 Review
Review

Affiliation:
1Medical Sciences Program, University of Cincinnati College of Medicine, Cincinnati, OH 45267, United States of America
Affiliation:
2Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229-3026, United States of America
Affiliation:
2Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229-3026, United States of America
3Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH 45229-3026, United States of America
4Tough Genes Foundation, Petersburg, MI 49270-9625, United States of America
Email: manoj.pandey@toughgenes.org
ORCID: https://orcid.org/0000-0002-0495-830X
Explor Neuroprot Ther. 2026;6:1004142 DOI: https://doi.org/10.37349/ent.2026.1004142
Received: September 10, 2025 Accepted: March 06, 2026 Published: April 02, 2026
Academic Editor: Jinwei Zhang, University of Exeter Medical School, UK
Neurodevelopmental Disorder with Regression, Abnormal Movements, Loss of Speech, and Seizures (NEDAMSS) is an ultra-rare, progressive neurological disorder, with more than 60 individuals described in the medical literature. It is caused by de novo mutations in the interferon regulatory factor 2 binding protein-like (IRF2BPL) gene, leading to early-onset symptoms including seizures, developmental delays, intellectual disability, and other severe neurological impairments, typically beginning in infancy or early childhood. This review aims to consolidate and refine current knowledge on NEDAMSS, focusing on the molecular functions of IRF2BPL, the spectrum of clinical features, and underlying disease mechanisms. A comprehensive understanding of NEDAMSS is essential for guiding the development of targeted interventions and therapeutic strategies to improve patient outcomes. By integrating current findings, we focus both on the progress made and the gaps that remain in research, providing a foundation for future studies to advance diagnosis, treatment, and overall patient care. We reviewed the published literature through studies available up to 2025 to synthesize current knowledge on clinical features, genetics, and proposed disease mechanisms. Reported phenotypes show substantial heterogeneity, and current genotype-phenotype correlations remain limited by small cohorts and inconsistent reporting. Key next steps include standardized phenotyping, natural history studies, and biomarker development to enable trial-ready outcome measures and accelerate targeted therapy development.
Neurodevelopmental Disorder with Regression, Abnormal Movements, Loss of Speech, and Seizures (NEDAMSS) is an ultra-rare genetic disorder caused by heterozygous, often de novo, variants in the interferon regulatory factor 2 binding protein-like (IRF2BPL) gene, which encodes the IRF2BPL protein [1, 2]. IRF2BPL is part of the IRF2BP family of transcriptional regulators, which includes IRF2BP1, IRF2BP2, and IRF2BPL. Members of this family are broadly implicated in transcriptional control and neural development [3–5]. NEDAMSS is characterized by progressive neurological decline, with just over 60 cases reported globally to date [6]. The clinical phenotype is heterogeneous and age-dependent. In childhood, common features include developmental delay, intellectual disability, seizures, abnormal movements, neurobehavioral symptoms, and ophthalmologic abnormalities. In adolescence and adulthood, patients are more likely to exhibit progressive cognitive decline, anarthria, psychiatric symptoms, epilepsy, movement disorders, and visual impairment [7, 8]. The disorder was first described in 2018 when several individuals with early-onset neurodevelopmental regression and seizures were identified with truncating mutations in IRF2BPL [1, 8, 9]. Since then, studies have expanded the known mutational spectrum of IRF2BPL and clarified the range of associated phenotypes [1, 2, 9, 10]. Importantly, mutations in IRF2BPL result in reduced production of functional IRF2BPL protein, ultimately leading to loss of its normal role in maintaining neuronal health. While many disease-associated mutations are truncating, producing a shortened, non-functional protein, others may disrupt the localization or stability of the full-length protein. Patient-derived cells often show lower expression levels of full-length IRF2BPL, supporting the hypothesis of a loss-of-function mechanism [1, 2, 11].
The IRF2BPL protein is believed to be critical for neuronal maintenance, and its dysfunction has downstream effects on cellular pathways. Notably, loss of IRF2BPL function has been linked to upregulation of the Wingless-related integration site (WNT/WG) signaling pathway, a crucial regulator of neural development and cellular communication. Dysregulation of this pathway is thought to contribute directly to the neurological manifestations observed in NEDAMSS [12, 13]. Functional similarities between IRF2BPL and its homologs IRF2BP1 and IRF2BP2 may offer further mechanistic insights, though the precise molecular cascades by which IRF2BPL loss leads to progressive neurodegeneration remain poorly understood. Given its severity, early onset, and defined genetic basis, NEDAMSS serves as a valuable model for exploring how single-gene dysfunction can drive complex neurodevelopmental regression.
This review consolidates current molecular and clinical knowledge of NEDAMSS and IRF2BPL, emphasizing key gaps that remain. We examine the function of genes, associated protein biology, clinical features, and emerging insights into disease mechanisms. By integrating genetic, molecular, and clinical perspectives, we aim to elucidate how studies of this ultra-rare disorder can inform broader understanding of neurodevelopmental disease and the consequences of transcriptional dysregulation in the brain.
