Review
Review

Affiliation:
1Department of Biotechnology and Genetic Engineering, Jahangirnagar University, Dhaka 1342, Bangladesh
Email: taslimsajib@gmail.com
ORCID: https://orcid.org/0009-0002-1696-8158
Affiliation:
2Army Medical College Chattogram, Chattogram 4210, Bangladesh
ORCID: https://orcid.org/0009-0006-8033-5215
Affiliation:
3Department of Neurology, Chittagong Medical College, Chattogram 4203, Bangladesh
ORCID: https://orcid.org/0009-0007-2967-7380
Affiliation:
4Department of Biomedical Engineering, Bangladesh University of Engineering and Technology, Dhaka 1205, Bangladesh
ORCID: https://orcid.org/0009-0009-2019-1186
Affiliation:
5Department of Biotechnology, BRAC University, Dhaka 1212, Bangladesh
ORCID: https://orcid.org/0009-0001-2171-8842
Affiliation:
6Rajshahi Medical College, Rajshahi 6000, Bangladesh
ORCID: https://orcid.org/0009-0004-0361-4726
Affiliation:
7American International University-Bangladesh, Department of Public Health, Dhaka 1229, Bangladesh
ORCID: https://orcid.org/0009-0007-3380-299X
Affiliation:
8Barind Medical College and Hospital, Rajshahi 6207, Bangladesh
ORCID: https://orcid.org/0009-0002-3086-1189
Affiliation:
9Department of Medicine, Sir Salimullah Medical College & Mitford Hospital, Dhaka 1100, Bangladesh
ORCID: https://orcid.org/0009-0006-0141-1866
Affiliation:
9Department of Medicine, Sir Salimullah Medical College & Mitford Hospital, Dhaka 1100, Bangladesh
ORCID: https://orcid.org/0009-0005-6594-1789
Affiliation:
10Department of Electronics and Communication Engineering, BRAC University, Dhaka 1212, Bangladesh
ORCID: https://orcid.org/0009-0004-1069-5341
Affiliation:
11Department of Psychology, National University, Gazipur 1704, Bangladesh
ORCID: https://orcid.org/0009-0004-3849-4955
Explor Neuroprot Ther. 2026;6:1004158 DOI: https://doi.org/10.37349/ent.2026.1004158
Received: February 11, 2026 Accepted: June 04, 2026 Published: June 08, 2026
Academic Editor: Guanghui Wang, Soochow University College of Pharmaceutical Sciences, China
The article belongs to the special issue Defending the Brain and the Mind: Exploring Neuroprotective Therapies for Mental Health Disorders
Microglia, the resident immune cells of the central nervous system, are increasingly recognized as key regulators of neural development, synaptic plasticity, and behavior. In addition to their classical role in immune surveillance, microglia actively shape neuronal circuits and influence cognitive and emotional functions across the life span. Accumulating evidence now implicates aberrant or chronic microglial activation as a central mechanism underlying neuroinflammation-associated behavioral and cognitive disturbances in a wide range of neuropsychiatric disorders, including major depressive disorder, schizophrenia, autism spectrum disorder, posttraumatic stress disorder, and attention-deficit/hyperactivity disorder. This narrative review synthesizes preclinical and clinical findings linking dysregulated microglial activation to brain pathology of neuropsychiatric disorders, with particular emphasis on cytokine signaling, oxidative stress, synaptic remodeling, and gut–brain–microglia interactions. This review discusses how sustained microglial priming and excessive inflammatory responses disrupt neurotransmitter systems, impair synaptic integrity, and alter neural network connectivity in brain regions critical for emotion regulation, cognition, and social behavior. Advances in neuroimaging, including TSPO-PET and multimodal approaches, have enabled in vivo assessment of microglial activation in humans, strengthening translational relevance. Furthermore, this review evaluates emerging therapeutic strategies aimed at modulating microglial function, including pharmacological immunomodulators, CSF1R-based depletion and repopulation approaches, lifestyle interventions, and novel cell-based and vesicle-based therapies. Finally, this review highlights key translational challenges, particularly species-specific differences between mouse and human microglia, and proposes future directions for precision neuroimmune interventions. Collectively, the evidence reviewed here positions microglia as both mechanistic drivers and therapeutic targets in neuropsychiatric disorders rooted in chronic neuroinflammation.
