 Review
Review

Affiliation:
1Department of Internal Medicine, Azienda Ospedaliero-Universitaria of Modena (–2023), 41100 Modena, Italy
†
Email: a.lonardo@libero.it
ORCID: https://orcid.org/0000-0001-9886-0698
Affiliation:
2Institute of Molecular Pathobiochemistry, Experimental Gene Therapy and Clinical Chemistry (IFMPEGKC), RWTH University Hospital Aachen, D-52074 Aachen, Germany
†
Email: rweiskirchen@ukaachen.de
ORCID: https://orcid.org/0000-0003-3888-0931
Explor Med. 2025;6:1001334 DOI: https://doi.org/10.37349/emed.2025.1001334
Received: April 13, 2025 Accepted: June 02, 2025 Published: June 18, 2025
Academic Editor: Alessandro Granito, University of Bologna, Italy
Obesity is a multifactorial chronic disease characterized by an excess of adipose tissue, placing a growing burden on individual health and public health systems worldwide. Here we aim to elucidate how obesity contributes to liver dysfunction and highlight the preventive, diagnostic, and management strategies that are most relevant to healthcare providers, researchers, and policy makers. To this end, a comprehensive literature search using major scientific databases was conducted. Various clinically heterogenous pathophenotypes, such as android, gynoid, sarcopenic, metabolically healthy and unhealthy obesity, exhibit variable associations with liver health in the context of chronic liver disease (CLD), including alcohol-related CLD, viral hepatitis B and C, and, particularly, metabolic dysfunction-associated steatotic liver disease (MASLD), which is the prototypic manifestation of obesity-associated CLD. Regardless of the etiology of CLD, obesity is a major risk factor for the progression to cirrhosis and hepatocellular carcinoma through a variety of lipotoxic, proinflammatory, pro-fibrotic, and carcinogenic pathomechanisms involving genetics and epigenetics, altered adipokine profile, oxidative stress, endoplasmic reticulum stress, apoptosis, intestinal dysbiosis, and altered gut-liver axis. Various strategies are available to address obesity-associated CLD, including lifestyle changes, endoscopic techniques, and metabolic/bariatric surgery. Integrative approaches bringing together clinicians, basic researchers, and public health experts will be crucial in developing a coherent, holistic framework to address, with a precision medicine approach, the rising tide of obesity-related CLD on a global scale.
