 Review
Review

Affiliation:
Stirchley Medical Practice, TF3 1FB Telford, UK
Email: thomas.dymond@nhs.net
ORCID: https://orcid.org/0000-0002-2276-9668
Explor Asthma Allergy. 2024;2:233–244 DOI: https://doi.org/10.37349/eaa.2024.00043
Received: January 17, 2024 Accepted: March 22, 2024 Published: June 12, 2024
Academic Editor: Garry M. Walsh, University of Aberdeen, UK
Recent data has resulted in an interest in the metabolic shift in cellular metabolism to aerobic glycolysis, increased reactive oxygen species (ROS), and mitochondrial dysfunction associated with asthma. There has been a push to better understand the immune and metabolic changes in allergy to improve understanding of disease pathology and treatment. Aerobic glycolysis seen in epithelial cells in asthma promotes chronic inflammation and the production of inflammatory cytokines. Asthma epithelial cells share a number of features proposed in the stages of cancer initiation including aerobic glycolysis and increased apoptosis with proliferation, all within a chronic inflammatory microenvironment. Metabolic reprogramming in malignant cells has been widely investigated since the glycolytic characteristics were first described last century. It is still debated whether these metabolic changes are the cause or consequence of carcinogenesis and oncogenic cell-selective pressures. Although historic results have been conflicting, recent data has found an increased lung cancer risk in asthma patients, independent of risk factors. A review of emerging research on the metabolic changes seen in asthma helps us to propose a pathway between the initiation of aerobic glycolysis and the selective pressures of the epithelial microenvironment and resulting malignant transformation risk.
Asthma is a complex respiratory disease characterized by chronic airway inflammation caused by excessive T-helper 2 responses. Pathological features include airway hyper-responsiveness and remodeling, eosinophilic inflammation, and mucus hypersecretion. Studies have repeatedly found the presence of metabolic anomalies and pathway variations in asthmatic patients, which have opened a further potential avenue for new treatments.
Epithelial cells in the airway of asthmatics play a key role in the pathogenesis of asthma [1]. Mitochondrial dysfunction in epithelial cells has recently been linked to the pathogenesis of asthma and other respiratory diseases [2]. Defects in mitochondrial metabolism are involved in the airway remodeling seen in asthma and also lead to increased oxidative stress, reduced adenosine triphosphate (ATP) synthase activity, and apoptosis [3]. Cell growth and division increase ATP and biosynthetic demands, therefore proliferating cells present an altered metabolic profile.
Normal lungs have been noted to generate more ATP through aerobic glycolysis than other tissues, with around 40% of glucose converted to lactate regardless of oxygen status in studies of rat lungs [4]. Recent data suggests asthma is associated with a change in cellular metabolism towards increased aerobic glycolysis in several cell types [5]. Over the last decade, an increased shift to glycolysis has been confirmed in bronchial epithelial cells in asthmatic patients [6]. The cell aerobic glycolysis seen in epithelial cells in asthmatics results in an increase in inflammatory cytokines and promotes a chronic airway inflammatory response and immune activation [7]. In the 1920s, it was described how malignant cells largely perform aerobic glycolysis despite the presence of oxygen. Warburg [8] proposed this damage in mitochondrial respiration was followed by an increase in glycolysis to make up for the loss in energy availability.
We now know that the working model of carcinogenesis proposes that oncogenic signalling is the underlying driver of malignant cell transformation, which in turn alters cell metabolism pathways. The tumour suppressor protein, p53, has now been decisively linked to the altered balance between oxidative phosphorylation (OXPHOS) and glycolysis seen in carcinogenesis [9]. This data helps corroborate malignant gene mutations to the Warburg effect on the level of tumour suppressors. Chandel [10] reported that mitochondrial metabolism and electron transport chain (ETC) function are required for malignant cell growth, again moving away from the notion of the role of damaged mitochondria in carcinogenesis proposed by Warburg [8]. Researchers have suggested that this metabolic shift could be caused by cancer-associated hypoxia, however, it is a feature in many cells without an oxygen limitation [11]. Other models suggest that increased glycolysis can shunt biosynthetic precursors into anabolic reactions that branch from this pathway. However, again this is challenged as glycolytic intermediates are not necessarily elevated in proliferating cells [12]. It is still debated whether these metabolic changes are the cause or consequence of carcinogenesis and oncogenic cell selective pressures [13].