By sustaining homeostasis, forming a synaptic architecture, and controlling neuroinflammatory reactions, microglia, the immune cells that dwell in the central nervous system (CNS), are essential for maintaining neuronal integrity [1, 2]. Microglia protect neurons through various mechanisms, including complement components 1q/3 (C1q/C3) signaling, phagocytosis, and the production of activity-dependent cytokines such as interleukin-1 beta (IL-1β) and tumor necrosis factor alpha (TNF-α), which help modulate synaptic plasticity. Additionally, microglia actively alter neuronal circuits in real time during neurodevelopment and following injury, highlighting the importance of these processes [3, 4]. Microglia are now recognized as extremely dynamic sentinels that constantly examine the brain environment. Previously, they were believed to be passive “resting” cells that became active only upon infection or injury [5, 6]. They make up between 10% and 15% of all brain cells, serve as the CNS’s main immunological effectors, and play crucial roles in neurogenesis, synaptic pruning, and the lifetime regulation of neuronal connections [7–9]. A significant paradigm change in neuroscience has resulted from this growing understanding: The immune system of the brain is an active architecture of cognition, emotion, and behavior rather than just a defense mechanism [10, 11]. However, when dysregulated, the same mechanisms that make microglia crucial for brain plasticity also make them potent drivers of pathology [12, 13]. The pathophysiology of several neuropsychiatric disorders, such as major depressive disorder (MDD), schizophrenia, autism spectrum disorder (ASD), and post-traumatic stress disorder (PTSD), has been linked to aberrant or chronic microglial activation, also known as microgliosis [14]. Under these circumstances, microglia may change from a homeostatic, balanced state to a persistent proinflammatory phenotype marked by oxidative stress, excessive cytokine release, and maladaptive synaptic remodeling. This is associated with aberrant synaptic pruning, persistent neuroinflammation, and disruption of neurotransmitter systems, all of which have the potential to significantly change how neural networks function [15, 16]. Multiple molecular processes, including chronic peripheral inflammation, stress-induced glucocorticoid exposure, age-related microglial priming, and hereditary vulnerability, contribute to this maladaptive shift in microglial function. These inputs trigger nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation and NOD-, LRR- and Pyrin domain-containing protein 3 (NLRP3) inflammasome priming, leading to increased cytokine release, oxidative stress, and synaptic dysfunction [17, 18].
Microglial signaling pathways can be sensitized or overactivated by peripheral inflammation, systemic infection, long-term psychological stress, aging-related microglial “priming,” and genetic susceptibility factors. Primed microglia typically release higher levels of proinflammatory mediators, including IL-1β, TNF-α, and IL-6, when responding to subsequent stresses [19–21]. These chemicals may link immune dysregulation to changes in mood, cognition, and social behavior by increasing oxidative stress and altering glutamatergic, dopaminergic, and serotonergic signaling [22]. Neural circuitry can be further compromised by excessive synaptic engulfment by activated microglia, particularly in areas such as the prefrontal cortex, hippocampus, and amygdala, which are crucial for emotion regulation and executive function [23]. Chronic exposure to cytokines and reactive oxygen species (ROS) intensifies these effects, including behavioral problems, neurotoxicity, and synaptic instability [24]. This new paradigm has given rise to the idea of neuroinflammation-induced behavioral pathology, which combines the immunological, neurological, and psychological aspects of mental illness [25]. Immune dysfunction is increasingly considered a mechanistic link between environmental stresses, genetic susceptibility, and clinical symptom presentation rather than as a peripheral or secondary condition [26]. In vivo correlations of these processes in psychiatric populations have started to emerge owing to developments in neuroimaging techniques such as positron emission tomography (PET) tracers that target microglial activation [27, 28]. Pharmacological techniques, including anti-inflammatory drugs, antioxidants, and immunomodulators that target microglia, are being investigated as possible therapeutic treatments to modulate microglial activity [29]. Thus, this review summarizes the most recent research that connects microglial activation to changes in behavior and cognition in various neuropsychiatric conditions. Important biological pathways that underlie these alterations, including oxidative stress mechanisms, cytokine signaling cascades, and microglia-mediated synaptic remodeling, are emphasized. Additionally, new developments in translation might make it possible to modify microglial activity selectively, potentially allowing for targeted interventions that could alleviate symptoms associated with neuropsychiatric conditions. Gaining more knowledge about the complicated relationship between microglial activation and behavioral outcomes could lead to the development of innovative neuroimmune-based treatments for mental illnesses caused by persistent neuroinflammation [30].
This review provides a comprehensive and integrative analysis of microglial activation’s impact on neuropsychiatric disorder behavioral and cognitive outcomes. It aims to consolidate experimental and clinical evidence linking dysregulated microglial function to neuroinflammation, synaptic remodeling, and neurotransmitter changes. The review also discusses cytokine signaling, oxidative stress, and gut–brain–microglia connections and critically evaluates microglia-targeted diagnostic and therapeutic approaches. This research seeks to discover obstacles and opportunities for precise neuroimmune interventions by combining mechanistic insights with translational relevance. The evidence linking microglial activation to behavior and cognition is growing; however, most research is correlational, cross-sectional, or preclinical. A definitive causal relationship between microglial dysfunction and neuropsychiatric symptoms is yet unknown. Microglial activation may cause and arise from intrinsic neuronal dysfunction. Causality and directionality of these correlations require longitudinal, interventional, and mechanistic research. This review integrates findings across preclinical and clinical domains while explicitly acknowledging the limitations associated with cross-level interpretation [31].
A structured literature search was performed on electronic databases such as PubMed, Scopus and Web of Science. We used combinations of keywords such as “microglial activation,” “neuroinflammation,” “neuropsychiatric disorders,” “depression” and “anxiety” with Boolean operators (AND, OR). Studies published in English up to 2025 were considered.
Original research articles and relevant review papers discussing microglial activation and its association with neuropsychiatric conditions were included. Studies without primary data, non-peer-reviewed articles, and studies not directly related to the topic were among the exclusion criteria. High quality original studies, particularly those based on humans, were given priority, with well-established review articles used to support broader contextualization. Quality of evidence was assessed based on study design, sample size, methodological rigor, and relevance to the research objective.