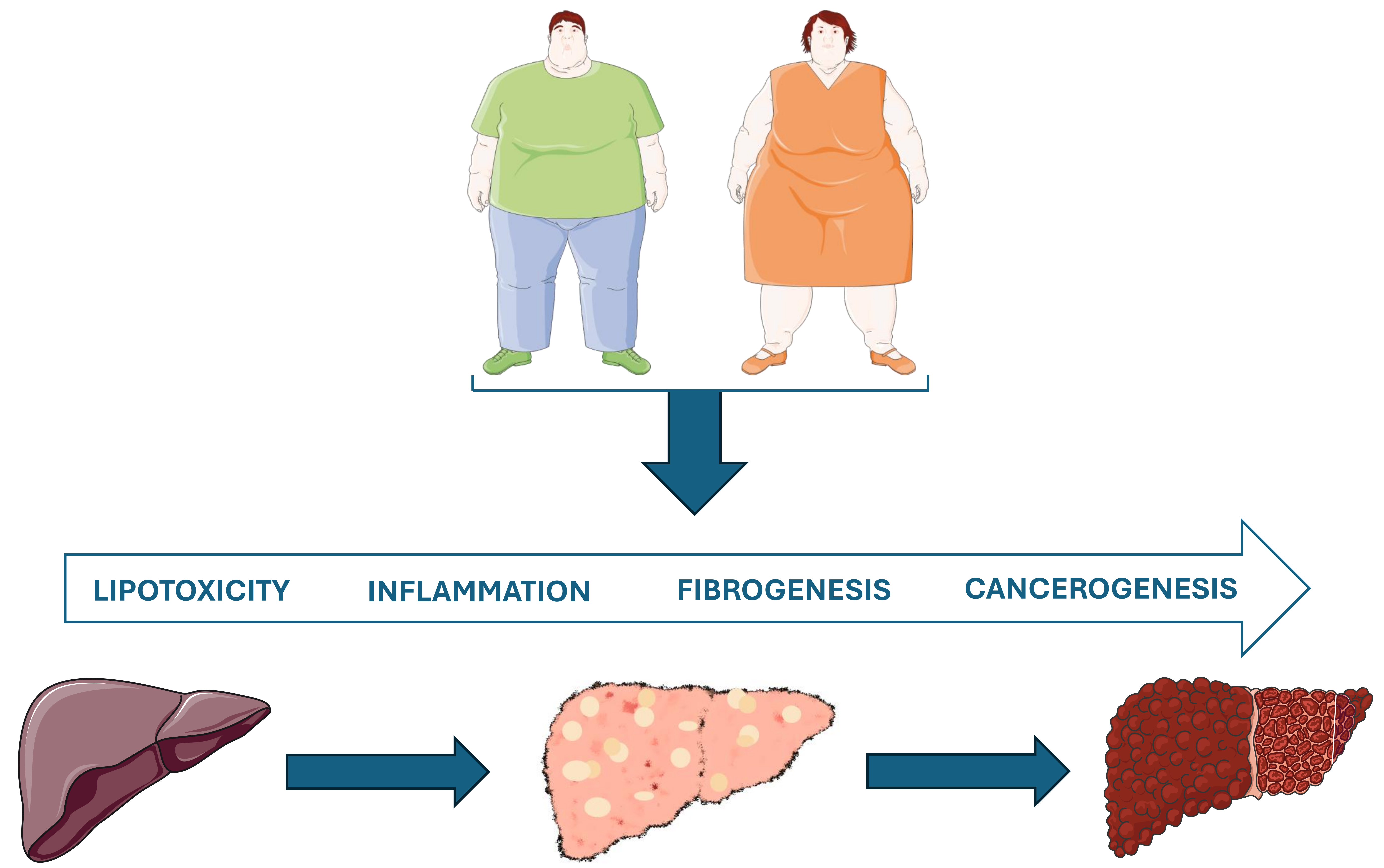
Obesity and the liver. Original image drawn with Servier Medical Art, licensed under CC BY 4.0
Obesity is a complex, multifactorial chronic disease characterized by excessive accumulation of adipose tissue, which places a significant burden on both individual health and public health systems worldwide. Obesity is traditionally defined as a body mass index (BMI) of ≥ 30 kg/m2 [1]. However, this cut-off value, although useful for large-scale epidemiological studies, does not fully capture the multiple genetic, environmental, and behavioral factors that contribute to obesity [2]. Additionally, certain populations develop metabolic dysfunction at lower values of BMI and therefore, ethnicity-specific definitions of obesity apply [3]. Among the organ systems most affected by obesity is the liver, which plays a central role in metabolism, detoxification, immunology, and the regulation of systemic energy homeostasis [4]. Abnormalities in liver function, often associated with obesity, affect the pathobiology of all types of chronic liver disease (CLD), including viral hepatitis, metabolic dysfunction-associated steatotic liver disease (MASLD), metabolic dysfunction-associated steatohepatitis (MASH), cirrhosis, and hepatocellular carcinoma (HCC) [5]. BMI, while a clinically utilizable metric, is neither the only one nor devoid of limitations (typically in athletes and in patients with states of fluid retention) [6]. The anatomic location where adipose tissue is positioned predicts metabolic dysfunction accurately and, accordingly, measurements of waist circumference (WC) are another clinically usable, sex- and ethnic-specific anthropometric measurement [7–9]. “Clinical obesity”, which defines a chronic, systemic illness characterized by impaired functions of tissues, organs, or the entire body owing to excess adiposity, is a major advancement.