This review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA)-informed principles, adapted for a mechanism and therapy-focused narrative synthesis, to ensure transparency, methodological rigor, and reproducibility. A comprehensive literature search was performed using PubMed, Scopus, and Web of Science, applying a dual search strategy to capture both the limited IRF2BPL-specific literature and the broader mechanistic blueprint relevant to neuroprotection and disease modification. Targeted searches were designed to identify publications directly addressing IRF2BPL and the IRF2BP family of transcriptional regulators (IRF2BP1, IRF2BP2), as well as the clinical entity NEDAMSS.
Search terms included combinations of IRF2BPL, IRF2BP1, IRF2BP2, NEDAMSS, developmental epileptic encephalopathy, progressive myoclonus epilepsy, and neurodevelopmental disorder. As expected for an ultra-rare genetic condition, these searches yielded a small but highly informative body of literature encompassing gene discovery, clinical phenotyping, and mechanistic studies. Non-targeted searches were conducted to contextualize IRF2BPL-related pathology within neuroprotective and disease-modifying paradigms. These searches included terms related to WNT/β-catenin signaling, transcriptional dysregulation, neuronal cell maintenance, synaptic dysfunction, cellular metabolism, neurodevelopmental disorders, epileptic encephalopathy, neurodegeneration, and neuroinflammation. Although these searches returned large numbers of publications, only studies providing clear mechanistic, conceptual, or translational relevance to IRF2BPL dysfunction or potential therapeutic targeting were considered for inclusion.
All research included studies published up to 2025. The combined database search identified 386 records (PubMed, n = 162; Scopus, n = 131; Web of Science, n = 93). After removal of 119 duplicate records, 267 unique articles remained for title and abstract screening. Of these, 175 articles were excluded due to lack of relevance to IRF2BPL biology, IRF2BP family function, neuroprotective mechanisms, or associated neurological phenotypes. The remaining 92 articles underwent full‑text review and 91 met all inclusion criteria. Studies were included if they satisfied one or more of the following criteria: (i) direct investigation of IRF2BPL or IRF2BP gene variants, IRF2BPL protein function, or regulatory mechanisms; (ii) clinical, genetic, or pathological characterization of IRF2BPL-related neurodevelopmental or neurodegenerative disorders, including NEDAMSS; or (iii) mechanistic studies providing disease-relevant insights into pathways implicated in neuronal vulnerability or protection, including WNT/β-catenin signaling, transcriptional regulation, neuronal cell maintenance, synaptic integrity, metabolic homeostasis, and neuroinflammatory processes. Eligible publication types included original research articles, case reports, case series, cohort studies, experimental mechanistic studies, Gene Reviews, and authoritative reviews. Studies were excluded if they (i) did not substantively involve IRF2BPL, IRF2BP family members, or closely related neuroprotective pathways; (ii) lacked sufficient clinical or mechanistic relevance; or (iii) were non-English publications without accessible translations.
Following full-text assessment, all 91 articles meeting eligibility criteria were included in the final qualitative synthesis and are cited in this review. Emphasis was placed on recent publications from the past decade to reflect advances in IRF2BPL genetics, disease phenotyping, and therapeutically relevant mechanisms, while seminal earlier studies were retained when foundational to gene discovery, transcriptional biology, or pathway regulation. In addition to IRF2BPL-focused studies, selected literature addressing WNT/β-catenin signaling, transcriptional co-regulators, neurodevelopmental and neurodegenerative disease mechanisms, and neuroinflammation was incorporated to support an integrative, neuroprotection-oriented interpretation. This approach enabled a comprehensive synthesis linking IRF2BPL dysfunction to transcriptional dysregulation, impaired neuronal maintenance, progressive neurodegeneration, seizure phenotypes, and candidate therapeutic pathways.
The IRF2BPL gene, positioned on chromosome 14q24, encodes IRF2BPL, a member of the IRF2BP family of transcriptional regulatory proteins involved in neuronal gene expression [9]. A key feature of IRF2BPL is that it is intron-less, consisting of a single continuous exon. Unlike most multi-exon genes, mutations introducing premature stop codons in IRF2BPL transcripts are not subject to nonsense-mediated decay (NMD). Consequently, truncated and nonfunctional protein fragments accumulate, which can disrupt cellular function and contribute to disease pathogenesis [1, 10, 14]. The IRF2BPL protein is approximately 796 amino acids in length and contains two major structural domains: an N-terminal coiled-coil domain, which facilitates protein-protein interactions, and a C-terminal RING finger domain, characteristic of E3 ubiquitin ligases [2]. The RING finger domain mediates ubiquitin-dependent protein degradation, a critical process for eliminating misfolded proteins and maintaining cellular proteotoxicity [15]. This domain architecture is shared with other family members, IRF2BP1 and IRF2BP2, which are established transcriptional repressors [2, 3, 5].