This phenotype is not unique to cancer and we now appreciate that aerobic glycolysis is exhibited by many proliferative non-transformed mammalian cells [14]. Along with this, activated T cells utilize aerobic glycolysis and it has been implicated in augmenting effector T cell response [15]. What drives aerobic glycolysis and why it is associated with proliferation in some cell populations have not yet been fully explained.
Large meta-analysis and cohort studies on adults with active asthma have found an increased lung cancer incidence, independent of smoking status [16]. We review the selective pressures of asthma epithelial chronic inflammatory microenvironment and the effect of this on apoptosis and metabolic pathway changes.
In a cell’s mitochondrial inner membrane, ATP is generated by the transport of electrons to a series of transmembrane protein complexes: the ETC. The reduced co-enzymes nicotinamide adenine dinucleotide + hydrogen (NADH) or FADH2 act as electron donors in this pathway. Complex I of the ETC accepts electrons from NADH and passes these onto complex II while oxidizing NADH to NAD+. As the electrons move through the ETC complexes I to IV, protons are moved from the mitochondrial matrix into the inter-membrane space by complexes I, III, and IV [16]. This results in an increased proton gradient across the mitochondrial membrane. Protons flow from the inner inter-membrane space crossing into the mitochondrial matrix through complex V causing the synthesis of ATP through the transmembrane protein ATP synthase. In this pathway, oxygen behaves as the terminal electron acceptor, as it is a strong oxidizing agent [17]. The reduction of oxygen does involve the production of potentially damaging intermediates [18]. Although the transfer of four electrons and four protons reduces oxygen to water, the transfer of one or two electrons produces superoxide or peroxide anions: reactive oxygen species (ROS). Oxidative stress can be the outcome of increased ROS production, an impaired antioxidant system, poor mitochondrial function, or a mixture of these [19].
Oxidative stress, the imbalance between production and accumulation of ROS in cells, plays a key role in the development of asthma [20]. Data has confirmed extensive free radical oxidative effects in airway tissue of ovalbumin-sensitized mice in asthma models [21]. Oxidative stress can disrupt calcium homeostasis and ETC activity, cause mitochondrial DNA mutations, and increase mitochondrial membrane permeability [22]. A fall in mitochondrial membrane potential results in a reduced proton gradient and therefore limits OXPHOS and is a hallmark of mitochondrial dysfunction [23]. Oxidative stress can inhibit ETC complex activity, thus limiting NADH oxidation at complex I [24, 25]. Mitochondrial dysfunction has been implicated in bronchial asthma epithelial cells [26]. Such dysfunction has also been seen in T2 low asthma and therefore is independent of immune profile [27]. Evidence over the last few years has been increasing indicating that mitochondrial dysfunction is involved in the pathogenesis of asthma [2]. Chellappan et al. [2] also noted drugs to target mitochondrial function could also be used in asthma treatment.
As a process, chronic inflammation in asthma generates oxidative stress in pulmonary cells. Oxidative stress initiates the production of mediators of inflammation and ROS in pulmonary epithelial cells and other inflammatory cells [28]. ROS can result in oxidative damage to bronchial epithelial cells in asthma, which promotes inflammation through the release of cytokines [29]. Damage to the airway epithelium may play a role in the increase of particles reaching the airway submucosa, causing increased inflammation and airway remodeling [30].
An interesting observation was made by Raby et al. [31] to link the maternally-inherited asthma risk factor to mitochondrial inheritance, which is known to be exclusively maternally inherited. This highlights the importance of mitochondrial DNA and mitochondrial dysfunction in asthma pathogenesis.
Glycolysis is a sequence of reactions that oxygen is used to reoxidise NADH to NAD+. This pathway starts with a molecule of glucose and ends up with two of pyruvate, which is usually only converted to lactate in the absence of oxygen. Glycolysis can occur in aerobic and anaerobic conditions. In aerobic conditions, pyruvate is directed towards the tricarboxylic acid cycle to ultimately undergo OXPHOS. In the first step of this in mitochondria, pyruvate is decarboxylated by pyruvate dehydrogenase (PDH) complex to form acetyl-CoA that is moved into the tricarboxylic acid cycle. PDH activity is limited by a low NAD+/NADH ratio, therefore PDH influences the extent a cell engages in this pathway [32]. In anaerobic conditions, pyruvate stays in the cytoplasm and is fermented to lactate by lactate dehydrogenase. In aerobic glycolysis, also known as the Warburg effect, pyruvate can be fermented to lactate even in the presence of oxygen or further oxidized. The pace of ATP generation in aerobic glycolysis is quicker than OXPHOS, which is thought to suit the energy demands of rapid proliferation [33]. Mechanism of regulation of glycolysis occurs through modification of rate-limiting enzymes, their allosteric activation or inhibition, and by hormonal control.