The increasing incidence of obesity can be attributed to rapid urbanization, sedentary lifestyles, and a global shift towards diets high in calorie-dense foods rich in fats and sugars [10]. These factors, combined with genetic predisposition, underlie the rise of obesity in both economically developed nations and countries undergoing rapid economic transition, affecting life expectancy, quality of life, healthcare expenditure, and workforce productivity [11]. It is estimated that billions of dollars are spent annually on healthcare costs related to overweight and obesity, putting a strain on healthcare systems worldwide [12]. In this context, the understanding of how obesity affects liver health is critical, as obesity may often perturb those crucial metabolic and detoxification processes for which the liver is responsible. In terms of pathophysiological mechanisms, obesity contributes to liver dysfunction through several interconnected pathways [13], including the excess release of free fatty acids (FFAs) and pro-inflammatory cytokines into the bloodstream, resulting in escalated reactive oxygen species (ROS) formation within the mitochondrial respiratory chain [14, 15]. Over time, systemic and hepatic insulin resistance (IR) dysregulate hepatic glucose output and fatty acid uptake [13], resulting in hepatic steatosis, i.e., the accumulation of fat in hepatocytes. Although simple steatosis can be relatively benign in some individuals, others progress to more severe forms such as MASH, cardiac structural and functional changes, as well as muscular and renal complications [14]. In addition to MASLD, obesity also affects the pathobiology of chronic viral hepatitis and cirrhosis, enhancing the risks of HCC with profound clinical implications and an urgent need for improved public health strategies and targeted therapeutic interventions [16].
Obesity develops when prolonged energy intake exceeds energy expenditure, leading to adipose tissue expansion through both hypertrophy (enlargement of existing adipocytes) and hyperplasia (increased adipocyte number). Hypercaloric diets and sedentary lifestyle cause lipids to accumulate not only in subcutaneous fat depots but also in visceral fat, liver, muscle, and other ectopic sites [13]. These lipid reservoirs release FFAs and pro-inflammatory cytokines [e.g., tumor necrosis factor-alpha (TNF-α), IL-6] that provoke low-grade, subclinical chronic inflammation and IR in both adipose tissue and peripheral organs [14, 15]. IR, in turn, worsens ectopic fat deposition and perpetuates a cycle of metabolic dysfunction, ultimately disrupting hepatic, cardiovascular, and endocrine homeostasis.
Beyond the liver, obesity exhibits key comorbidities including type 2 diabetes mellitus (T2DM), arterial hypertension, dyslipidemia, and atherosclerosis, all of which substantially elevate the risk of cardiovascular disease (CVD) [9]. Furthermore, obesity is linked to chronic kidney disease, obstructive sleep apnea, osteoarthritis, certain cancers, and cognitive decline, underscoring the multifactorial burden of this condition.
Obesity affects over 1.9 billion people worldwide [17]. The burden of liver disease associated with obesity is compounded by overlapping risk factors such as metabolic syndrome (MetS), which includes central obesity, hyperglycemia, dyslipidemia, and hypertension, and is strongly associated with MASLD and MASH [18]. Additionally, the prevalence of obesity in high-income areas is inversely correlated with life expectancy in adults (Figure 1).