Expression of IRF2BPL is tissue-specific, with particularly high levels in the central nervous system (CNS). Studies in humans and Drosophila indicate that IRF2BPL expression changes across developmental stages, implying roles in both brain maturation and long-term neuronal maintenance [1, 2, 12]. Functional studies suggest that IRF2BPL acts as a transcriptional repressor in neurons, regulating neuronal survival, synaptic homeostasis, and axonal integrity [2, 12]. Loss-of-function experiments in Drosophila have linked IRF2BPL deficiency to progressive neurodegeneration [12]. Mechanistically, these effects appear to involve dysregulation of WNT signaling. Normally, IRF2BPL helps regulate β-catenin turnover through ubiquitination, thereby constraining WNT pathway activity. Mutations that impair this function result in aberrant WNT signaling, protein accumulation, and neuronal dysfunction [1, 12, 15].
Pathogenic variants in IRF2BPL are typically de novo nonsense, frameshift, or missense mutations (gene), often clustering within the C-terminal RING finger region [9, 10]. These mutations impair the E3 ligase function and destabilize the protein. Because IRF2BPL is intron-less, NMD cannot correct these defects, leading to the accumulation of toxic protein fragments. This pathogenic mechanism distinguishes IRF2BPL from many other neurodevelopmental genes [10].
Although much of IRF2BPL function is cytoplasmic, where it contributes to protein degradation, autophagy, and neuronal homeostasis, evidence also supports a nuclear role in transcriptional regulation [1, 12, 15]. Immunofluorescence and subcellular fractionation experiments demonstrate dual localization of IRF2BPL in cytoplasmic and nuclear compartments [1, 12, 15]. In the lysosome and cytoplasm, deficiency leads to the buildup of polyubiquitinated proteins, lysosomal dysfunction, and neuronal stress [1, 12, 16–18]. In the nucleus, IRF2BPL acts as a transcriptional repressor, likely through recruitment of histone deacetylases (HDAC1/2) and chromatin remodeling complexes [3, 5, 17, 18]. Nuclear localization appears to be mediated by an N-terminal nuclear localization signal (NLS), though the precise sequence has not yet been fully mapped. Once in the nucleus, IRF2BPL represses transcription of genes critical for neurodevelopment and neuroendocrine regulation [3, 5].
Several transcriptional targets of IRF2BPL have been identified, and together they suggest that this protein plays an essential role in integrating neurodevelopmental and neuroendocrine regulation. One of the best-characterized targets is the gonadotropin-releasing hormone (GnRH). IRF2BPL, also known as enhanced at puberty 1 (EAP1), directly represses GnRH promoter activity in hypothalamic neurons, linking it to neuroendocrine regulation and the initiation of puberty [16, 17]. This finding establishes a mechanistic connection between IRF2BPL dysfunction and possible disturbances in reproductive development.
In addition to GnRH, IRF2BPL has been shown to repress immediate early genes (IEGs) such as c-Fos and Egr1. These genes are activity-dependent transcription factors that play crucial roles in synaptic plasticity and neuronal activity regulation. By limiting their expression, IRF2BPL helps maintain appropriate levels of neuronal excitability and activity-dependent gene programs. Dysregulation of these pathways could contribute to abnormal synaptic function and progressive neurodegeneration, as observed in NEDAMSS [16].
Beyond these established targets, other genes have been proposed as plausible downstream regulators of IRF2BPL activity. For instance, brain-derived neurotrophic factor (BDNF), a neurotrophin essential for neuronal cell survival, synaptic function, and long-term potentiation, may be regulated directly or indirectly by IRF2BPL. While evidence for this remains preliminary, such a connection would align with the observed phenotypes of neuronal cell vulnerability and impaired plasticity in IRF2BPL-related disorders [19]. Similarly, neurogenic differentiation 1 (NEUROD1), a transcription factor that drives neuronal differentiation and lineage commitment, represents another compelling candidate target. Dysregulation of NEUROD1 could help explain the neurodevelopmental regression that characterizes NEDAMSS [20]. The influence of IRF2BPL may also extend into neuroendocrine signaling beyond GnRH. The genes KISS1 and TAC3, which are essential for puberty and regulation of the reproductive axis, are plausible downstream targets of IRF2BPL repression [21, 22]. If validated, these links would broaden the role of IRF2BPL in coordinating the interface between neuronal development and endocrine function.
At the mechanistic level, IRF2BP family members, including IRF2BPL, undergo SUMOylation, a post-translational modification that enables their recruitment to promyelocytic leukemia nuclear bodies (PML-NBs). Within these nuclear compartments, IRF2BPL interacts with HDACs and co-repressor complexes to enforce transcriptional silencing. Mutations that impair SUMOylation or disrupt these protein-protein interactions could result in uncontrolled activation of developmental and neuroendocrine genes. Such a loss of transcriptional repression provides a plausible mechanism for the progressive neurological decline observed in NEDAMSS [3, 5, 23].