While a lot of research has been carried out in the area of cancer, we know that aerobic glycolysis is exhibited by many proliferative non-transformed mammalian cells. Current research suggests asthma to be associated with a shift in cell metabolism to aerobic glycolysis. Increased glycolysis was reported in bronchial epithelial cells from both obese and control asthma patients [6]. These changes have been linked to an overall increase in oxidative stress [34]. Inhibition of epithelial aerobic glycolysis was found to significantly decrease inflammatory cytokines, suggesting it is involved in the chronic inflammation seen in asthma [7]. A shift to aerobic glycolysis has emerged as a key feature of allergic asthma and has also been proposed as a feature of the development of mucus metaplasia, airway hyper-reactivity, and inflammation [35, 36]. Glycolysis generally occurs in oxygen-deficient conditions but in these studies cells or in vivo disease models were examined under normal oxygen conditions, or the oxygen consumption rates were monitored.
It has been demonstrated that the glycolytic phenotype is observed in human cancers and is often seen early in carcinogensis [37]. The glycolytic shift caused by oncogene activation and loss of tumour suppressors in carcinogenesis is thought to result in a selective advantage for tumours, by providing essential precursors for building the products to sustain growth and proliferation. The current working models of carcinogenesis propose DNA damage occurs, activating oncogenes with a subsequent metabolic shift [38].
The traditional thought that healthy proliferating cells require increased aerobic glycolysis does not always seem to be the case. A study of fibroblasts triggered into a quiet state compared to proliferating cells found no increase in glucose uptake in the proliferating population [39]. Recent findings in this field have found aerobic glycolysis is not just limited to supporting proliferation. Cells can alter their metabolism to adjust to energy requirements and signalling events [40].
Why some cells engage in aerobic glycolysis remains poorly understood. Early proposals of mitochondrial dysfunction or hypoxia being the cause of these changes in carcinogenesis have been strongly challenged with a better understanding of the role of the functional ETC in malignant cells. Additionally, it is noted tumours have functional mitochondria, which are required for growth and progression. Healthy endothelial cells, in the process of vessel sprouting, need to differentiate and proliferate in a synchronized way. These healthy cells have been found to have high rates of aerobic glycolysis that results in lactate production rather than OXPHOS, regardless of the fact they are in direct contact with oxygenated blood, which again challenges the role of hypoxia as the cause of this metabolic change [41]. An additional previous proposal is that aerobic glycolysis provides essential biosynthetic precursors for anabolic reactions branching from glycolysis. However, conflicting data has noted most of the biomass from proliferating cells is derived from amino acids rather than glucose derivatives, which again challenges this notion [42]. Other authors have proposed that aerobic glycolysis may protect against the increased oxidative stress seen in a proliferating cell [43]. Aerobic glycolysis has recently been found to be a rate-limiting factor for the proliferation and survival of human bronchial epithelial cells [44].
Why aerobic glycolysis is commonly associated with proliferation remains an ongoing question. However, a recent model of aerobic glycolysis has led to some promising data to help explain why some cells shift their metabolism. This showed that aerobic glycolysis reflects a state in which the demand for NAD+ for oxidation reactions is greater than the cell demand for ATP [45]. The authors found that when NAD+ regeneration by mitochondrial respiration became limited, fermentation was increased, despite available oxygen. They confirmed their hypothesis that mitochondrial respiration and ATP synthase activity were insufficient to support NAD+ regeneration so cells engaged in aerobic glycolysis. It was also noted that suppressing aerobic glycolysis decreased proliferation in both malignant and control cell populations. Inhibiting aerobic glycolysis decreased the NAD+/NADH ratio in cells and increased dependency on the mitochondrial ETC for NAD+ regeneration. The decreased NAD+/NADH ratio observed in aerobic glycolysis-inhibited cells suggested that NAD+ regeneration by mitochondrial respiration was not able to support increased proliferation. This research was consistent with previous evidence that the rate of NADH oxidation could limit biomass synthesis in aerobic conditions if proliferation is quick enough, in the context of impaired mitochondrial OXPHOS capacity [46]. This also supports previous proposals on the role of aerobic glycolysis in protecting against oxidative stress in proliferating cells [43].
