 Perspective
Perspective

Affiliation:
1Centro Científico y Tecnológico de Excelencia Ciencia & Vida, Fundación Ciencia & Vida, Santiago 8580702, Chile
2Doctorado en Biotecnología y Bioemprendimiento, Facultad de Medicina, Universidad San Sebastián, Santiago 7510156, Chile
ORCID: https://orcid.org/0000-0001-9695-0155
Affiliation:
1Centro Científico y Tecnológico de Excelencia Ciencia & Vida, Fundación Ciencia & Vida, Santiago 8580702, Chile
3Facultad de Medicina, Universidad San Sebastián, Santiago 7510156, Chile
Email: rpacheco@cienciavida.org; rodrigo.pacheco@uss.cl
ORCID: https://orcid.org/0000-0001-8057-9806
Explor Neuroprot Ther. 2026;6:1004151 DOI: https://doi.org/10.37349/ent.2026.1004151
Received: December 30, 2025 Accepted: March 31, 2026 Published: May 12, 2026
Academic Editor: Ta-Yuan Chang, Geisel School of Medicine at Dartmouth College, USA
Short-chain fatty acids (SCFAs) are microbial-derived metabolites produced primarily through the fermentation of dietary fibre by the intestinal microbiota. Current evidence indicates that they play a key role in modulating nociception and pain processing across immune, metabolic, and neural pathways. The prevailing view that SCFAs suppress pain has been challenged by emerging evidence demonstrating that these same metabolites can also drive hyperalgesia. This apparent “SCFA paradox” persists because most studies have examined individual metabolites in isolation rather than considering them within their broader biological context. Here, we propose an integrative framework in which SCFAs function within a competitive receptor triad, and pain outcomes are dictated by the balance among three signalling axes: a pro-inflammatory immune axis driven by acetate acting through G protein-coupled receptor 43 (GPR43), a pro-resolutive metabolic axis mediated by butyrate via histone deacetylase (HDAC) inhibition and activation of GPR109A, and a direct neural sensing axis triggered by propionate through olfactory receptor 78 (OLFR78). Chronic pain, therefore, does not arise simply from the presence or absence of SCFAs, but from the pathological dominance of one of these axes shaped by specific dysbiosis profiles. This framework moves beyond correlation by providing a mechanistic basis for precision interventions designed to rebalance SCFA signalling, offering novel therapeutic opportunities for neuropathic and inflammatory pain conditions.
For over a decade, the role of short-chain fatty acids (SCFAs) as mediators of the microbiota-host communication in the regulation of pain has been explored. Particularly, butyrate has been described as an analgesic and anti-hyperalgesic mediator of microbiota-host communication [1]. This narrative, however, is incomplete. Although most studies exploring this issue point to an anti-inflammatory and analgesic role of butyrate, other studies reveal contradictory conclusions: the same microbial metabolites, butyrate in this example, might also exacerbate hyperalgesia [2]. Similarly, evidence shows that propionate predominantly exerts anti-nociceptive effects, reducing hyperalgesia in inflammatory, trigeminal/migraine, and visceral pain models through anti-inflammatory, antioxidant, and neuroprotective mechanisms [3]. However, when present as part of a sustained, elevated SCFA milieu (rather than administered alone), it can contribute to microglia-driven pain sensitisation in neuropathic contexts [4]. Acetate displays a more ambivalent and context-dependent profile: at high systemic or local concentrations, it can be clearly pronociceptive (e.g., headache models and chemical irritation-induced visceral pain), whereas under conditions of microbiota-mediated physiological modulation, moderate acetate levels have been associated with reduced visceral hypersensitivity. In neuropathic settings, sustained increases in acetate as part of a combined SCFA pool can also participate in the maintenance of chronic pain [4].