Together, these findings suggest that IRF2BPL is a multifunctional protein that operates in both the cytoplasm and nucleus. In the cytoplasm, it maintains proteostasis through ubiquitin-mediated degradation and autophagy, whereas in the nucleus, it acts as a transcriptional repressor of genes critical for neurodevelopment and neuroendocrine regulation. Pathogenic variants, particularly those affecting the RING finger domain, compromise its E3 ligase activity and destabilize its transcriptional repression function. The combined effects, such as accumulation of toxic protein aggregates, impaired autophagy, dysregulated WNT signaling, and aberrant gene expression, cause the neuronal vulnerability and progressive neurological deterioration characteristics of NEDAMSS.
The earliest and most prominent feature of NEDAMSS is developmental regression, which typically manifests between the ages of 2 and 6 years. Children who had previously achieved developmental milestones begin to lose speech, motor abilities, and cognitive skills. In some cases, regression can begin later, even after the age of 10 [9, 10, 12, 24–26]. The regression process is variable, where some children experience a rapid decline over a few months, while others show a slower, more gradual deterioration. Despite this heterogeneity, the overall path is a similar progressive loss of skills that were once mastered [24, 27–31]. This regression is irreversible, though supportive therapies may help stabilize or modestly slow its progression [1, 2].
Speech is often the first function affected, initially presenting as slurred or unclear speech, followed by gradual language loss [9, 10, 24]. Early presentations of loss of speech typically include slurred speech to significant language and speech decline [24]. Over time, many patients develop severe expressive language decline, progressing in some cases to complete anarthria [24, 31–33]. Alongside speech decline, motor regression is another defining feature. Patients gradually lose coordination, ambulation, and fine motor skills, ultimately increasing dependence on caregivers for daily activities [24, 33]. Severe cases may present early and profound motor impairment. Abnormal movements are frequently observed during or after the regression phase and represent a hallmark of NEDAMSS. These manifestations include dystonia, a neurological movement disorder characterized by sustained or intermittent involuntary muscle contractions that produce twisting, repetitive movements or abnormal postures; chorea, which involves rapid, irregular, and unpredictable “dance-like” movements often affecting the hands, feet, and face; ataxia, defined by impaired coordination that leads to unsteady gait, clumsiness, balance difficulties, and sometimes dysarthria; and tremors, involuntary, rhythmic oscillatory movements affecting one or more body parts. The severity of these motor abnormalities can vary widely, ranging from subtle postural changes to profound, disabling movement disorders that substantially impair mobility. Collectively, these abnormal movements, alongside progressive motor decline, significantly compromise independence, posture, and overall quality of life [7, 9, 24–26, 31–33].
Seizures are another major clinical feature and often appear after speech and motor regression have begun. They typically emerge in middle childhood but may also develop during adolescence or, in rare cases, early adulthood. The types of seizures observed include focal, myoclonic, and atonic seizures [7, 25–28, 30]. As with other symptoms, severity varies; some patients develop refractory epilepsy, while others respond relatively well to anti-seizure treatments [24, 29, 33]. The onset of seizures often coincides with or accelerates cognitive and behavioral decline, suggesting shared mechanistic reinforcements between electrogenesis and neurodegeneration in NEDAMSS. In addition to the four cardinal features (e.g., regression, loss of speech, abnormal movements, and seizures), patients exhibit a broader spectrum of neurological and systemic symptoms. Cognitive impairment ranges from mild to severe intellectual disability, and many patients demonstrate behaviors resembling autism spectrum disorder (ASD), including irritability, hyperactivity, and ASD-like social difficulties [24, 33, 34]. Sleep disturbances are common, often linked to painful dystonia and tremors during the night, which lead to insomnia and daytime fatigue [25]. As oral-motor dysfunction progresses, feeding difficulties emerge, sometimes necessitating gastrostomy tube placement. In advanced cases, respiratory complications develop and may become life-threatening [9, 24].
Ophthalmologic manifestations are also documented. Strabismus is relatively common, while cortical visual impairment has been reported in some patients, reflecting deficits in visual processing despite intact ocular structures. More rarely, optic nerve hypoplasia has been observed. These visual problems contribute to learning difficulties, mobility challenges, and increased dependence [9, 24]. Genotype-phenotype correlations are still emerging, but early evidence suggests that truncating variants of IRF2BPL could be associated with earlier onset and more severe disease, whereas some missense variants may lead to later onset or milder progression [9, 10, 24]. Importantly, NEDAMSS is caused by de novo heterozygous variants in IRF2BPL, consistent with an autosomal dominant mechanism. While recurrence risk for siblings is generally low, it cannot be excluded if parental mosaicism is present [9, 10].
Because NEDAMSS was first described in 2018, its rarity and phenotypic overlap with other disorders make diagnosis challenging [9, 10]. Its core features can be mistaken for Rett syndrome, ASD, cerebral palsy, or mitochondrial and metabolic diseases [9, 31]. However, certain distinguishing features can help differentiate NEDAMSS, which is the later age of onset compared to Rett syndrome or cerebral palsy; the lack of dysmorphic features, which are common in mitochondrial disorders; and the absence of progressive vision loss, which is often seen in neuronal ceroid lipofuscinoses [9, 24, 31–33].