Of note, lung mitochondrial NAD+/NADH ratios have been found to be significantly lower in models of airway inflammation [47]. The authors of this study also confirmed a shift to aerobic glycolysis and a much-reduced ETC complex I activity in asthmatics in a model of allergic airway inflammation. In asthma, recovering mitochondrial function, such as through physical exercise, could potentially reduce aerobic glycolysis as seen in malignancy [48].
In apoptosis, the intrinsic and extrinsic pathways mediate the cell death signalling cascade. The intrinsic pathway is mitochondrial-mediated and triggered by a number of cellular stresses, including oxidative stress. The extrinsic pathway refers to a death receptor-mediated activation pathway. Both pathways come together at the point of initiator and effector caspase activation. In the pathogenesis of asthma, epithelial cells respond to allergens by producing inflammatory cytokines. Epithelial cells can undergo apoptotic cell death in response to allergen exposure, which further drives the inflammatory cascade. Asthma and T-helper 2-mediated airway inflammation are associated with airway epithelial cell apoptosis [49]. Whilst epithelial apoptosis is commonly described, it has not universally been identified in models of asthma [50, 51]. Its relevance to asthma pathogenesis remains uncertain. Despite this, a number of interesting observations of apoptosis in asthma, particularly in severe disease, have been made. A number of factors, including infection and excess oxidative stress, have been described in mediating apoptosis in asthma [52]. Apoptotic epithelial cell clusters have been found in sputum and bronchoalveolar lavage samples in asthmatics [53]. An apoptosis pan-caspase inhibitor was found to decrease airway inflammation in an asthma model [54]. It has been described how T cells and eosinophils induce apoptosis in epithelial cells through the secretion of IFN-gamma and TNF-alpha [55]. A number of studies have demonstrated epithelial cell apoptosis after allergen exposure [56, 57].
There are multiple signals that initiate apoptosis including death receptors, cytokines, and oxidative stress. One such death receptor identified in epithelial cells is CD95 [58], which may also be increased in the asthmatic epithelium linking directly to the increase in apoptosis seen in asthma [49].
The intrinsic pathway of apoptosis is also strongly linked to asthma, as there is a significant overlap between asthma, mitochondrial dysfunction, oxidative stress, and apoptosis [59]. Epithelial cell apoptosis is closely linked with increased cell proliferation and may be stimulated by the same signals to maintain homeostasis [60]. Allergen insults have been found to stimulate epithelial cell proliferation in asthmatics [61]. It has been found that apoptosis and proliferation may both be elevated in chronic inflammatory disease states such as asthma [62]. More significant apoptosis, cell cycling, and proliferation are seen in asthma airway epithelium and may be linked to disease severity [62, 63].
Apoptosis-induced proliferation has been widely described in wound healing, cancer, and other disease processes, where activated caspases trigger compensatory proliferation [64]. Whilst the role of caspases in apoptosis is well understood, recent studies have found they can promote proliferation through autonomous regulation of the cell cycle as well as signaling neighbouring tissue [65]. Apoptosis-induced proliferation is believed to be used to maintain tissue morphology following unexpected cell population loss, where apoptotic cells trigger surviving cells to proliferate. It also involves a number of other mediators including increased oxidative stress and immune cell recruitment [66]. An increase in epidermal growth factor receptor expression has been found in the epithelium of asthmatic patients [67]. This plays a key role in the epithelial repair process by mediating cell proliferation and differentiation to maintain tissue morphology in the airway. While this is still not conclusive in current asthma models, increased apoptosis and increased epithelial proliferation have been observed in a number of studies, particularly related to the severity of asthma [62, 68]. Further work is needed to fully establish this pathway in asthma pathogenesis.
From the evidence we have reviewed so far, we can make several observations (Table 1). Firstly, mitochondrial dysfunction and oxidative stress are now recognized to play a key role in asthma pathogenesis. There is a strong link between oxidative stress and the increase in apoptosis via the intrinsic pathway, particularly linked to the severity of asthma inflammation. Increased epithelial proliferation is observed in asthma, but it is not certain whether this is a result of increased apoptosis or a combination of other pathway pressures. However, apoptosis-induced proliferation has been appreciated in a number of chronic inflammatory diseases. Finally, the asthma epithelium has been shown to display increased oxidative stress, mitochondrial dysfunction, and a shift in metabolism to aerobic glycolysis.
