined in a heavy walled glass pressure vessel equipped with a stir bar (Note: To prevent undesired adsorption of the silane surface modifier onto the glass surface of the vessel, the pressure vessel was pretreated overnight with 1/20 v/v octadecyltrichlorosilane in anhydrous toluene prior to this synthesis). The pressure vessel was then transferred into a glove box kept inert (N2) and anhydrous (P2O5). Trichlorosilane (0.100 mL) was added cautiously to the reaction mixture inside the pressure vessel, which was then closed tightly. The reaction mixture was then stirred at room temperature for no less than 24 h. The excess trichlorosilane was then evaporated under vacuum, which yielded CES-EG2-SiCl3 as an orange yellow oil (0.007 g). Due to the anticipated difficulties in performing high-vacuum distillation/column chromatography whilst keeping the compound intact, the crude oil was used for the surface modifications without any further purification (i.e., with the catalyst inside) (Figure 3D).
1H and 13C NMR spectra were recorded at room temperature on Agilent DD2 500 MHz or Bruker Avance III 400 MHz spectrometers (available at the University of Toronto, Department of Chemistry) using deuterated chloroform with tetramethylsilane (CDCl3 with TMS) as the NMR solvents. 1H and 13C NMR spectra are referenced to the residual solvent peak (CDCl3: 7.26 and 77.16 ppm, respectively).
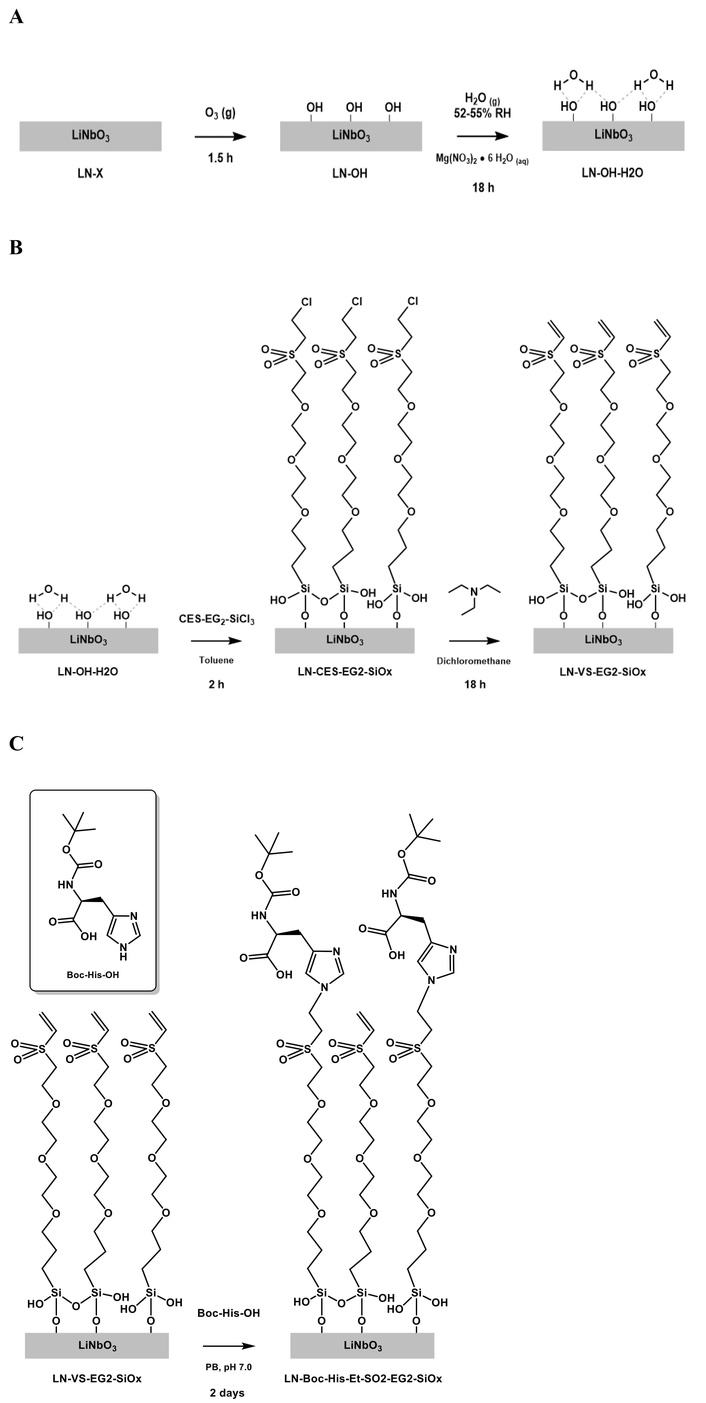
Bare black LN wafers (LN-X) were first sonicated in aqueous cleaning solution (15 min) and then rinsed thoroughly in hot tap water, followed by distilled water. The substrates were then sonicated successively in ACS grade methanol (5 min) and ACS grade chloroform (5 min). After a final rinse with ACS grade chloroform (3×) followed by drying with a gentle stream of nitrogen, the substrates were transferred to a custom built gas chamber (hollow glass tube, fitted with 24/40 female ground glass joint on both ends, both covered with 24/40 male gas adapters). Ozone was flown through this chamber for 1.5 h. The LN wafers were then sonicated successively in distilled water (5 min), ACS grade methanol (5 min), and ACS grade chloroform (5 min). They were then each rinsed with ACS grade chloroform (3×) and then dried under a gentle stream of nitrogen. The nitrogen dried samples were either stored under nitrogen awaiting surface characterization (LN-OH) or used immediately for subsequent water physisorption (Figure 4A).