This controversy persists because research has treated SCFAs as isolated mediators rather than as components of an integrated signalling system. Since many SCFA receptors might bind the same ligands, but with different affinities (see Table 1), and are expressed in different cell types involved in regulatory circuits of pain (immune cells, nociceptive neurons, epithelial cells, and others), we propose here an integrative model for the role of these bacterial mediators in the regulation of pain. We hypothesize that SCFAs operate within a competitive triad of receptors whose internal balance determines pain outcomes. This triad consists of three context-dependent axes: i. A pro-inflammatory immune axis, ii. a resolution-promoting metabolic axis, and iii. a direct neuronal sensing axis. Their dynamic interplay, which we refer to as triad competition, functions across multiple biological levels. At the ligand level, SCFAs with overlapping receptor affinities compete for binding. At the cellular level, distinct cell populations differentially sequester and metabolise these ligands.
The SCFA receptor triad and its functional axes in pain regulation.
| Triad axis | SCFA receptor (gene) | Primary ligands & relative affinities | Key cell types expressing receptor in vivo | Output effect on pain (based on in vivo models) | Key supporting references |
|---|---|---|---|---|---|
| Immune axis (pro-algesic; primary driver of inflammatory pain) | GPR43 (FFAR2) | Acetate ≈ propionate > butyrate | Neutrophils, mast cells, colonic macrophages, colonic T-cells, enteroendocrine cells, adipocytes | Pro-nociceptive: Drives inflammatory and visceral pain. GPR43 activation promotes neutrophil recruitment, mast cell degranulation, and M1 macrophage polarisation, leading to the release of algogenic that sensitise nociceptors. | [5–8] |
| Neuronal axis (dual modulation; primary driver of direct neural sensing) | GPR41 (FFAR3) | Propionate ≈ butyrate > acetate | Neurons (DRG, sympathetic, enteric), adipocytes, endocrine cells, T-cells | Context-dependent (bidirectional): In visceral pain models, high propionate levels are pro-nociceptive, sensitising afferent neurons. Under homeostatic conditions, GPR41 signalling may exert an inhibitory tone. It is a central node for direct neural sensing of SCFAs. | [5, 9] |
| OLFR78 (OR51E2 in humans) | Acetate ≈ propionate | Enteroendocrine cells, vascular smooth muscle cells, vagal sensory neurons (evidence emerging) | Pro-nociceptive (proposed mechanism): As a Gαs-coupled receptor, its activation by high propionate levels in DRG neurons would increase cAMP/PKA signalling, leading to phosphorylation and sensitisation of ion channels (e.g., TRPV1, Nav). Direct in vivo pain studies are limited. | [10, 11] | |
| Metabolic axis (analgesic; primary driver of resolution) | GPR109A (HCAR2) | β-Hydroxybutyrate > butyrate | Macrophages, keratinocytes, Langerhans cells, T-cells, neutrophils, microglia, astrocytes, neurons, satellite cells, hepatocytes, colonic epithelial cells, adipocytes | Anti-nociceptive: Activation of microglial GPR109A attenuates chronic pain by suppressing p38 MAPK activity, NLRP3 activation, and pro-inflammatory cytokines. Its deficiency leads to chronic neuroinflammation. | [8, 12, 13] |
SCFA: short-chain fatty acid; GPR43: G protein-coupled receptor 43; DRG: dorsal root ganglia; OLFR78: olfactory receptor 78; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; TRPV1: transient receptor potential vanilloid 1; Nav: voltage-gated sodium channels; NLRP3: NOD-like receptor family pyrin domain-containing 3.
Finally, at the network level, their downstream transcriptional and metabolic programmes exert reciprocal regulation, whereby activation of one axis can suppress or enhance signalling through the others. In this framework, pain does not result simply from the presence or absence of SCFAs, but from a disruption in the balance among the axes of the triad. Understanding how this competition is regulated enables movement beyond correlative associations towards a predictive, mechanistic understanding of how microbial metabolism shapes nociception.