Summary of selected supporting literature
| Relevant findings | Author [Ref] | Date of publication | Study design |
|---|---|---|---|
| Mitochondrial dysfunction is increased in the asthmatic airway epithelium | Zhao et al. [27] | 2023 | Gene analysis of airway epithelial brushings from healthy and asthmatic patients |
| Increased oxidative stress and damage in asthmatic patients | Gazdík et al. [20] | 2002 | Levels of co-enzyme Q10 measured in asthmatic and control patients |
| Bronchial epithelial cells display increased glycolysis in asthmatics | Winnica et al. [6] | 2019 | Airway epithelial cells isolated from healthy and asthmatic patients |
| Aerobic glycolysis is up-regulated in airway epithelial cells in asthma | Yu et al. [7] | 2022 | Ovalbumin-sensitized mouse model and in vitro investigation of human bronchial epithelial cell lines |
| Asthma increases epithelial cell apoptosis | Iwata et al. [54] | 2003 | Ovalbumin-sensitized mouse model |
| Trautmann et al. [55] | 2005 | Evaluated epithelial layer of bronchial biopsies from asthma patients | |
| Truong-Tran et al. [56] | 2002 | Ovalbumin-sensitized mouse model | |
| Dorscheid et al. [57] | 2003 | Ovalbumin-sensitized mouse model | |
| Proliferation is increased in asthma airway epithelial cells after allergen challenge | Hastie et al. [61] | 2002 | In vitro study of human epithelial cells from asthmatic and non-asthmatics |
| Epithelial cell proliferation contributes to airway remodeling in severe asthma | Cohen et al. [62] | 2007 | Epithelial cells examined from bronchial biopsy from asthmatic and healthy patients |
| Reduced ETC complex I and III activity in airway inflammation in asthmatics | Niu et al. [47] | 2020 | Lung tissue and monocytes from ovalbumin-sensitized mouse model evaluated |
| Increased demand for NAD+ drives up-regulation of aerobic glycolysis | Luengo et al. [45] | 2021 | In vitro investigation of malignant and non-transformed cell populations (airway cell lines not involved in the study) |
| Aerobic glycolysis rate limiting factor for proliferation in airway epithelial cells | Park et al. [44] | 2024 | In vitro study of bronchial epithelial cells |
| Inflammation-induced cell proliferation potentiates DNA damage-induced mutations | Kiraly et al. [69] | 2015 | In vivo mouse model examining overall mutation rate |
| Asthma significantly associated with increased lung cancer risk | Qu et al. [16] | 2017 | Meta-analysis |
ETC: electron transport chain
From these facts and the discussion on the metabolic switch to aerobic glycolysis in proliferating cells in health and cancer, we propose a pathway to the metabolic changes seen in the asthma epithelium and subsequent lung cancer risk (Figure 1).
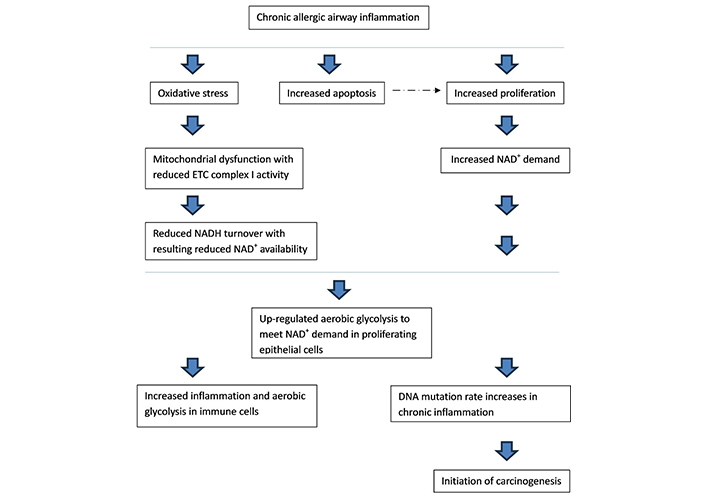
The pathway from chronic airway inflammation in asthma to the up-regulation in aerobic glycolysis in airway epithelial cells, and resulting lung cancer risk. ETC: electron transport chain; NADH: nicotinamide adenine dinucleotide + hydrogen. ---→ : discussed under “
Initial allergen and subsequent immune activation increases inflammatory pressures, cytokine production, and oxidative stress in epithelial cells and the airway microenvironment. This increases oxidative stress and ROS production in epithelial cell mitochondria. Subsequently, rates of cell apoptosis increase with resulting increased survivor epithelial cell proliferation, as found with current chronic inflammation models. NADH turnover decreases due to mitochondrial dysfunction in the ETC from oxidative stress [70]. ROS and oxidative stress can also affect complex I of the ETC to generate more free radicals and result in complex I dysfunction [70]. Complex I is thought to be the most susceptible to oxidative stress [71]. Chronic ROS exposure causes oxidative damage to mitochondrial proteins, whilst acute ROS exposure can deactivate iron-sulphur centres of ETC complex I and III resulting in mitochondrial dysfunction at these complexes [72]. Oxidative stress depletes cellular NAD+ further reducing availability for its role as a co-factor [73]. To further support this statement, monocytes from asthmatic patients, as well as lung tissue from mouse models, displayed greatly reduced ETC complex I and III activities [47].