Laboratory evaluations for metabolic disorders are typically unremarkable, providing limited diagnostic direction in most individuals. Neuroimaging is similarly nonspecific in the early stages, as MRI scans may appear normal before gradually evolving to show mild cerebral or cerebellar atrophy over time [35]. Electroencephalography tends to offer more consistent evidence of neurological dysfunction, frequently demonstrating background slowing and epileptiform discharges [36]. Because these routine clinical assessments often yield nondiagnostic or only subtly abnormal findings, genetic testing becomes essential for identifying rare neurodevelopmental conditions [37]. In this context, whole-exome sequencing remains the most reliable and informative approach, enabling definitive confirmation of pathogenic IRF2BPL variants that underlie the disorder [10, 26, 27, 30, 32].
Despite increasing recognition, NEDAMSS remains substantially underdiagnosed. At the time of its initial description, only about 34 cases had been reported, and although the number has since grown to more than 60 confirmed individuals worldwide, the true prevalence is almost certainly higher [9, 24, 31, 33]. The marked phenotypic variability, ranging from early developmental delay to later-onset regression, movement disorders, and epilepsy, further complicates clinical recognition. In regions with limited access to genomic testing, these challenges are amplified, increasing the likelihood that additional individuals remain undiagnosed or misdiagnosed.
Currently, there are no disease-modifying therapies for NEDAMSS, and treatment is primarily supportive, requiring a multidisciplinary approach [2]. Seizure management is central, with anti-seizure medications such as levetiracetam or valproate commonly prescribed, though drug resistance is frequent [30, 33]. Movement disorders may be treated with medications such as baclofen or botulinum toxin, and physical therapy is often employed to preserve mobility as long as possible. Speech therapy and augmentative communication devices help maintain communication, while feeding support, including swallow assessments and gastrostomy feeding, becomes necessary as oral-motor skills decline [24]. Behavioral and cognitive challenges are managed with occupational therapy, behavioral interventions, and, when appropriate, medications to address irritability or sleep disturbances [34]. In advanced disease stages, proactive respiratory support is critical, including vigilant monitoring for aspiration and implementation of respiratory therapy when clinically indicated [9].
Supportive care remains a central component of management for patients with NEDAMSS, extending not only to patients but also to their families. Multidisciplinary coordination across neurology, physical and occupational therapy, nutrition, and, when appropriate, palliative care services is essential for optimizing day-to-day functioning and long-term well-being [1, 12]. Because NEDAMSS is a progressive neurodevelopmental disorder, families often experience substantial emotional, logistical, and caregiving burdens, drawing attention to the importance of ongoing counseling and psychosocial support.
Alongside supportive care, active research efforts are advancing potential therapeutic strategies. Preclinical studies in animal and cellular models have evaluated WNT-signaling inhibitors and autophagy-enhancing agents as approaches to mitigate the downstream molecular consequences of IRF2BPL dysfunction [1, 2, 12]. Early exploratory work in gene-directed therapies, including antisense oligonucleotides and adeno-associated virus (AAV) mediated gene delivery, is also underway [2, 23]. Although these approaches remain experimental, they represent promising steps toward future disease-modifying interventions. Until such therapies mature, clinical management continues to focus on maximizing quality of life through comprehensive, anticipatory, and family-centered supportive care.
Although the pathogenic mechanism underlying NEDAMSS is not yet fully resolved, multiple converging lines of evidence indicate that IRF2BPL loss-of-function represents the primary driver of disease. Developing observations further suggest that mislocalized or truncated IRF2BPL species may impose an additional gain-of-burden effect through aberrant protein interactions, impaired proteostasis, or cytoplasmic aggregation, but these secondary processes appear to amplify, rather than replace, the core loss-of-function mechanism. Collectively, these insights provide a mechanistic bridge between the period of early normal development in affected children and the subsequent onset of progressive neurological decline. Central to this line is a de novo pathogenic IRF2BPL variant that yields truncated or dysfunctional protein fragments, which compromise neuronal cell maintenance, destabilize proteostasis networks, and ultimately promote neurodegeneration [1, 9, 10, 12, 24].
Under physiological conditions, IRF2BPL functions as an E3 ubiquitin ligase, a role essential for maintaining neuronal proteostasis given the high metabolic demands and vulnerability of neurons to protein misfolding [1, 15]. By ubiquitinating damaged or misfolded proteins, IRF2BPL facilitates their proteasomal degradation. Pathogenic variants impair this function, reducing ubiquitination efficiency and leading to the accumulation of misfolded and polyubiquitinated proteins within neurons. The resulting proteostatic burden disrupts axonal transport, perturbs lysosomal function, and imposes chronic cellular stress [1, 12]. IRF2BPL deficiency disrupts multiple cellular processes, contributing to the progressive neurodegeneration characteristic of NEDAMSS [1, 2, 12].