OH-functionalized LN wafers (LN-OH) were then transferred into a humidity chamber (maintained at 52–55% relative humidity in room temperature with a saturated aqueous solution of magnesium nitrate hexahydrate) for 18 h to induce water vapor physisorption, in preparation for surface functionalization with the CES-EG2-SiCl3 surface modifiers. After 18 h, the water-adsorbed LN wafers (LN-OH-H2O) were then removed from the chamber and promptly transferred into a glovebox (kept inert using nitrogen and anhydrous with phosphorus pentoxide) and used immediately for modification with CES-EG2-SiCl3.
Neat CES-EG2-SiCl3 surface modifier was diluted with anhydrous toluene (1 μL linker to 1 mL toluene). Next, these stock solutions are portioned in appropriate amounts (1,000 μL) to scintillation vials, into which the water-adsorbed hydroxylated LN wafers (LN-OH-H2O) were then individually soaked. The vials were then closed tightly with screw caps, taken out of the glove box, and agitated on a spin plate for 2 h. The CES-EG2-SiCl3 treated wafers were then rinsed with ACS grade toluene (2×) and then sonicated in another portion of toluene (5 min). After a final rinse with toluene following the sonication, the above rinse and sonication steps were repeated with ACS grade chloroform. Following subsequent drying with a gentle stream of nitrogen, the CES-EG2-SiCl3 modified LN wafers were either stored under nitrogen awaiting surface characterization (LN-CES-EG2-SiOx) or used immediately for the next step.
CES-EG2-SiCl3 modified LN wafers (LN-CES-EG2-SiOx) were immediately placed in a solution of triethylamine (5 μL) diluted in dichloromethane (1,000 μL) and stirred for 18 h. The treated LN substrates were then rinsed with dichloromethane before being dried with a gentle stream of nitrogen. The dried LN wafers (supposedly now bearing the VS head functions) were either stored under nitrogen, awaiting surface characterization (LN-VS-EG2-SiOx) or used immediately for subsequent treatment with Boc-His-OH (Figure 4C).
Dried VS functionalized LN substrates were immediately placed in a solution of Boc-His-OH in PB (pH 7.0) for 2 days. The treated black LN substrates were then rinsed successively with deionized water, followed by chloroform, before finally being dried with a gentle stream of nitrogen. The dried Boc-His-OH treated LN wafers were stored under nitrogen, awaiting surface characterization (LN-Boc-His-Et-SO2-EG2-SiOx) (Figure 4C).
LN wafers at the various aforementioned surface modifications (LN-X, LN-OH, LN-CES-EG2-SiOx, LN-VS-EG2-SiOx, and LN-Boc-His-Et-SO2-EG2-SiOx) were characterized using X-Ray Photoelectron Spectroscopy (XPS) and Contact Angle Goniometry (CAG) techniques.