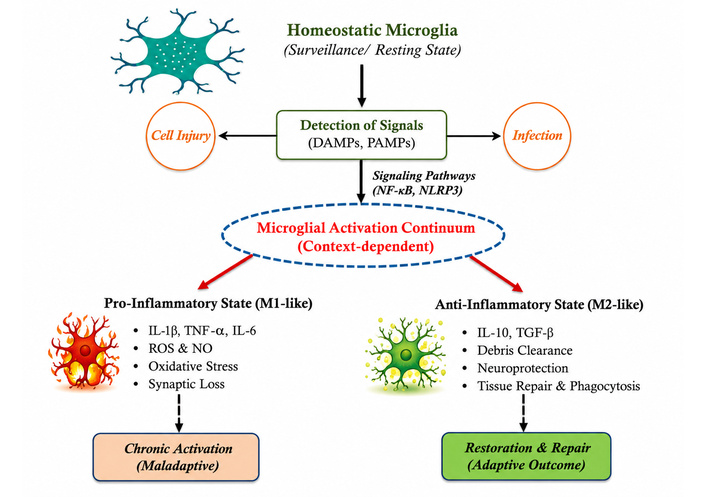
Because of their amazing morphological and functional flexibility, microglia can react quickly to changes in their surroundings. They are specialized cells that function within the CNS milieu as a component of the immune system [32]. As the brain’s resident immune cells, they maintain neuronal homeostasis by continuously scanning the CNS, improving synaptic connections, and phagocytosing cellular waste. These processes dynamically regulate multiple signaling pathways triggered in response to protein aggregation, neuronal death, tissue damage, or infection [33]. Microglia extend highly motile processes that continuously survey the surrounding brain tissue when they are in their surveillant (commonly referred to as “resting”) condition. Microglia become activated when damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) are identified [34–36]. Pathways such as the NLRP3 inflammasome, which needs a priming signal before full activation, mediate this change [37, 38]. IL-1β, IL-6, TNF-α are among the proinflammatory mediators released by activated microglia, which also exhibit distinctive morphological hypertrophy and proliferation [39, 40]. Microglial activation is frequently reduced to two polarization states, although it occurs along a functional continuum. Microglia that are in the M1 (pro-inflammatory microglial phenotype) state release ROS, nitric oxide (NO), and cytokines that increase oxidative stress and cause damage to neurons [41, 42]. On the other hand, in the M2 (anti-inflammatory microglial phenotype) state, microglia promote the resolution of inflammation, improve the phagocytosis of cellular debris, and aid in tissue repair [43]. Microglia can be biased toward an anti-inflammatory phenotype by bioactive lipids such as the ganglioside GM1, suggesting possible treatment options for neuroinflammatory diseases [44]. Additionally, nucleotides produced from injured neurons quickly attract microglia to sites of tissue injury. They consume extracellular detritus and apoptotic cells via scavenger receptors and adaptor protein-mediated signaling. Neurodegenerative diseases, including Alzheimer’s disease (AD), are significantly linked to impairment of these clearance pathways [45, 46]. However, microglial activation may become dysregulated and persistent in long-term neurodegenerative and neuropsychiatric disorders [47]. In the end, this persistent inflammatory state contributes to behavioral and cognitive impairment by causing abnormal synapse remodeling, prolonged cytokine release, and disruption of neural connections [48].
The traditional classification of microglial activation into M1 (pro-inflammatory) and M2 (anti-inflammatory) polarization is currently considered an oversimplification [49]. Recent data from transcriptome, single-cell, and functional research suggests that microglia exist on a highly dynamic and context-dependent continuum of activation states rather than discrete phenotypes. These states are affected by developmental stage, regional brain environment, disease setting, and the attributes and duration of stimuli [50]. Recent consensus initiatives, such as the publication “Microglia states and nomenclature: A field at its crossroads” (2022), advocate for the shift from the M1/M2 paradigm to a more nuanced classification based on molecular markers, functional outputs, and environmental context. This revised approach elucidates microglial responses as diverse and adaptive, encompassing intersecting transcriptional pathways associated with immune surveillance, synaptic remodeling, metabolic regulation, and neuroinflammation [51, 52]. This review conceptualizes microglial activation as a continuum of functional states, preserving M1/M2 terminology mainly for historical context. Homeostatic microglia respond to injury or infection via DAMPs/PAMPs-mediated signaling pathways (e.g., NF-κB, NLRP3), as shown in Figure 1. According to the situation, microglia can be pro- or anti-inflammatory (M1- or M2-like). Such responses can produce chronic neuroinflammation or repair tissue [53].
We first showed how crucial microglia are for preserving brain homeostasis before looking at the consequences of their dysregulation. This section critically examines the data linking aberrant microglial activation to a range of behavioral alterations in patients with neuropsychiatric diseases. Microglia were formerly believed to be passive observers, but it is becoming increasingly evident that they actively participate in the formation of the neural circuits that regulate emotion, behavior, and thought [13, 54, 55]. When their delicate balance is disrupted, these innate immune cells can transform from guardians to major perpetrators of neuropathology, which directly may contribute to the behavioral phenotypes observed in conditions such as depression, schizophrenia, and ASD. We discussed the mechanisms from excessive synaptic pruning to inflammatory cytokine release and presented the preclinical and clinical data that bolster this connection [56].
It is essential to acknowledge that the existing evidence base encompasses various tiers of investigation, including in vitro experiments, animal models, and human studies. While synthesizing these findings offers a holistic view, it simultaneously presents intrinsic constraints. In particular, mechanistic insights obtained from preclinical models may not completely correspond to human pathophysiology. Consequently, the conclusions derived from this review must be regarded with due caution, particularly in relation to clinical implications [57].