Distinct pain disorders exhibit characteristic microbial signatures that move beyond mere correlation and increasingly suggest causality. Chronic visceral pain conditions, particularly irritable bowel syndrome (IBS), are consistently associated with fermentative dysbiosis, characterised by increased abundance of Prevotella and Bacteroides species. These bacteria generate high levels of propionate and acetate, a profile that correlates with symptom severity [14]. Conversely, chronic neuropathic pain states frequently display a depletive dysbiosis marked by reduced microbial diversity and lower abundance of butyrate-producing genera such as Roseburia [15]. In inflammatory and post-surgical pain models, broad-spectrum antibiotics have been shown to either attenuate or exacerbate pain, directly implicating microbial communities in both the initiation and resolution of pain [16]. Together, these clinical and experimental patterns provide a critical conceptual bridge between the microbial ecosystem and the pain phenotype, underscoring the need for mechanistic explanations linking microbiota dynamics to nociceptive regulation.
This axis functions as an active danger-sensing network that is primarily driven by acetate. Its pro-inflammatory nature is defined by its capacity to prime, recruit, and activate innate immune effectors, while simultaneously biasing adaptive immunity towards pathological phenotypes. Dominance of this axis depends on the convergence of three conditions. First, substrate availability: fermentative dysbiosis generates excessive acetate, saturating receptor signalling capacity. Secondly, inflammatory priming: pre-existing inflammation upregulates G protein-coupled receptor 43 (GPR43) expression and provides the necessary co-stimulatory signals. Thirdly, failure of resolution: suppression of the butyrate-driven resolution axis removes essential regulatory brakes. When this delicate equilibrium is disrupted, acetate-driven pathways amplify inflammatory signalling, undermine regulatory mechanisms, and lock the system into a state of chronic activation.
Through activation of the GPR43, acetate regulates innate immunity by promoting chemotaxis and functional activation across multiple myeloid lineages. In neutrophils, where GPR43 is the predominant SCFA receptor, its stimulation induces intracellular calcium influx, priming cells for chemotaxis, production of reactive oxygen species (ROS), and the release of algogenic mediators that contribute to pain [17]. In mast cells, GPR43 activation provides an immunoglobulin E (IgE)-independent pathway for degranulation, leading to the release of histamine, tryptase, and nerve growth factor (NGF), which collectively reduce the activation threshold of adjacent C-fibres [18]. In macrophages, acetate signalling acts synergistically with canonical inflammatory cues to reinforce a pro-inflammatory M1 phenotype, enhancing the production of interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), and nitric oxide (NO), while concurrently suppressing M2 repair-associated programming [19].
This axis extends beyond innate immunity. Acetate and propionate function as immunometabolic modulators that recalibrate T-cell differentiation. In T-helper 17 (Th17) cells, they act as metabolic substrates that increase the availability of acetyl coenzyme A (acetyl-CoA), thereby promoting histone acetylation at the retinoic acid receptor-related orphan receptor C (RORc) locus and facilitating Th17 lineage commitment [20]. Simultaneously, signalling through GPR43 in dendritic cells (DCs) enhances the production of IL-6 and IL-23, further consolidating Th17 polarisation [20]. Tissue-resident gamma-delta (γδ) T-cells, which are pivotal amplifiers of nociception through the release of IL-17, are also modulated by SCFAs via GPR43, which regulates their activation threshold and cytokine output [21]. Persistent exposure to SCFA-driven cytokine networks weakens immune resolution by impairing the suppressive capacity of regulatory T-cells (Treg). Whereas butyrate supports Treg induction under homeostatic conditions, chronic GPR43-dependent inflammation disrupts these regulatory pathways, shifting adaptive immunity towards pathological dominance [20].