XPS was conducted using a ThermoFisher Scientific K-Alpha Instrument (ThermoFisher Scientific) located at Open Centre for the Characterisation of Advanced Materials (OCCAM) (Toronto, Ontario, Canada). Samples were analyzed with monochromated Al Kα X-rays focused to elliptical spots at a take-off angle of 90 degrees relative to the surface. The binding energy scale was calibrated to the main C 1 s signal at 285 eV. Data processing and analysis of characteristic elements were performed using Avantage software. The Smart Background subtraction method (based on the traditional Shirley background function with the additional constraint that the background should not be of a greater intensity than the actual data at any point in the energy range) was applied to the raw XPS spectra. The background subtracted spectra for each of the characteristic elements were included in the Supplementary materials and also referred to as appropriate in the Results and discussion section.
CAG measurements are conducted using a CAM101 goniometer (KSV Instruments Ltd.) and ultrapure water as the test liquid. The generated images were further processed using ImageJ software equipped with Low Bond Axisymmetric Drop Shape Analysis (LB-ADSA) plug-in, in which static contact angles values were determined by superimposing carefully fitted Young-Laplace curves upon the droplet images obtained from the contact angle instrumentation [30].
NMR signals were included below for the intermediate compounds and final product of the synthesis (VS-EG2-ene, HO-Et-SO2-EG2-ene, CES-EG2-ene and finally CES-EG2-SiCl3) (Note: NMR spectra were provided in Figures S1 to S7 from the Supplementary materials). The abbreviations d, t, dd, dt, ddt, and m respectively stand for doublet, triplet, doublet of doublet, doublet of triplet, doublet of doublet of triplet, and multiplet.
VS-EG2-ene
1H NMR (500 MHz, CDCl3): δ 6.81 (1H, dd), 6.39 (1H, d), 6.07 (1H, d), 5.90 (1H, ddt), 5.30–5.23 (1H, m), 5.20–5.16 (1H, m), 4.01 (2H, dt), 3.89 (2H, t), 3.66–3.62 (6H, m), 3.61–3.57 (2H, m), 3.25 (2H, t). 13C NMR (125 MHz, CDCl3): δ 138.12, 134.75, 128.83, 117.39, 72.39, 70.71, 70.68, 70.40, 69.56, 64.79, 55.19.
HO-Et-SO2-EG2-ene
1H NMR (500 MHz, CDCl3): δ 5.90 (1H, ddt), 5.32–5.24 (1H, m), 5.23–5.18 (1H, m), 4.08–4.03 (2H, m), 4.02–3.99 (2H, m), 3.96–3.92 (2H, m), 3.68–3.62 (6H, m), 3.61–3.57 (2H, m), 3.42–3.33 (4H, m). 13C NMR (125 MHz, CDCl3): δ 134.44, 117.76, 72.38, 70.60, 70.31, 70.21, 69.39, 65.08, 57.63, 56.86, 54.79.
CES-EG2-ene
1H NMR (500 MHz, CDCl3): δ 5.91 (1H, ddt), 5.31–5.24 (1H, m), 5.23–5.16 (1H, m), 4.02 (2H, dt), 3.95–3.86 (4H, m), 3.68–3.58 (10H, m), 3.29–3.24 (2H, m). 13C NMR (125 MHz, CDCl3): δ 134.77, 117.41, 72.40, 70.84, 70.77, 70.41, 69.57, 64.88, 57.10, 54.91, 35.85.
CES-EG2-SiCl3
1H NMR (500 MHz, CDCl3): δ 3.99–3.84 (4H, m), 3.79–3.72 (2H, m), 3.71–3.54 (10H, m), 3.32–3.26 (2H, m), 1.32–1.18 (2H, m), 0.90–0.85 (2H, m).
NMR confirmed the successful preparation of VS-EG2-ene from HO-EG2-ene, as indicated by the presence of 6.81 ppm, 6.39 ppm, and 6.07 ppm on the 1H NMR and 138 ppm and 129 ppm on the 13C NMR, all of which, together, were consistent with the presence of a newly introduced C=C double bond from the VS moiety.
HO-Et-SO2-EG2-ene was also prepared successfully from VS-EG2-ene, as suggested by the disappearance of 6.81 ppm, 6.39 ppm, and 6.07 ppm signals on the 1H NMR and 138 ppm and 129 ppm signals on the 13C NMR for HO-Et-SO2-EG2-ene, which was consistent with the consumption of the C=C moiety from the VS moiety. This was further corroborated by the appearance of newly appeared signals at 56.86 ppm, 54.79 ppm signals on the 13C NMR spectra which were expected for a C=C moiety, the VS group, being formerly sp2 hybridized carbons, being converted to sp3 hybridized carbons upon forming the 2-hydroxyethyl sulfone moiety.