The prevalent and debilitating mental illness known as MDD is characterized by persistent low mood, disinterest, cognitive decline, and physiological alterations. It is becoming more widely accepted that MDD is a neuroinflammatory illness [58, 59]. Stress stimulates the hypothalamic-pituitary-adrenal (HPA) axis, leading to sustained glucocorticoid elevation that directly affects microglial activity. In preclinical models such as chronic social defeat stress, microglia exhibit reduced process complexity, enlarged somas, and a transition to an amoeboid morphology. They also have elevated levels of activation markers such as Iba1 and CD68. These alterations are associated with an increase in the synthesis of pro-inflammatory cytokines (such as IL-1β and TNF-α), which disrupt synaptic plasticity in the hippocampus and prefrontal cortex, and is associated with behavioral changes resembling depressive-like states in preclinical models [60, 61]. Studies using translocator protein (TSPO) tracers in PET imaging have shown that individuals with depression have increased higher microglial activation in areas such as the prefrontal cortex and anterior cingulate cortex [62–64].
Preclinical studies indicate that prolonged stress induces microglial proliferation and IL-1β production, leading to diminished hippocampal neurogenesis and anhedonia-like behavior. These conditions are linked to low motivation, cognitive rigidity, and poor emotional regulation [65–67]. Moreover, pharmacological attenuation of microglial activation (e.g., via minocycline or P2X7 receptor antagonists) has been shown to diminish depressive behaviors in animal models, underscoring a causal relationship [68]. Although microglial activation has been observed in depression, its direct role in behavioral symptoms remains unclear. Experimental studies using microglial depletion or pharmacological inhibition (e.g., minocycline) suggest partial reversal of behavioral phenotypes, providing preliminary support for a causal contribution of microglial activity [69].
Neurodevelopmental and neurodegenerative theories of schizophrenia raise the possibility of microglial dysregulation. It has been suggested that the disorder’s characteristic synaptic and anatomical alterations in the brain are also influenced by aberrant microglial activation [70, 71]. Microglia carry out a process called synaptic pruning that optimizes neuronal connectivity during the healthy developmental stages of the brain. However, excessive or inappropriate pruning has been linked to altered gray matter volume and disrupted cortical connections in patients with schizophrenia [72, 73]. TSPO ligand-based PET has been linked to psychotic symptoms and cognitive deficits and has shown a substantial increase in microglial activity, particularly in the prefrontal and temporal cortices [27, 30, 74]. Complement component 4 (C4) overexpression increases microglial synaptic engulfment, leading to synapse overelimination and neuronal circuit dysfunction, supporting the importance of genetic risk factors. Thus, immunological dysregulation and the pathological cognitive, emotional, and perceptual deficits associated with schizophrenia are mechanistically linked by this “microglia–synapse–behavior” axis [75, 76].
ASD is characterized by alterations in microglia. Postmortem brain examinations, particularly in the cerebellum and temporal cortex, have revealed microglial hyperactivation and structural changes [77–79]. Increased inflammatory cytokines such as IL-6, TNF-α, and MCP-1 (monocyte chemoattractant protein-1) are also linked to these patterns of aberrant activation, suggesting that ASD brains exhibit persistent neuroinflammation [80, 81]. The delicate balance between excitatory and inhibitory (E/I) brain circuits can be disrupted by these neuroimmune abnormalities, which are thought to potentially contribute to ASD-related symptoms such as repetitive behaviors, poor social interaction, and abnormal sensory processing [82, 83]. Furthermore, because microglia are crucial for synaptic pruning and refinement during development, dysregulation of these cells may lead to aberrant connections and excessive synapse density features that are frequently observed in ASD [84, 85].
Research on animals further supports these conclusions. Prenatal exposure to inflammatory stimuli (such as poly I:C or LPS) primes microglia toward a hyperreactive phenotype, which is associated with persistent neuroinflammation and behavioral consequences similar to those of ASD in offspring, according to maternal immune activation (MIA) models [86–88]. Pharmacological manipulation or elimination of overactivated microglia in these animals improves social behaviors and restores synaptic density, highlighting their causal significance and therapeutic potential in ASD [89].
The neurobiological effects of trauma exposure are also mediated by microglial activation. High levels of neuroinflammatory markers, such as increased microglial reactivity, are observed in limbic and cortical regions associated with fear processing and memory consolidation, according to postmortem and neuroimaging results [90, 91]. The process of traumatic stress involves the activation of peripheral immunological pathways and central microglial priming, which increases vulnerability of the brain to stresses in the future and intensifies pathological anxiety [21, 92]. This procedure provides significant mechanistic support from preclinical models. Recurrent encounters with hostile conspecifics result in significant microglial activity in the hippocampus, amygdala, and prefrontal cortex, according to social defeat stress models [93, 94]. The models consistently predict behavioral effects such as social behavior withdrawal, hypervigilance, and anxiety-related behavioral symptoms that are identical to those of PTSD in individuals. Similarly, fear-conditioning studies have shown that microglial priming is associated with greater release of TNF-α and IL-1β during the formation of fear memories, which is associated with overconsolidation of traumatic memories. In these paradigms, rodents are trained to condition a neutral signal with an unpleasant stimulus [95–97].