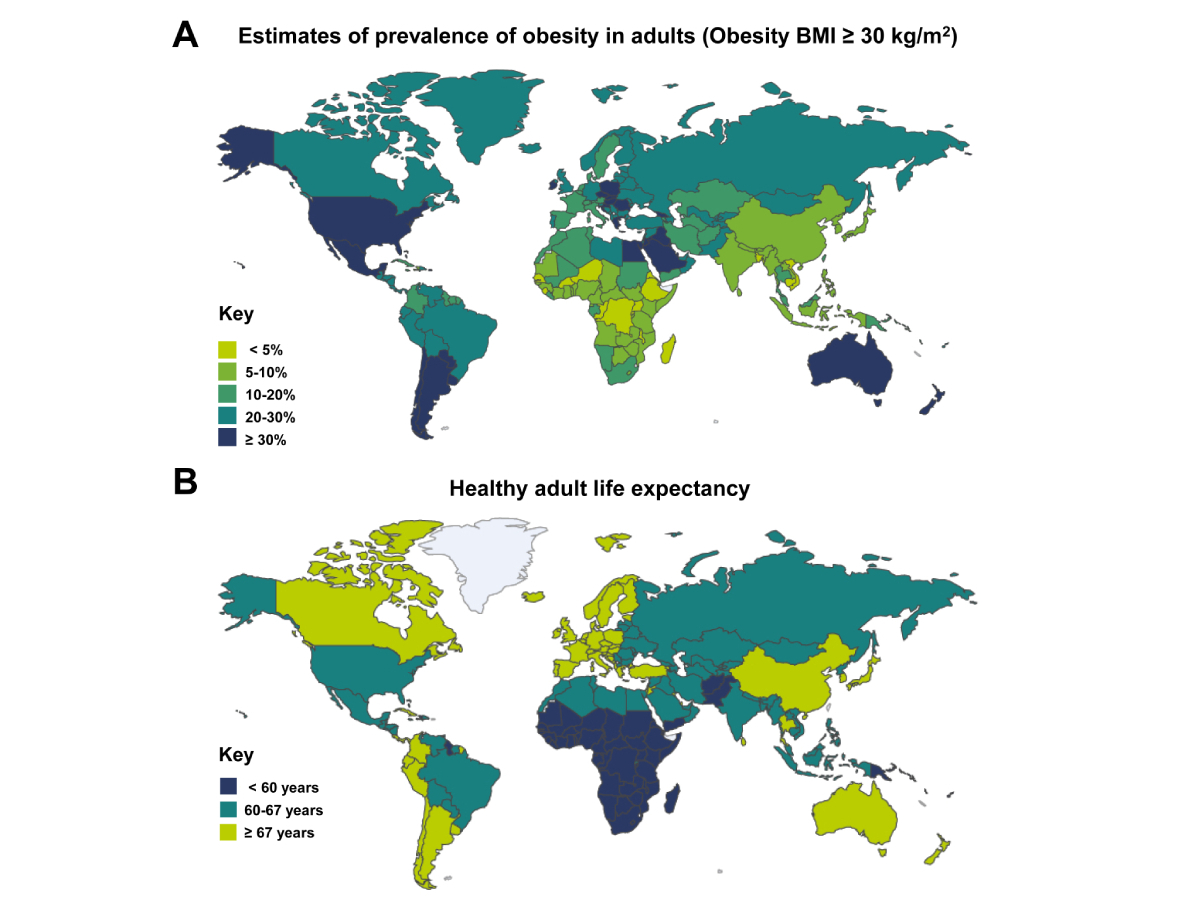
Estimates of the prevalence of obesity and healthy adult life expectancy. (A) The prevalence of adults with obesity was published in 2024 as assessed using data from 1990 to 2022 from 3,663 representative studies involving 222 million children, adolescents, and adults. Obesity was defined as a body mass index (BMI) greater than 30 kg/m2, adapted from the NCD Risk [19]; (B) world adult life expectancy data published by the WHO Global Health Observatory in January 2021 is presented, printed with permission from the WHO Global Health Obesity Observatory [20]
Moreover, the prevalence of adult overweight and obesity varies significantly between low- and high-income areas and can differ between males and females (Supplementary material). According to the regularly updated information provided by the World Obesity Observatory, which was last updated on April 8, 2025, the lowest incidence is currently found in Ethiopia (1%), while the highest incidence is found in American Samoa (80.2%). The growing clinical burden associated with obesity requires early detection, appropriate staging of liver disease, and careful consideration of interventions to prevent progression to cirrhosis or liver failure for individuals and their healthcare providers. Lifestyle modifications play a crucial role in improving clinical outcomes by reducing the number, activity, and composition of circulating pro-inflammatory blood cells [21]. In more advanced cases or in instances of morbid obesity, pharmacotherapy and bariatric surgery offer additional treatment options, although the evidence on their long-term impact on liver health is still evolving [22, 23]. Understanding the precise interplay between excess adipose tissue and liver function remains a challenge owing to heterogeneity of patient populations, variable genetic predisposition, and differences in environmental factors, which collectively hinder standardized treatment guidelines and targeted therapeutics [24]. Furthermore, there is still disagreement about the optimal diagnostic thresholds for MASLD and MASH, and screening guidelines [25]. As a result, clinicians often face uncertainty about the best possible approach to prevent or treat obesity-related liver disease in individual patients.