CES-EG2-ene was prepared successfully, as indicated by the presence of 57.10 ppm, 54.91 ppm, and 35.85 ppm signals from 13C NMR, which, after comparison to other reported 2-chloroethyl sulfone based compounds [31, 32], are consistent with the presence of the methylene carbons from the 2-chloroethylsulfonyl moiety. Despite being a trivial reaction which involves the conversion of OH to Cl, due to the moisture sensitivity of the SOCl2 reagent used, this step of the overall synthesis was achieved only with great difficulty after careful drying of the reaction solvent (benzene).
The final step of the synthesis of CES-EG2-SiCl3 also proved to be quite challenging due, in large part, to the reactivity of trichlorosilyl based linkers in general to even trace amounts of moisture. Indeed, while the observed absence of expected alkene peaks at 5.90 ppm, 5.26 ppm, and 5.18 ppm on 1H NMR was consistent with the consumption of the terminal alkene from CES-EG2-ene during the hydrosilylation reaction, the newly observed peaks at 1.32–1.18 ppm and 0.90–0.85 ppm were not typical for 1H nuclei belonging to newly formed methylene groups in α and β positions relative to the trichlorosilyl moiety, as similar EG based trichlorosilane linkers synthesized in our laboratory have displayed peaks characteristic of the aforementioned methylene 1H nuclei at ~1.85 ppm and ~1.50 ppm after careful comparison against theoretically predicted values. The discrepancy observed for CES-EG2-SiCl3 was consistent with a partial displacement of chlorine atoms black from trichlorosilyl moiety by moisture (forming Si–OH bonds in the process) after the initial formation of the trichlorosilyl moiety, which would result in the shifting of said 1H nuclei from the expected 1.85 ppm and 1.50 ppm.
Background subtracted XPS narrow scans for characteristic elements (O 1s, N 1s, C 1s, Nb 3d, Cl 2p, S 2p, Si 2p, Li 1s) are included in the Supplementary materials (Figures S8 to S15) and also highlighted as appropriate in the Results and discussion section. Table 1 lists the water contact angles of the LN samples (averaged over triplicate samples with specific surface modifications). The separate replicates of the water contact angle measurements can be found in Figure S16 of the Supplementary materials.
Water contact angle values for black LN at the various steps of surface modification (Averaged over three trials with standard deviation for each step of surface modification).
| Sample(Surface Chemistry on LN) | Contact angle(average ± standard deviation) |
|---|---|
| LN-X(Bare, untreated) | 72.9° ± 6.4° |
| LN-OH(Hydroxylated) | 56.5° ± 7.7° |
| LN-CES-EG2-SiOx(CES-EG2-SiCl3 modified) | 68.6° ± 2.3° |
| LN-VS-EG2-SiOx(VS-bearing) | 64.8° ± 5.8° |
| LN-Boc-His-Et-SO2-EG2-SiOx(Boc-His-OH treated) | 70.3° ± 0.9° |
For bare untreated LN (LN-X), along with the expected oxygen (O 1s at 530 eV), niobium (Nb 3d at 209.5 eV and 206.5 eV, Nb 4s at 60 eV), and lithium (Li 1s at 54 eV) XPS peaks (Figures 5A, 5C, and 5D) a nitrogen signal was observed (N 1s at 395 eV) (Figure 5B) which is consistent with the use of nitrogen during the conversion of LN to its black form. In practice, surface modification of inorganic materials (of the biosensor materials) with organotrichlorosilane molecules requires the material surface to be saturated with hydroxyl groups. In the context of LN, this was achieved by a combined cleaning/surface oxidation protocol, which involved successive exposures to aqueous solution of multi-purpose cleaner and various organic solvents, followed by subsequent ozonolysis. This procedure gave rise to an expected increase for the main oxygen signal (O 1s at 530 eV), which was consistent with surface saturation with hydroxyl groups on the black LN, post-ozonolysis (Figure 5A). An increase in the N-Nb signal (N 1s at 395 eV) was also observed following the surface oxidation protocol (Figure 5B) which was consistent with the N-Nb binding modes being more exposed after the cleaning procedure. Further careful examination of the main niobium (Nb 3d) XPS profile reveals noticeable shifts (0.1–0.2 eV) towards higher binding energies, which were indicative of a more oxidized LN surface post-ozonolysis (Figure 5C).