Direct trauma exposure is not necessary to activate microglia; observed or vicarious trauma can also cause stress sensitivity and neuroinflammation. Research shows that when mice see stress inflicted on their cage mates, they develop PTSD-like symptoms and microglial hyperactivation, indicating that direct and indirect trauma overlap with immunological mechanisms. Additionally, increased TSPO binding, an indicator of microglial activation in PTSD patients, is supported by clinical PET imaging. Neuroinflammation is linked to increased arousal, intrusive memory, and fear extinction [98]. These converging lines of evidence highlight microglia as both biomarkers and potential therapeutic targets in trauma-related disorders.
The role of microglial dysregulation in the neurodevelopmental route of attention deficit hyperactivity disorder (ADHD) is becoming more widely acknowledged [99]. The brain regions responsible for executive function and attention regulation, the prefrontal and orbitofrontal cortices, have shown an increase in microglial activity via PET imaging. This suggests that neuroimmune signaling in networks linked to ADHD is disrupted [100, 101]. Inattention, impulsivity, and poor cognitive control may result from excessive microglial activation interfering with synapse refinement, dopaminergic transmission, and cortical maturation. Preclinical models of ADHD also corroborate this neuroinflammatory concept [101, 102]. Rats with spontaneous hypertension have elevated Iba1+ microglia, elevated proinflammatory cytokines such as TNF-α, and altered hippocampal and prefrontal cortex blood–brain barriers, all of which are indicative of cognitive deficits in ADHD. The importance of microglia-mediated inflammatory pathways in the onset and maintenance of symptoms is further supported by evidence showing that early-life immunological activation and increased levels of peripheral inflammatory markers may increase the risk for ADHD [103].
Molecular and biological pathways that link microglia to behavior beyond their conventional function in immune monitoring, microglia influence behavior through an elaborate network of interrelated molecular and cellular pathways. A growing body of research indicates that microglial activation modifies circuit dynamics and neuronal function via oxidative stress, inflammatory signaling, synaptic remodeling, and connections with peripheral metabolic systems [104–106]. These processes operate together to form an integrated neuroimmune network that can sustain behavioral changes long after the initial shock has subsided.
Activated microglia emit cytokines that change synaptic transmission and neuronal excitability. Cytokine-mediated regulation of synaptic function: The release of proinflammatory cytokines such as IL-1β, TNF-α, and IL-6 is one of the most direct ways that microglia alter behavior [72, 107]. Activated microglia release cytokines such as IL-1β, TNF-α, and IL-6, which alter astrocytic function and impair pre- and postsynaptic signaling [108]. Low-level cytokine signaling supports learning and synaptic plasticity in physiological settings. However, if microglial activation persists for an extended period of time, cytokines may remain in the body for an extended period of time, upsetting the delicate equilibrium required for regular synaptic transmission. By altering NMDA receptor signaling and intracellular calcium dynamics, interleukins such as IL-1β has been shown to impair hippocampal long-term potentiation, hence impairing memory formation and cognitive flexibility [109–111]. Conversely, TNF-α modifies synaptic scaling by decreasing the intensity of inhibitory GABAergic transmission and increasing the number of surface AMPA receptors. Neural networks become less stable as a result of this shift toward excitatory dominance, and people are more prone to behave in ways associated with stress and anxiety [112, 113].
Concurrently, astrocytic glutamate absorption is disrupted by inflammatory cytokines, leading to increased extracellular glutamate levels and excitotoxic stress [114, 115]. Anhedonia, decreased motivation, attentional impairments, and emotional dysregulation basic traits shared by depression, schizophrenia, and trauma-related disorders are behavioral manifestations of these biological disturbances.
Owing to microglia and oxidative stress, long-term generation of reactive oxygen and nitrogen species is linked to sustained microglial activation, which is physiologically demanding. Although long-term redox imbalance negatively impacts neuronal integrity, transient oxidative signaling is essential for host defense [116, 117]. Because of their high energy needs, neurons are particularly vulnerable to oxidative stress from microglia. Reduced neural resilience and synaptic inefficiency are the results of oxidative damage to respiratory complexes and mitochondrial membranes, which impairs ATP synthesis and calcium buffering capacity [118, 119]. Large-scale brain networks fall out of sync as a result, making it more difficult to think and regulate one’s emotions. Fatigue, psychomotor slowness, executive dysfunction, and depressive-like phenotypes are among the behavioral signs of mitochondrial malfunction that are clinically important [120–122]. Fatigue, cognitive decline, and depressive behavior have all been connected to oxidative imbalance. In animal models, antioxidant therapy decreases microglial activation and restores normal behavior, suggesting that redox balance is a therapeutic axis [123].
Through immunological signaling and microbial metabolites, including short-chain fatty acids (SCFAs), the gut microbiome affects microglial maturation and function: Microglia may be primed toward proinflammatory states by dysbiosis, which may contribute to behavioral symptoms such as anxiety, autism, and depression [79, 124, 125]. According to recent studies, the gut microbiota is an essential upstream regulator of immunological regulation and microglial maturation [126]. Metabolites generated from microbes, particularly SCFAs, influence the transcriptional programs, morphology, and inflammatory levels of microglia [124, 127]. Figure 2 illustrates how immunological signals and gut-derived metabolites affect microglial activation states via vagal and systemic circulation routes.