Here, we aim to provide a comprehensive understanding of the relationship between obesity and liver health, bridging the gap between general pathophysiological concepts and specific clinical implications. We seek to elucidate how obesity contributes to liver dysfunction and highlight the preventive, diagnostic, and management strategies that are most relevant to healthcare providers, researchers, and policy makers. By synthesizing the latest research findings and discussing future directions, we hope to provide guidance that can inform clinical decision making, promote effective patient counseling, and drive research into novel interventions. In addition, we explore the wider societal and economic burden of obesity-related liver disease, as understanding the cost-effectiveness of different intervention programs will be critical for policy makers seeking to contain costs while improving patient outcomes.
The review will discuss the pathophysiological interplay between adipose tissue, the gut microbiome, and the liver. We cover how obesity exacerbates alcohol-related liver damage and chronic viral hepatitis, underlining the ways in which excess adiposity can accelerate disease progression. We also highlight the negative impact of obesity on cirrhosis development and progression, as well as how it heightens the risk of HCC. By examining these intersections, we aim to shed light on how various forms of CLD converge in the context of obesity, thereby underscoring the broader clinical and societal significance of this epidemic. Our comprehensive approach spanning from epidemiology, clinical science, and pathobiology, together with an emphasis on liver damage in the context of obesity, render the present review article novel and different from similar published articles.
In preparing this review article, we conducted a comprehensive literature search using major scientific databases, including PubMed (https://pubmed.ncbi.nlm.nih.gov/), Scopus (https://www.scopus.com/home.uri), and Web of Science (https://www.webofknowledge.com/). We selected these databases because of their broad coverage of biomedical and clinical research, ensuring a thorough retrieval of relevant publications. The search terms used included various combinations of “obesity”, “liver”, “non-alcoholic fatty liver disease”, “metabolic dysfunction-associated steatotic liver”, “MASLD”, “metabolic dysfunction associated steatohepatitis”, “MASH”, “NAFLD”, “nonalcoholic steatohepatitis”, “NASH”, “metabolic syndrome”, and “steatosis,” using Boolean operators (AND, OR) to refine the results. We also reviewed relevant guidelines and position statements from professional societies such as the European Association for the Study of the Liver (EASL), the American Association for the Study of Liver Diseases (AASLD), and other authoritative bodies. In addition to these database searches, our methodology included screening the reference lists of selected articles to identify further relevant studies that may not have been included in the initial search.
Articles were considered for inclusion if they focused on the conceptual links between obesity and liver disease, provided clinical data on diagnostic or therapeutic strategies, or offered novel insights into pathophysiological mechanisms. Priority was given to studies published within the last 10 years to ensure the timeliness of the review. However, seminal papers published earlier were also included if they addressed fundamental concepts that remain critical to current understanding. Studies involving pediatric populations were included where relevant, given the alarming rise in childhood obesity and its potential to catalyze early-onset liver disease. Following an initial screening of titles and abstracts, full-text articles were assessed for scientific quality, relevance to the aims of the review, and contribution to the understanding of the role of obesity in liver pathology. The methodological rigor of each study was assessed, including design [e.g., randomized controlled trial (RCT), cohort study, case-control study, or cross-sectional design], statistical power, and potential sources of bias. Finally, we synthesized the findings into key thematic categories that shape the structure of this review: (i) definitions and epidemiology, (ii) pathophysiology and mechanisms of disease, (iii) diagnostic approaches and disease assessment, (iv) clinical management strategies, and (v) future research directions and gaps.