This axis represents the most immediate interface between microbial metabolites and nociception. Propionate directly modulates sensory neuronal excitability and drives local neuroimmune crosstalk. Primary sensory neurons express GPR41 and olfactory receptor 78 (OLFR78), forming an integrated signalling network. At physiological concentrations, activation of the Gi/o-coupled receptor GPR41 by propionate may exert a mild inhibitory effect. However, at pathologically elevated concentrations, such as those observed in small intestinal bacterial overgrowth, propionate becomes strongly pronociceptive. It potentiates transient receptor potential vanilloid 1 (TRPV1) and transient receptor potential ankyrin 1 (TRPA1) channels. Moreover, propionate-mediated OLFR78 activation elevates cyclic adenosine monophosphate (cAMP) levels, leading to protein kinase A (PKA)-dependent phosphorylation of voltage-gated sodium channels (Nav) Nav1.7 and Nav1.8 [9, 10]. Sensitised nociceptors release substance P (SP) and calcitonin gene-related peptide (CGRP), activating mast cells and establishing a paracrine amplification loop. Satellite glial cells, which closely envelope the neuronal soma, may also respond to SCFAs via GPR43, potentially modulating neuroglial coupling [22]. Pathological dominance of this axis requires a permissive niche characterised by fermentative dysbiosis generating excess luminal propionate, neuronal vulnerability with upregulated receptor expression, and failure of compensatory dampening mechanisms due to butyrate deficiency.
In innate immunity, butyrate suppresses activation of the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome and promotes macrophage polarisation towards an anti-inflammatory/tissue repairing M2 phenotype through histone deacetylase (HDAC) inhibition [23]. Simultaneously, activation of GPR109A reinforces this programme and induces additional anti-inflammatory mediators [24]. In adaptive immunity, butyrate functions as a critical epigenetic regulator of peripheral Treg differentiation and function by stabilising forkhead box P3 (Foxp3) expression, while concurrently inhibiting Th17 and Th1 differentiation [25]. Its HDAC inhibitory activity amplifies pre-existing transcriptional landscapes, meaning that its pro-resolution effects depend on a cellular context already primed for repair. This underscores its role within the competitive triad whose integrated dynamics are represented in our model (Figure 1).
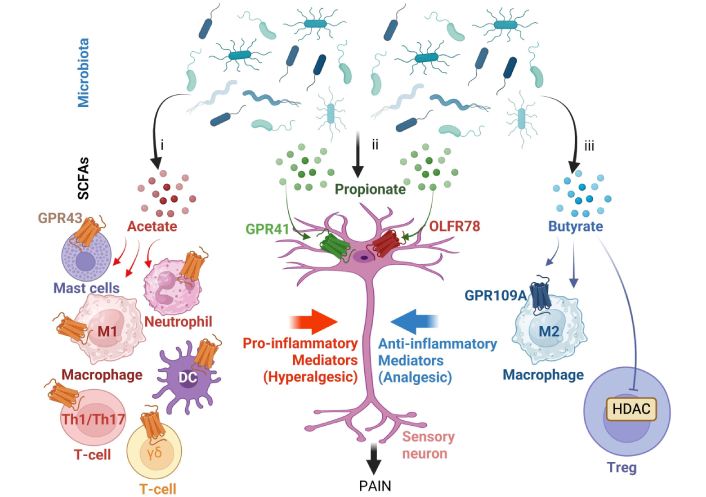
The short-chain fatty acid (SCFA) receptor triad balance system and its dysregulation in chronic pain. The effect of SCFAs on pain is governed by dynamic competition among three signalling axes. i. The pro-inflammatory immune axis, mainly stimulated by an excess of acetate via GPR43 in innate immune cells (neutrophils, mast cells, DC, and M1 macrophages) and T-cells (Th1, Th17, and γδ T-cells). ii. The direct neural sensing axis, stimulated mainly by propionate via OLFR78 and GPR41, which sensitises nociceptors. iii. The resolution-promoting metabolic axis, stimulated mainly by butyrate through the inhibition of HDAC and activation of GPR109A, which promotes anti-inflammatory function in macrophages (M2) and T-cells (Treg). GPR43: G protein-coupled receptor 43; Th1: T-helper 1; DC: dendritic cell; γδ: gamma-delta; OLFR78: olfactory receptor 78; HDAC: histone deacetylase; Treg: regulatory T-cells. Created in BioRender. Pacheco, R. (2026) https://BioRender.com/8v3otn9.