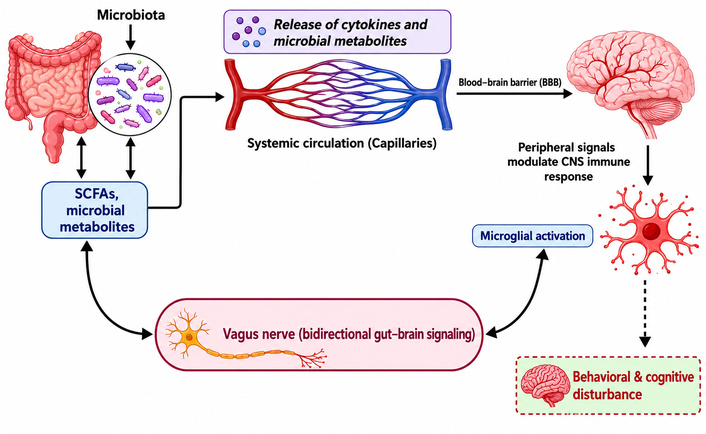
Integrated gut–brain–microglia axis and neuroimmune regulation. This diagram illustrates the complex communication network between the gastrointestinal tract and the central nervous system.
While preclinical studies provide important mechanistic insights, their direct translation to human conditions remains limited. Differences in microglial biology, experimental conditions, and disease complexity necessitate cautious interpretation when extrapolating these findings to clinical contexts [128].
Instead of depending solely on static imaging, neuroinflammation is now assessed in actual individuals using dynamic, multimodal monitoring. Diffusion tensor imaging (DTI), magnetic resonance spectroscopy (MRS), and second-generation TSPO-PET biomarkers make it possible to evaluate neuroinflammation in humans in vivo [129]. The TSPO, which is produced on activated microglia, is more efficiently bound by modern tracers, increasing its binding affinity [130]. They produce significantly clearer and more dependable images because of their superior brain penetration (uptake). Clinicians can now identify “low-grade” neuroinflammation, which was previously undetectable with outdated technologies, owing to these advancements [131, 132]. While TSPO-PET imaging is widely used to investigate neuroinflammation, its interpretation remains challenging due to variability in findings across disorders, methodological limitations, and concerns regarding biological specificity [133, 134]. While DTI detects white matter damage caused by chronic inflammation, which is crucial for multimodal integration, MRS monitors changes in glial-specific metabolites such as myo-inositol [135]. However, interindividual variability in TSPO binding and the lack of cell specific markers remain challenges [136].
Specific pharmacologically adjusted M1-like microglial polarization is inhibited by minocycline. In addition to its antibacterial qualities, minocycline efficiently reduces the “cytokine storm” in the CNS and alleviates unpleasant symptoms in neuropsychiatric patients by inhibiting the p38 MAPK and NF-κB pathways [137, 138]. Medications that target microglial activation are being developed or repurposed for use in mental health. Minocycline has been suggested to alleviate depressive and negative symptoms in patients with schizophrenia by reducing the production of proinflammatory cytokines [139]. Ibudilast (AV411), also referred to as a phosphodiesterase inhibitor, exhibits early promise in treating mood disorders by suppressing microglial activation. In substance use disorders, ibudilast has been shown to be effective in reducing inflammation in brain reward circuits [140, 141]. Inhibitors of the colony-stimulating factor 1 receptor (CSF1R) allow for the regulated depletion and repopulation of microglia, which may reset the immunological tone [142, 143]. Following treatment, “naïve” microglia repopulate the CNS, potentially halting the development of tauopathies and chronic inflammation [144].
By altering SCFAs, which regulate microglial priming via the vagus nerve and metabolic pathways, dietary modifications can affect neuroinflammation [145]. M2-like phenotypes are altered by aerobic exercise, which increases the levels of anti-inflammatory cytokines, including brain-derived neurotrophic factor (BDNF) and IL-10 [146, 147]. Resveratrol and curcumin are examples of compounds that block microglial activation by acting on the sirtuin 1 (SIRT1) pathway. By lowering peripheral CRP (C-reactive protein) and IL-6 levels, mindfulness exercise decreases the overall inflammatory burden in the brain [148]. Exercise, dietary polyphenols (curcumin, resveratrol), and mindfulness techniques improve behavioral results and lower neuroinflammatory indicators, potentially by changing microglial polarization to an anti-inflammatory phenotype. In a comprehensive neuroimmune therapy strategy, these nonpharmacological techniques could support medication-based tactics [149].
Despite accumulating evidence linking microglial activation and cognitive changes, most human data is correlational. Thus, causal assumptions should be tested carefully, especially in clinical population. Microglia are now recognized as primary causative agents rather than merely secondary responders in neuropsychiatric disorders owing to recent breakthroughs in neuroimmunology. This review explores the “causation versus consequence” conundrum, may help explain the special roles of microglia in conditions such as schizophrenia and depression, and evaluates novel therapeutic strategies, including modified microglia and precision biomarkers. Determining whether microglial activation is a cause or a result of neuronal injury is a significant difficulty in neuroimmunology. To restore equilibrium, it was previously believed that microglial activation is a response to damaged or overactive neurons. Genetic research suggests that the C4 gene is a direct risk factor for schizophrenia because it is linked to excessive microglial synaptic pruning prior to the onset of clinical symptoms [150, 151]. According to research on experimental validation, injecting “primed” microglia into healthy mice can result in depressive-like behaviors, indicating that immunological dysregulation alone may be the cause of disease [66]. Currently, some contend that categorizing microglia as “M1” (toxic) or “M2” (helpful) microglia is oversimplified since it ignores the significant variations in their transcriptional patterns [152]. Because TSPO is increased under both protective and detrimental circumstances, current PET imaging methods employing TSPO ligands are unable to differentiate between them. Single-cell RNA sequencing (scRNA-seq) is being used by researchers to identify distinct “immunopsychiatric” signatures [153, 154]. Certain indicators, such as CD68, tell cells to actively phagocytize items, whereas TREM2 is a typical indicator of debris removal and cellular defense [155, 156]. The following Table 1 illustrates how microglia can be both beneficial and detrimental under extreme circumstances:
The beneficial and detrimental effects of microglia under extreme circumstances.