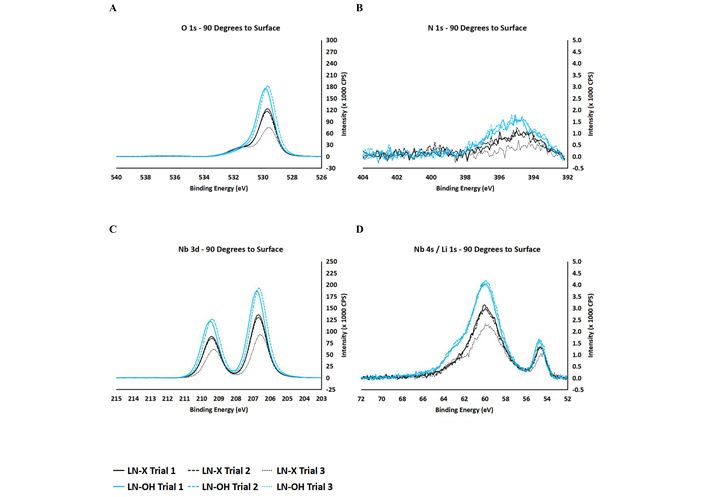
XPS narrow scans for untreated LN (LN-X) and hydroxylated LN (LN-OH) for the following: O 1s (A), N 1s (B), Nb 3d (C), Nb 4s (D), and Li 1s (D).
An overall drop in contact angle values is observed from LN-X to LN-OH across the triplicate measurements, which is indicative of LN surfaces becoming more hydrophilic following the cleaning/oxidation protocol (Table 1). This was consistent with the LN surface being cleaned and becoming saturated with OH groups, both of which largely contributed to the observed increase in hydrophilicity.
The most noticeable change resulting from this step of surface treatment relative to LN-OH was the observed increase of Si-O XPS signal (Si 2p at 103 eV) for silicon (Figure 6E) which was consistent with newly formed Si-O bonds from polysiloxane network resulting from the newly-deposited CES-EG2-SiOx adlayer. A new chlorine signal (Cl 2p at 198 eV) as well as oxidized sulfur signals (S 2p at 169 eV) correspond respectively to the primary alkyl chloride and sulfonyl moieties from the on-surface CES head function (Figures 6C and 6D). The main oxygen (O 1s at 530 eV) and niobium (Nb 3d at 209.5 eV and 206.5 eV) decreased as expected (Figure 6A and 6B), but indicated from their magnitude that LN-OH is still exposed to a degree. A plausible explanation for this is that the CES-EG2-SiCl3 linker used in the surface modification experiments was already partially degraded due to moisture sensitivity during storage prior to immobilization on the black LN substrate, which can potentially compromise the quality of the organotrichlorosilane surface layer. Indeed, this explanation was in line with the observed 1H NMR for the CES-EG2-SiCl3 linker, which suggested that the trichlorosilyl group from the linker was partially degraded (due to the moisture sensitivity), leading to the presence of Si-OH groups.
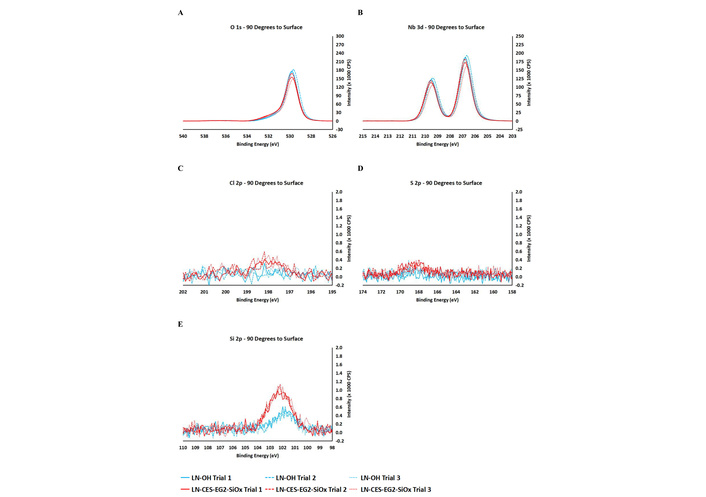
XPS narrow scans for hydroxylated LN (LN-OH) and CES-EG2-SiCl3 modified LN (LN-CES-EG2-SiOx) for the following: O 1s (A), Nb 3d (B), Cl 2p (C), S 2p (D), and Si 2p (E).