NAD+ is an essential electron acceptor and is a key requirement to increase biomass during proliferation [74]. Proliferating cells are unable to meet their NAD+ needs through OXPHOS, so show a metabolic switch to aerobic glycolysis, as seen in current working models [45]. Recent data proposed a role in NAD+ boosting therapy to suppress mast cell degranulation in a model of allergy, which is relevant to this discussion [75].
Chronic inflammation-induced cell proliferation is thought to increase the frequency of DNA mutations and drive cancer initiation [69]. A cell metabolic shift to aerobic glycolysis can inhibit pathways that halt proliferation, including p53, again possibly increasing selection for cells with a suppressed p53 phenotype and a high rate of proliferation in the airway epithelium [76].
Asthma is associated with an increased incidence of lung cancer, independent of smoking risks [16]. This risk is directly linked to the severity of presenting asthma. A significant percentage of lung cancers are thought to originate from epithelial cells so the metabolic microenvironment in this setting is likely to play a key role in carcinogenesis [77]. As we have reviewed, there is considerable overlap between the metabolism of epithelial cells in asthma and the malignant microenvironment with mitochondrial dysfunction. This has interesting implications for the metabolic shift prior to malignant transformation in this setting.
The evolving understanding of the metabolic pathways seen in the bronchial epithelium and in other diseases provides an opportunity for the development of new strategies in asthma treatment. We have proposed a pathway to the metabolic changes seen in asthma from the inflammation in the epithelial microenvironment and the resulting increase in oxidative stress and mitochondrial dysfunction. Reduced ETC NADH turnover in the inflammatory airway environment means cells are unable to meet increased NAD+ demand in proliferation, which results in a subsequent shift to aerobic glycolysis; this is supported by current models. Increased apoptosis and proliferation are observed in bronchial asthmatic epithelial cells, resulting in airway remodeling. In vivo studies have found that inflammation-induced cell proliferation potentiates the frequency of DNA mutations. Higher rates of cancer are seen in adults with active asthma, linked to disease severity. It would be easy to propose how inflammation, subsequent shift to aerobic glycolysis and proliferation, then DNA mutation, would lead to the initiation of carcinogenesis in this setting, especially as a large proportion of lung cancers are thought to originate from epithelial cells in the airways.
Interestingly, this puts cell metabolic shift to aerobic glycolysis and proliferation before DNA mutation in the pathway of carcinogenesis in the asthma microenvironment. The Warburg effect is currently believed to be an early event in carcinogenesis that is a consequence of an initial oncogenic mutation. Therefore, this is a significant change when compared to current working models of carcinogenesis by proposing the Warburg effect precedes DNA mutation in at least this setting. This also highlights the importance of controlling airway inflammation in asthma management in line with recent shifts in the Global Initiative for Asthma treatment guidelines to earlier use of inhaled steroids [78].
This hypothesis is obviously dependent on the current working models of aerobic glycolysis activation translating to pathways in healthy airway cell populations, related to proliferation and increased NAD+ demand with mitochondrial dysfunction. Caution also has to be taken applying in vitro data, in the context of abundant nutrients, to the underlying changes in asthmatic epithelial cells in a very different micro-environment. Further investigation is required in this area to further define these metabolic pathways and establish this proposal.
ATP: adenosine triphosphate
ETC: electron transport chain
NADH: nicotinamide adenine dinucleotide + hydrogen
OXPHOS: oxidative phosphorylation
PDH: pyruvate dehydrogenase
ROS: reactive oxygen species
TD: Conceptualization, Writing—original draft, Writing—review & editing.
The author declares that he has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 4505
Download: 34
Times Cited: 0