| Condition | Microglial role in pathology | Therapeutic potentials | References |
|---|---|---|---|
| Anxiety/Depression | Stress-induced “parainflammation” may contribute to synaptic atrophy in the hippocampus. | Shifting polarization via H3R (histamine 3 receptor) or SSRIs (selective serotonin reuptake inhibitors). | [65, 157] |
| Schizophrenia | Excessive C4-mediated synaptic pruning and inflammation in white matter are associated with schizophrenia. | Inhibiting excessive synaptic pruning or correcting the maturation of glial cells may be beneficial. | [72, 158] |
| Autism (ASD) | Early-life immune activation (MIA) disrupts synaptic refinement and neuronal connectivity. | Early intervention to restore synaptic homeostasis. | [159] |
| Addiction | Activation in reward circuits reinforces behavior. | Targeted neuroimmunomodulation to reduce withdrawal-related anxiety. | [160, 161] |
Furthermore, human microglia show notable species-specific variations in gene expression, notably Siglec receptor patterns, which are far more relevant in terms of translational gaps, even though many studies have employed mouse models [162]. Figure 3 provides an overview of the major translational gaps between mouse and human microglia, the consequences of chronic microglial hyperactivation, and the potential risks associated with microglial depletion.
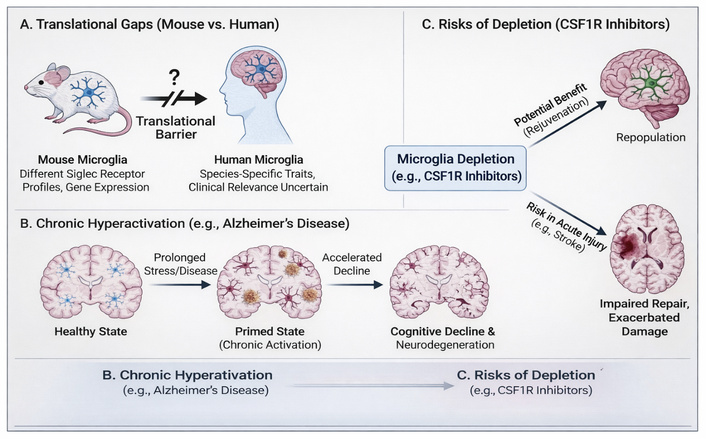
Overview of key challenges in translating microglial research. (A) Differences in microglial biology between species impede direct translation from mouse models to humans [163]. (B) Chronic microglial activation in neurodegenerative diseases may contribute to a “primed” state and accelerated cognitive decline. (C) While microglial depletion strategies may offer rejuvenation, they pose significant risks in the context of acute brain injury [164, 165].
Regarding possible therapeutic developments, “engineered microglia” refers to the use of human iPSC-derived microglia as “cellular couriers” to carry proteins that change the course of disease directly to the sites of injury. To facilitate neural repair, extracellular vesicles (EVs) generated from microglia can be altered to transport miRNAs across the blood–brain barrier. These innovative approaches illustrate the potential of leveraging the body’s intrinsic biological mechanisms to combat neurodegenerative disease. Researchers expect to improve patient outcomes and therapeutic efficacy in diseases such as Parkinson’s disease and AD by focusing on these strategies [166–168].
EVs generated from microglia are becoming important intercellular communication mediators in the brain. These vesicles (MVs) contain a variety of biological “cargo,” such as proteins, enzymes, DNA, and RNA, as shown in Figure 4. These vesicles can transfer toxic pathologies, including tau and amyloid-beta proteins, to nearby neurons in neurodegenerative diseases such as AD [169, 170]. To facilitate neuronal repair, these same vesicles can be altered or designed to transport therapeutic substances (such as miRNAs) across the blood–brain barrier, thereby potentially improving treatment outcomes for neurodegenerative diseases like AD. Profiling these microvesicles provides a method for identifying accurate precision biomarkers to predict the onset and progression of disease disorders because their payload reflects the internal condition of the microglia [171, 172].