IRF2BPL functions as a critical regulator of WNT/WG signaling, a pathway essential for neuronal differentiation, synaptic development, and circuit maturation [1, 12, 38]. WNT/WG pathway dysregulation is a shared pathogenic feature across Alzheimer’s disease, ASD, Parkinson’s disease, and major neuropsychiatric disorders, where it contributes to synaptic failure, neurodevelopmental disruption, dopaminergic neuron vulnerability, and impairments in higher-order cognitive function (Table 1). Mechanistically, IRF2BPL normally represses WNT target gene expression and promotes β-catenin ubiquitination, thereby constraining pathway activity [1, 12]. When IRF2BPL is defective, β-catenin escapes ubiquitination and accumulates, leading to pathological overactivation of WNT signaling. This dysregulated signaling environment drives aberrant synaptic growth, heightened neuronal excitability, and progressive cellular dysfunction and degeneration [1, 12]. Together, these findings support a model in which excessive WNT/β-catenin signaling represents a central driver of NEDAMSS-associated brain pathology.
Dysregulation of WNT/WG signaling in neurological diseases and its potential relevance to NEDAMSS-associated brain pathology.
| Disease | Role of WNT/WG signaling | Key references | Implications for NEDAMSS |
|---|---|---|---|
| Alzheimer’s disease (AD) | Promotes synaptic plasticity, neuronal survival, neurogenesis, and BBB integrity; suppresses amyloid-β and tau pathology. Dysregulation accelerates synaptic loss and neurodegeneration. | [39–42] | Suggests that WNT impairment in NEDAMSS may drive synaptic dysfunction, neuronal vulnerability, and cognitive-motor decline. |
| Autism spectrum disorder (ASD) | Abnormal WNT/β-catenin signaling disrupts neurodevelopmental programs, synaptic connectivity, and behavioral outcomes. | [43–47] | Supports the role of WNT dysregulation in NEDAMSS-related developmental delay, atypical motor control, and cognitive phenotypes. |
| Parkinson’s disease (PD) | Regulates dopaminergic neuron survival and synaptic function; restoration of WNT activity improves neuronal resilience. | [48, 49] | Provides a mechanistic basis for WNT-linked dopaminergic vulnerability in NEDAMSS, potentially explaining dystonia, tremor, and Parkinsonian features. |
| Neuropsychiatric disorders (NSD) | WNT pathway dysregulation is associated with schizophrenia, bipolar disorder, and others, affecting cortical development and higher-order brain function. | [50–53] | Suggests that WNT disruption may contribute to cognitive, affective, and behavioral disturbances reported in NEDAMSS. |
BBB: blood-brain barrier; NEDAMSS: Neurodevelopmental Disorder with Regression, Abnormal Movements, Loss of Speech, and Seizures; WNT/WG: Wingless-related integration site.
IRF2BPL also exerts key nuclear functions, acting as a transcriptional co-repressor through its N-terminal coiled-coil and C-terminal RING finger domains [1]. It regulates genes involved in neuroendocrine signaling and neuronal cell activity. For example, IRF2BPL normally represses GnRH, and loss-of-function may impair this regulatory axis in NEDAMSS [18]. In addition, IRF2BPL represses IEGs, which are critical for neuronal cell plasticity, learning, and memory [5, 25]. Loss of IRF2BPL-mediated repression leads to IEG overexpression, excessive synaptic activity, heightened excitability, and dysregulated calcium dynamics [5, 25]. This hyperexcitability lowers the seizure threshold, predisposing patients to epilepsy, a clinical hallmark of NEDAMSS [1, 25]. Seizures, in turn, exacerbate neuronal stress by amplifying excitotoxic glutamate release and calcium influx, creating a self-reinforcing cycle of oxidative stress, metabolic strain, and neuronal injury [1, 2, 14]. This impaired metabolic resilience likely accelerates neuronal cell vulnerability and degeneration [1, 2, 12].
Taken together, the emerging pathogenic architecture of NEDAMSS reflects a multilayered convergence of nuclear, cytoplasmic, and neuroimmune dysfunction, with dysregulated WNT/β-catenin signaling increasingly recognized as a central mechanistic hub. Loss of IRF2BPL function disrupts transcriptional repression and proteostatic surveillance, promoting cytoplasmic mislocalization and aggregation of IRF2BPL and related neuronal proteins, aberrant activation of WNT/β-catenin dependent transcription, and secondary mitochondrial and metabolic impairment [1, 9, 12]. These primary defects destabilize neuronal cell maintenance programs, lower the threshold for progressive neurodegeneration, and prime the CNS for maladaptive inflammatory responses.
Viewed within this broader mechanistic architecture, the cascade observed in NEDAMSS aligns with a well-recognized paradigm across other CNS diseases, in which abnormal accumulation of neuronal proteins and glycolipids triggers protein misfolding and is tightly interconnected with disease progression [54–64]. In neurodegenerative disorders defined by pathogenic protein aggregation, including α-synuclein in Parkinson’s disease, amyloid-β and tau in Alzheimer’s disease, and combined α-synuclein/tau pathologies in dementia with Lewy bodies and multiple system atrophy, misfolded protein species act as potent activators of neuronal and glial cell sensing pathways [65–74]. Their accumulation and aberrant conformations trigger innate immune receptors, amplify neuroinflammatory cascades, and accelerate the progression of neurodegeneration [55, 56, 65–75]. These parallels focus on a unifying principle that the biochemical identity, conformational state, and cellular handling of aggregated or misfolded IRF2BPL proteins likely determine how neural cells detect, interpret, and respond to injury-associated signals. This background suggests that similar protein-aggregation-driven mechanisms may be operative in NEDAMSS, shaping both the inflammatory milieu and the trajectory of neurodegeneration.