The phenotypic heterogeneity of obesity poses a challenge in assessing cardiometabolic risk associated with a specific BMI [26]. Similar considerations may also apply to the risks of liver disease associated with different types of obesity. In the early 1950s, researchers discovered that the distribution of adipose tissue played a significant role in determining cardiometabolic risk [27]. This idea was further supported by the identification of subsets of patients who showed metabolic dysfunction despite having a lean phenotype, and conversely, individuals with obesity who appeared to be protected from metabolic dysfunction [27]. Imaging techniques have revolutionized the classification of obesity by enabling the quantitative assessment of the regional distribution of adipose tissue [26]. However, simple clinical observation, including measuring WC and other basic anthropometric indices, allows for the quick identification of android and gynoid patterns of obesity [28].
It is widely accepted that individuals with an excess of visceral adipose tissue (VAT), also known as android, central, truncal, visceral, or “apple type” obesity, have the highest probability of ectopic fat deposition and the most elevated cardiometabolic risk [26]. Conversely, subcutaneous obesity affecting the gluteo-femoral region, also known as gynoid or “pear type” obesity, is typically considered to be at low cardiometabolic risk [26]. The different cardiometabolic outcomes underlie variable attitudes of subcutaneous, visceral, and ectopic adipose tissue to store and release fatty acids and to synthesize and secrete adipokines [26]. Therefore, sex and reproductive status are major modifiers of the various obesity phenotypes [29].
Sarcopenic obesity is characterized by a bidirectional, pathogenic interaction between obesity and the loss of skeletal muscle mass and function. This ultimately leads to a synergistically increased cardiometabolic risk and functional impairment compared to the risks posed by each condition separately [30]. While these classifications remain clinically significant, other important factors influencing cardiometabolic risk include cardiorespiratory fitness (higher level is associated with lower accumulation of VAT), diet quality (poor diet increases cardiometabolic risk) and, in the context of precision medicine approaches, lifestyle habits should complement traditional anthropometric indices in the field of obesity, which should be referred to as ‘obesities’ [26].
Although there are various definitions of metabolic health [31], it is generally accepted that metabolically healthy obesity (MHO) is defined by a BMI of ≥ 30 kg/m2 and a healthy metabolic status identified by the absence of components of the MetS [31, 32].
Approximately one in three individuals with obesity has MHO [32], and a continuous linear increasing trend in metabolically unhealthy obesity (MUO) has been reported among US adults from 1999 to 2018 [33]. Mechanistically, adult subjects with MHO present more brown adipose tissue (BAT) and higher thermogenesis than their metabolically unhealthy counterparts [34], suggesting that BAT presence and activity affect a healthier phenotype in subjects with overweight or obesity. Additionally, individuals with the MHO phenotype exhibit slightly increased bone turnover activity, probably as an initial skeletal compensatory response to the increased BMI [35].
Typically, MUO is considered a temporary and potentially evolving condition, situated between metabolically healthy lean individuals and MUO. Studies show that over half of individuals with MHO develop MUO within a 10-year period [32, 36]. Moreover, MHO is linked to a higher risk of cancer [37], CVD [38–40], chronic kidney disease [41], and depression, especially in women and individuals from North America and Europe, who may benefit from personalized interventions [42]. While the group of individuals with consistently stable MHO is a topic of great scientific interest, it is recommended to focus on weight management to prevent the transition from MHO to MUO status [32]. This is further discussed in Principles of treatment of obesity-associated CLD in this review.
The factors that determine a metabolically unhealthy phenotype among individuals with obesity are of significant scientific interest. Experimental evidence suggests that pro-inflammatory macrophages, the most common immune cells in adipose tissues, may act as a switch from MHO to MUO in a mouse model [36].
The orexigenic neuropeptide Y (NPY) plays a major role in humans, as exceeding the critical threshold of 471.5 pg/mL leads to an 18% increased risk of MUO for each 10 pg/mL increment in NPY serum levels, independently of confounding factors [43]. This study provides a compelling framework for other studies that document how sex, cultural achievements, lifestyle habits and exogenous factors (such as diet quality, sedentary behavior, smoking, alcohol, and drugs) impact intestinal dysbiosis, levels of general or visceral obesity, and contribute to an increased risk of MUO [44].