This “receptor triad” model reaches its full potential when conceptualised as a dynamic signalling balance system. Chronic pain does not arise from the simple presence or absence of SCFAs, but from pathological imbalances among three interdependent axes: a pro-inflammatory immune axis, a resolution-promoting metabolic axis, and a direct neuronal sensing axis, each shaped by distinct dysbiosis profiles. Within this framework, the adaptive immune component, particularly colonic T-cells, emerges as a pivotal mediator that translates microbial metabolism into long-term neuroimmune outcomes.
In disorders such as IBS, fermentative dysbiosis generates excess propionate and acetate. Propionate saturates GPR41 and OLFR78 on visceral afferents, directly potentiating the activation of TRPV1, TRPA1, and Nav [26]. Acetate activates mucosal mast cells via GPR43, leading to the release of histamine and NGF, which synergistically sensitise sensory neurons [8]. The resulting acidic environment suppresses butyrate-producing bacteria, thereby impairing the resolution axis and producing severe pain in the absence of major tissue pathology.
In systemic pain syndromes such as fibromyalgia, diabetic neuropathy, or chemotherapy-induced pain, failure of the resolution-promoting axis permits the persistence of chronic neuroinflammation. Depletive dysbiosis results in critical butyrate deficiency, suppressing GPR109A signalling and HDAC inhibition. In the absence of these signals, microglia and macrophages fail to sustain an anti-inflammatory M2 phenotype, while Treg lose suppressive stability. Physiological levels of acetate maintain glial activation through GPR43 stimulation, establishing a state of chronic neuroinflammation. Neurons become sensitised by cytokines released from activated glia and Th17 cells, effectively hijacking the neuronal sensing axis through immune feedback rather than through direct exposure to luminal metabolites [27].
Acute tissue damage recruits neutrophils and M1 macrophages, which release ATP, damage-associated molecular patterns (DAMPs), and cytokines that activate nociceptors, while acetate amplifies these responses via GPR43. Recovery depends on timely, butyrate-driven resolution mechanisms, including activation of GPR109A, HDAC inhibition, macrophage polarisation towards an anti-inflammatory M2 phenotype, and expansion of Foxp3+ Treg that suppress IL-17-mediated responses. When dysbiosis or metabolic stress attenuates butyrate signalling, this transition fails, resulting in persistent pain [12].
The therapeutic implications of the balance theory extend beyond simple SCFA supplementation, reframing treatment as the strategic restoration of equilibrium among the three signalling axes. Since each pain phenotype reflects the dominance or failure of a specific axis, therapeutic strategies should be designed to selectively rebalance the system rather than broadly increasing SCFA exposure. For the dominance of the neuronal sensing axis (as in visceral pain), strategies should focus on reducing propionate load [for example, low-fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAP) diets or rifaximin] and dampening neuronal excitability, potentially through GPR41 antagonism [28]. For the collapse of the resolution axis (as in neuropathic pain), restoring butyrate signalling through targeted-release formulations, slow-fermenting prebiotics, or GPR109A-agonists is central. For dominance of the inflammatory axis, transient inhibition of GPR43 may help contain inflammatory surges, while butyrate-promoting strategies ensure adequate resolution.