If this happens as predicted here, immunotherapy development against such abnormal neuronal protein could be a direct potential treatment for NEDAMSS patients, as such therapies, like anti-alpha synuclein and anti-amyloid β therapy, are currently in place for the treatment of patients with Parkinson’s and Alzheimer’s diseases [76–82]. Active and passive α-synuclein immunotherapies are already being explored as disease-modifying approaches in Parkinson’s disease [76, 77, 83–88]. Similarly, substantial progress has been achieved in Aβ-directed therapies, with agents such as aducanumab, lecanemab, and donanemab demonstrating robust amyloid plaque clearance and potential clinical benefit in Alzheimer’s disease [78–82, 89]. These advances suggest the feasibility of designing targeted immunotherapies against disease-defining protein species and provide a compelling conceptual foundation for future therapeutic development in NEDAMSS.
Despite meaningful progress in defining IRF2BPL-related neurodevelopmental disorders, substantial challenges remain in timely diagnosis, mechanistic resolution, and therapeutic development for NEDAMSS. Clinical overlap with other early-onset regression syndromes continues to delay recognition, reinforcing the need for heightened clinical awareness and early genomic evaluation. Ensuring equitable access to sequencing technologies and coordinated multidisciplinary care will be essential for improving diagnostic yield and long-term outcomes. The absence of validated biomarkers, quantitative functional readouts, and standardized outcome measures continues to limit clinical monitoring and impede trial readiness. Coordinated efforts, including multicenter natural history studies, harmonized phenotyping principles, and prospective patient registries, will be critical for defining disease paths, refining genotype-phenotype correlations, and supporting the development of mechanism-based interventions.
Mechanistic gaps also persist. Current evidence indicates that IRF2BPL dysfunction primarily reflects loss of nuclear function and dysregulated WNT signaling, while the cytoplasmic puncta observed in some model systems likely represent mislocalized, unstable, or truncated IRF2BPL protein species [1, 2, 10–12]. An important emerging concept is that, under conditions of chronic stress or impaired proteostasis, these species may interact, coalesce, and potentially evolve into higher-order aggregated structures, thereby adding a second layer of toxicity beyond simple loss of function. Future studies should define the biochemical composition, aggregation propensity, and cellular handling of these cytoplasmic species and determine whether they act as seeds for broader disturbances in protein homeostasis or neuroinflammatory mechanism activation. Parallel efforts should clarify how IRF2BPL loss alters neuronal signaling, synaptic maintenance, and vulnerability to injury, and how downstream consequences, such as WNT pathway overactivation, impaired transcriptional repression, and secondary neuroinflammatory responses, shape disease progression. Additional priorities include identifying selectively vulnerable neuronal cell populations, mapping disruptions in cellular signaling and protein homeostasis, and integrating these insights with rational therapeutic strategies. By linking mechanistic understanding with early diagnosis, structured natural history data, and coordinated clinical care, the field is increasingly well-positioned to accelerate the development of precision-based therapies for NEDAMSS.
NEDAMSS is an ultra-rare neurological disorder marked by substantial clinical heterogeneity, contributing to delayed diagnosis and persistent under-recognition [9, 24, 33]. Variation in age of onset, disease severity, and rate of progression is likely shaped by mutation type, residual IRF2BPL activity, and additional genetic modifiers; however, clear genotype-phenotype correlations remain elusive [9, 10, 24]. This uncertainty continues to impede accurate prognostication and the rational design of targeted therapeutic strategies [2, 31]. Converging evidence supports loss of IRF2BPL function as the central pathogenic mechanism in NEDAMSS. IRF2BPL is essential for maintaining neuronal homeostasis, and disruption of its transcriptional and regulatory roles [1, 2, 38, 90] precipitates neurodevelopmental regression, progressive neurodegeneration, and frequently refractory seizures [24, 31].
Although the precise molecular intermediates linking IRF2BPL dysfunction to WNT hyperactivation, emerging protein aggregation, and neuroimmune engagement remain incompletely defined, the integrated model illustrated in Figure 1 provides a coherent systems-level explanation for how a single-gene defect can drive developmental regression, seizures, and progressive neurodegeneration. In this operational model, loss of IRF2BPL function initiates a cascade in which nuclear depletion and cytoplasmic mislocalization of mutant IRF2BPL promote aggregate formation that aberrantly activates WNT/β-catenin signaling, leading to dysregulated gene expression and neuronal cell vulnerability. Within this model, WNT/β-catenin signaling and its downstream inflammatory mediators emerge as central, pathway-targeted points of intervention. Therapeutic strategies that restore proteostasis, recalibrate aberrant WNT activity, or attenuate chronic neuroinflammation may therefore hold promise for modifying the disease pathway in NEDAMSS, rather than merely mitigating downstream manifestations.