As expected, the water contact angles increased from LN-OH to LN-CES-EG2-SiOx across the triplicate measurements (Table 1). This is indicative of the LN being slightly more hydrophobic following adlayer formation and is consistent with newly introduced CES groups decreasing the LN affinity to water. However, the values for the modified surface were lower than that of an ideally hydrophobic surface (closer to 90°), which was expected for an LN surface totally covered with the adlayer in place. This further supports the fact that the LN surface is not completely covered by CES-EG2-SiOx.
For this surface reaction, the chlorine (Cl 2p at 198 eV) is expected to decrease in intensity in keeping with the removal of chlorine from the surface during the conversion of the CES head group to VS head function. However, no noticeable drop in intensity was observed (Figure 7). We attribute this result to residual solvent assured on the material surface, an effect we have witnessed in previous work. As anticipated, the water contact angle revealed no change in wettability for conversion of LN-CES-EG2-SiOx to LN-VS-EG2-SiOx (Table 1).
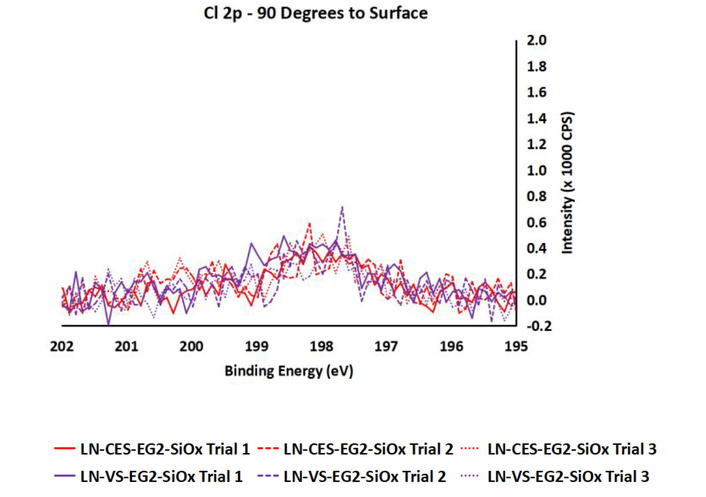
Cl 2p XPS narrow scans for CES-EG2-SiCl3 modified LN (LN-CES-EG2-SiOx) and VS-functionalized LN (LN-VS-EG2-SiOx).
In the final step of the overall surface modification, the adlayer-modified LN substrates were exposed to N-Protected Histidine at pH 7.0. The most noticeable change resulting from this surface modification was the clear appearance of a new nitrogen XPS signal (N 1s peak at 400 eV) (Figure 8B), which we attribute to nitrogen atoms introduced by the imidazole side chain of Histidine. This constitutes the most conclusive evidence of histidine residue being immobilized onto the black LN surface following adlayer deposition. The presence of Boc-His-OH was further supported by small but noticeable shifts to higher binding energies (0.1–0.2 eV) observed for oxygen (O 1 s) (Figure 8A), which is consistent with the introduction of carbonyl moieties (from the C-terminal carboxylic acid and the tert-butyloxycarbonyl groups from Boc-His-OH). The chlorine (Cl 2p at 198 eV) profiles and Si-O peak (Si 2p at 103 eV) also witnessed noticeable drops in intensities following Boc-His-OH immobilization (Figures 8D and 8E) as expected for a more buried polysiloxane adlayer due to being shielded by the surface-adsorbed Boc-His-OH residues. In contrast to this strong evidence, the niobium (Nb 3d) profiles displayed no drop in intensity (Figure 8C). One plausible explanation for this result lies in the instigated orientation of the Boc-His-OH on the surface, in which the electrostatic interaction between carbonyl oxygen atoms (from Boc-His-OH) and positively charged niobium and lithium atoms (from the LN surface) can potentially cause the added molecule to adopt a “lateral” orientation, which would “partially expose” the LN surface. To be thorough, however, this observation of the Nb 3d XPS profile can also be indicative of poor surface coverage (of the CES-EG2-SiCl3 adlayer or the Boc-His-OH model probe) for the LN, which can potentially counterbalance the otherwise expected decrease in Nb 3d intensity even though the LN surface is being more buried by the Boc-His-OH.