The relative abundance of the three major SCFAs in the gut—acetate, propionate, and butyrate—is primarily determined by the interaction between dietary substrates reaching the colon, the composition of the gut microbiota, and host-related factors such as antibiotic exposure and intestinal transit. Among these, the chemical structure and availability of fermentable carbohydrates are the most important determinants of SCFA production. Human intervention studies have shown that specific dietary fibres differentially modulate SCFA profiles. For instance, resistant starch markedly increases butyrate production, whereas other substrates, such as fructooligosaccharides or arabinoxylans, can differentially affect acetate and propionate levels depending on the microbial community present [29, 30]. The baseline microbiota strongly influences these responses; increases in butyrate following resistant starch supplementation are associated with the presence or expansion of key degraders such as Ruminococcus bromii and butyrate-producing bacteria, including Eubacterium rectale [29]. Clinical conditions that disrupt the microbiota, particularly broad-spectrum antibiotic exposure, can markedly reduce SCFA levels, an effect that may be partially restored through fibre supplementation or microbiota restoration strategies such as faecal microbiota transplantation [31]. Targeted approaches have also been developed to modify specific SCFAs, including inulin-propionate ester to selectively increase colonic propionate [32] and butyrylated resistant starch to deliver butyrate directly to the colon [33].
Butyrate produced by Roseburia intestinalis has been shown to activate the vagus nerve via GPR41 and to modulate brain circuits that suppress pain in mouse models of neuropathic pain induced either by oxaliplatin or by spared nerve injury [1]. Propionate has also been reported to alleviate neuropathic pain by inhibiting the inflammatory RIG-I–NF-κB signalling pathway in a mouse model of sciatic nerve transection [34].
Regarding inflammatory pain, butyrate has been shown to reverse the hyperexcitability of trigeminal ganglion neurons induced by inflammation in a rat model of orofacial inflammation triggered by complete Freund’s adjuvant [35]. Furthermore, in a mouse model of nitroglycerin-induced migraine, both propionate and butyrate reduce pain and promote the restoration of the intestinal epithelial barrier [36].
In the context of visceral pain, however, results concerning butyrate remain controversial. Using a rat model of colorectal distension, one study reported that butyrate promotes pain. Mechanistic analyses indicated that butyrate acts on mast cells via GPR43, thereby promoting the sensitisation of dorsal root ganglion neurons [18]. Therefore, the triad model appears to be limited to neuropathic and inflammatory pain and does not seem to adequately account for visceral pain.
The SCFA receptor triad model resolves the longstanding paradox by reconceptualising SCFA signalling as a competitive balance system. Chronic pain arises from specific axis imbalances shaped by underlying dysbiosis. This framework provides more than mechanistic insight; it offers a precision medicine roadmap in which identifying dysbiosis profiles and dominant axes can guide targeted therapeutic interventions. By moving beyond correlation towards mechanistic prediction, it opens new avenues for treating pain through the strategic rebalancing of microbial-host communication.
Foxp3: forkhead box P3
GPR43: G protein-coupled receptor 43
HDAC: histone deacetylase
IBS: irritable bowel syndrome
IL-6: interleukin-6
Nav: voltage-gated sodium channels
NGF: nerve growth factor
OLFR78: olfactory receptor 78
SCFAs: short-chain fatty acids
Th17: T-helper 17
Treg: regulatory T-cells
TRPA1: transient receptor potential ankyrin 1
TRPV1: transient receptor potential vanilloid 1
LRC: Conceptualization, Formal analysis, Writing—original draft, Writing—review & editing. RP: Supervision, Project administration, Funding acquisition, Writing—review & editing. Both authors read and approved the submitted version.
Rodrigo Pacheco, who is the Editorial Board Member of Exploration of Neuroprotective Therapy, had no involvement in the decision-making or the review process of this manuscript. The other author declares no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by “Financiamiento Basal para Centros Científicos y Tecnológicos de Excelencia de ANID” Centro Ciencia & Vida, FB210008 (to Fundación Ciencia & Vida). This study was also supported by grants from the Michael J. Fox Foundation (MJFF-021112) and from ANID (FONDECYT-1250021). LRC holds a scholarship for PhD studies from ANID (2131921240578). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