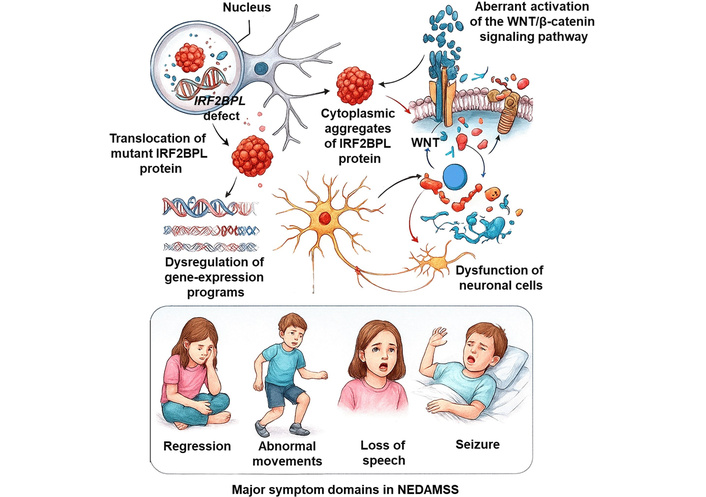
Proposed mechanistic model linking IRF2BPL loss of function to NEDAMSS pathophysiology. This figure was conceptualized and produced by M.K.P. using original artistic expertise, integrating mechanistic insights derived from the literature. Interferon Regulatory Factor 2 Binding Protein-Like (IRF2BPL) gene defects and the resulting translocation of mutant IRF2BPL protein from the nucleus to the cytoplasm trigger the formation of protein aggregates that aberrantly activate WNT/β-catenin signaling. This cascade leads to dysregulated gene expression, neuronal dysfunction, and the characteristic features of Neurodevelopmental Disorder with Regression, Abnormal Movements, Loss of Speech, and Seizures (NEDAMSS).
As depicted in Figure 1, pathogenic IRF2BPL variants disrupt transcriptional repression and proteostatic surveillance, enabling truncated or mislocalized IRF2BPL species normally unstable and rapidly degraded to accumulate under conditions of cellular stress. These species may aberrantly interact and assemble into higher-order aggregated structures that impose a secondary gain-of-burden effect, amplifying stress-response pathways, perturbing protein homeostasis, and further sensitizing neurons to injury. In this view, aggregation-linked toxicity operates in parallel with the primary loss of nuclear IRF2BPL function, together creating a multilayered vulnerability across neuronal compartments that ultimately converges on WNT dysregulation and neuroimmune activation.
Despite several such advances, major challenges remain, including delayed diagnosis, limited access to comprehensive genomic testing, and the absence of validated biomarkers for disease monitoring or therapeutic response [1, 2, 26–28, 30, 31, 33, 91]. Addressing these gaps while deepening mechanistic understanding of IRF2BPL dysfunction, WNT pathway dysregulation, proteostasis failure, and neuroimmune activation will be essential for translating emerging biological insights into effective therapies. Continued progress in defining NEDAMSS pathogenesis and natural history provides a critical foundation for therapeutic development and integrating these clinical and mechanistic insights to advance rational, pathway-directed interventions. As the field moves toward targeted strategies that restore proteostasis, modulate WNT signaling, or temper chronic neuroinflammation, a more complete understanding of disease biology will be indispensable for shaping the next generation of treatments for this disturbing disorder.
ASD: autism spectrum disorder
CNS: central nervous system
GnRH: gonadotropin-releasing hormone
HDAC: histone deacetylases
IEGs: immediate early genes
IRF2BPL: interferon regulatory factor 2 binding protein-like
NEDAMSS: Neurodevelopmental Disorder with Regression, Abnormal Movements, Loss of Speech, and Seizures
NEUROD1: neurogenic differentiation 1
NMD: nonsense-mediated decay
WNT/WG: Wingless-related integration site
The authors acknowledge the support of the Tough Genes Foundation (Petersburg, MI, USA). As our first scientific contribution on behalf of the Foundation, this review reflects our commitment to advancing understanding of NEDAMSS and informing future diagnostic and therapeutic efforts. We also acknowledge the foundational contributions of Dr. Manoj Pandey’s previous laboratory in the Division of Human Genetics at Cincinnati Children’s Hospital Medical Center (Cincinnati, OH, USA). We thank Vian Pandey for assistance with manuscript editing and express our gratitude to the patients and families whose experiences continue to guide progress in this ultra-rare disorder.
MJSC: Writing—original draft. AFM: Data curation, Formal analysis. MKP: Conceptualization, Methodology, Supervision, Resources, Writing—original draft, Writing—review & editing, Data curation, Visualization, Project administration. All authors reviewed and approved the final manuscript and agreed to its submission.
The authors declare that there are no competing interests.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2232
Download: 56
Times Cited: 0