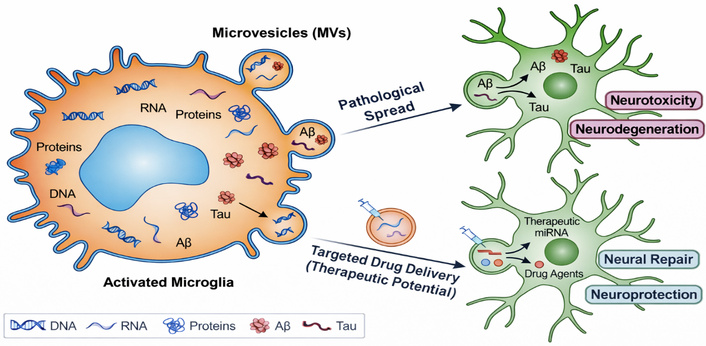
The main goal of this review to elucidate the intricate role that microglia play in behavioral pathology. This field opens prospects for cutting-edge neurological and psychiatric treatments while navigating several crucial challenges. In behavioral pathology, microglia play a crucial role in both substantial problems and exciting prospects. Chronic neuroinflammation, aberrant synaptic pruning, ambiguous microglial subtypes, and restricted medication delivery across the blood–brain barrier are some of the main obstacles. Growing knowledge of microglial biology also opens up new possibilities, including therapeutic modulation, precision immune interventions, developmental-stage-specific tactics, and bolstering connections between neuroimmunology and psychiatry, all of which contribute to the advancement of focused treatments for neurological and psychiatric disorders [173–176].
There is strong evidence that microglial activation is connected to neuropsychiatric symptoms, but causation and association are difficult to distinguish. Many human investigations use imaging markers like TSPO-PET, which show microglial activity but not temporal or mechanistic directionality [177]. Preclinical investigations provide stronger causal inference, but interspecies differences limit direct applicability, making it challenging to translate findings from animal models to human conditions. Neuronal failure, systemic inflammation, and stress can activate microglia. Thus, data support a bidirectional, context-dependent link rather than a causal one [178].
Despite notable progress, the existing evidence base exhibits several critical limitations. First, a large part of the data comes from preclinical models, which may not fully represent human microglial biology because gene expression, receptor profiles, and functional responses can be different between species. These variations restrict direct translational applicability. Second, clinical evidence primarily derives from cross-sectional and imaging studies, especially TSPO-PET, which lacks cellular specificity and fails to reliably differentiate between protective and harmful microglial states. Such evidence makes it difficult to understand how things work [179, 180]. Third, a lot of research is correlational, which makes it challenging to show that microglial activation is associated with neuropsychiatric symptoms. Changes in microglia could be both causes and effects of problems with neurons [178, 181]. To overcome these limitations, it is essential to conduct longitudinal, multi-modal, and human-centered studies that integrate molecular, imaging, and clinical data.
Microglia have emerged as central orchestrators of the neuroimmune processes that shape behavior, cognition, and emotional regulation. Microglia do more than just respond to neuronal injury; they also play an active role in synaptic refinement, circuit maturation, and neurotransmission when the body is healthy. When they are not working properly, they may contribute to the development of neuropsychiatric disorders. The evidence reviewed here supports a unifying framework in which chronic microglial activation, priming, and maladaptive inflammatory signaling disrupt neural homeostasis, and is associated with persistent behavioral and cognitive impairment. Importantly, this body of work underscores that microglial activation exists along a dynamic continuum, with context-dependent protective or detrimental effects. While transient activation may support repair and plasticity, prolonged or exaggerated responses promote oxidative stress, aberrant synaptic pruning, and network instability. Translational efforts are further complicated by species-specific differences in microglial gene expression and receptor profiles, highlighting limitations in directly extrapolating findings from animal models to humans, as summarized in Figure 3. Despite these challenges, rapid advances in neuroimaging, single-cell profiling, and immunomodulatory therapies offer promising opportunities to refine microglia-targeted interventions. Strategies aimed at selectively resetting or reprogramming microglial states rather than broadly suppressing immune activity may provide more effective and safer therapeutic outcomes. Integrating neuroimmune biomarkers with clinical phenotyping and developmental timing will be critical for precision medicine approaches, as it will enable tailored interventions that account for individual variability in disease progression and treatment response. In conclusion, enhancing our comprehension of microbial biology at the molecular, cellular, and systems levels is crucial for reconciling neuroinflammation with behavioral pathology. Targeting microglia holds considerable promise for redefining therapeutic paradigms in psychiatry and neurology, moving toward mechanism-based interventions for disorders driven by chronic neuroimmune dysregulation. These findings position microglia as potential contributors to disease mechanisms and promising therapeutic targets.
AD: Alzheimer’s disease
ADHD: attention deficit hyperactivity disorder
ASD: autism spectrum disorder
C4: complement component 4
CNS: central nervous system
DAMPs: damage-associated molecular patterns
DTI: diffusion tensor imaging
EVs: extracellular vesicles
HPA axis: hypothalamic-pituitary-adrenal
IL-1β: interleukin-1 beta
MDD: major depressive disorder
MRS: magnetic resonance spectroscopy
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
NLRP3: NOD-, LRR- and Pyrin domain-containing protein 3
PAMPs: pathogen-associated molecular patterns
PET: positron emission tomography
PTSD: post-traumatic stress disorder
ROS: reactive oxygen species
SCFAs: short-chain fatty acids
TNF-α: tumor necrosis factor alpha
TSPO: translocator protein
TU: Conceptualization, Supervision. CH: Resources, Investigation. AA: Writing—original draft, Resources. AZ: Resources, Writing—review & editing. AN: Supervision, Writing—review & editing. RSN: Methodology, Formal analysis. JF: Investigation, Writing—original draft. RS: Investigation, Methodology. MMM: Writing—review & editing. ST: Supervision. JAR: Supervision, Data curation. NSS: Writing—original draft. All authors read and approved the submitted version.
The authors declare no conflict of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This research received no specific grant from any funding agency.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2049
Download: 24
Times Cited: 0
Elizabeta B. Mukaetova-Ladinska ... Qadeer Arshad