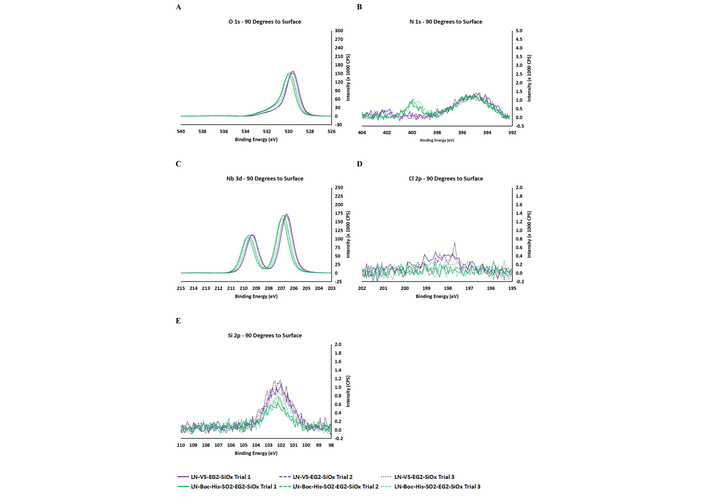
XPS narrow scans for VS-bearing LN (LN-VS-EG2-SiOx) and Boc-His-OH treated LN (LN-Boc-His-Et-SO2-EG2-SiOx) for the following elements: O 1s (A), N 1s (B), Nb 3d (C), Cl 2p (D), and Si 2p (E).
Corroborating evidence for successful Boc-His-OH attachment is provided by the noticeable increase in water contact angles from LN-VS-EG2-SiOx to LN-Boc-His-Et-SO2-EG2-SiOx (Table 1). This result is expected for an LN surface now capped by the hydrophobic tert-butyloxycarbonyl group from the immobilized Boc-His-OH, leading to an overall decrease in hydrophilicity of the LN surface.
In summary, we have described, in the present work, the development of a novel organotrichlorosilane surface modifier incorporating the EG backbone and VS group (protected as CES) as a head group designed to immobilize the imidazole side chain of histidine. To the best of our knowledge, the work described here comprises the first example in which the VS group was adapted successfully for histidine attachment via the very popular silane surface chemistry whilst incorporating, in tandem fashion, the well-studied antifouling EG backbone for future biosensor applications. In this case, the surface chemistry was applied to LN with a fugue view to application in AW biosensor technology. Given that the His-tag motif is very commonly engineered onto proteins and peptides during their expression, the work described herein could lead to better control of the orientation of protein-based probes on the prospective LN AW device. Implementation of this surface chemistry for future immobilization of His-tag containing probes onto LN is the object of current investigation.
AW: acoustic wave
BTS: benzenethiosulfonate
CAG: Contact Angle Goniometry
CES: 2-chloroethylsulfone
EG: ethylene glycol
LN: lithium niobate
PB: phosphate buffer
TFE: 2,2,2-trifluoroethyl ester
VS: vinyl sulfone
XPS: X-ray Photoelectron Spectroscopy
The supplementary figures for this article are available at: https://www.explorationpub.com/uploads/Article/file/101366_sup_1.pdf.
The authors are grateful to Dr. Peter Brodersen (Ontario Centre for the Characterization of Advanced Materials, formerly Surface-Interface Ontario) for assistance with XPS measurements, Dr. Sonia Sheikh for valuable scientific discussion and constructive feedback, and Professors Robert Batey and Andrei Yudin (University of Toronto) for the provision of critical chemicals.
EC: Conceptualization, Investigation, Visualization, Writing—review & editing. AK: Writing—review & editing. MT: Writing—original draft, Writing—review & editing, Validation, Supervision. All authors read and approved the submitted version.
Michael Thompson, who is the Editorial Board Member of Exploration of BioMat-X, had no involvement in the decision-making or the review process of this manuscript. The other authors declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
All data are available from the corresponding author on reasonable request.
This research was funded in part by the Natural Sciences and Engineering Research Council of Canada (NSERC) [MT 9522] and by Thompson Surface Innovations of Toronto. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 764
Download: 11
Times Cited: 0