A large study involving 3,351,989 people identified a significant association between the transition from MHO to MUO and general characteristics, health behaviors, and metabolic components [45]. Male participants had a 1.30 times higher risk of progressing to MUO compared to females, and individuals in the lowest economic status were also at risk for this transition [odds ratio (OR): 1.08; 95% confidence interval (CI): 1.05 to 1.1]. Smoking, consuming more than 30 g of alcohol, and lack of regular exercise were also associated with this transition [45, 46].
In humans, higher adherence to unhealthier dietary patterns (i.e., consuming refined and processed foods) is associated with a reduced probability of having a healthy metabolic phenotype, while the opposite was observed for healthier dietary patterns [47]. Consistently, a one-unit increase in macronutrient quality index is associated with a 5% lower incident MUO (HR = 0.95; 95% CI: 0.92 to 0.99) [48]. In addition to a poor diet, the use of proton pump inhibitors may cause gut dysbiosis, which may predispose to MUO through a “leaky gut”, systemic low-grade inflammation, and reduced amounts of short-chain fatty acids such as butyrate that promote metabolic health [49]. Obesity, in its turn, is also a driver of gut dysbiosis [50], giving rise to a cause-and-effect vicious circle. A study from Korea analyzing data in 5,191 adults with obesity (BMI ≥ 25 kg/m2) found that food insecurity, older age, higher BMI, lower educational level, and lower physical activity were associated with MUO [51].
BMI-associated hyperuricemia is a risk factor for MUO compared to MHO in Chinese adults [52]. Advanced age and higher WC are significantly correlated with all metabolically unhealthy states [53], and both general and central obesity are associated with the risk of transitioning from MHO to MUO in a study conducted among women in Iran [54]. Regardless of underlying risk factors, transitioning from a metabolically healthy state to MUO is associated with higher risks of CVD and mortality [55, 56] and mortality owing to cancer [57].
Men and women store excess triglycerides differently, both in the adipose tissue and in ectopic sites (such as the liver and skeletal muscle). This is due to sex-specific differences in messenger RNA expression, protein content, and enzyme activities of skeletal muscle and hepatic lipid metabolic pathways [58]. As a result, MASLD is more prevalent in men than in women. Women tend to store more intramyocellular lipids (IMCL) than men, yet their risk of T2DM is not greater. During exercise, women rely more on lipid metabolism than men do, due to upregulated pathways of lipid oxidization [58]. Additionally, android fat predisposes individuals to MASLD, while gynoid fat protects against MASLD [59].
MUO is strongly associated with MASLD and the progressive variant of MASLD, MASH. A study from Iran involving 8,360 adults found that the risk of MASLD in individuals with the MHO phenotype increased by 8.92 times (95% CI: 2.20 to 15.30), and those with the MUO phenotype increased by 32.97 times (95% CI: 15.70 to 69.22) compared to the metabolically healthy-non-obese phenotype [60]. Another study conducted in Germany on 141 patients undergoing bariatric surgery found that, among the components of the pathophysiology of MUO, glucose metabolism predicts MASH more accurately than parameters of lipid metabolism, inflammation or the presence of CVD [61] indicating that IR is the main driver of progressive liver disease in these MUO subjects. Analysis of the liver biopsy database of the NASH Clinical Research Network has shown that low relative muscle mass independently predicted MASH in males (OR: 0.550; 95% CI: 0.312 to 0.970), while a high android to gynoid ratio was the independent risk factor for NASH in females (OR: 1.694; 95% CI: 1.073 to 2.674) [62]. Collectively, the data support a major role for body fat distribution and sarcopenic obesity in the development and progression of MASLD.
Epidemiological studies strongly support the notion that obesity plays a major role in the pathobiology of various types of CLD, ranging from alcohol-related liver disease (ALD), and chronic viral hepatitis, to MASLD, cirrhosis, and HCC. The impact of obesity is analyzed separately in each of the above conditions.
ALD defines a spectrum spanning from alcohol-associated steatosis, steatohepatitis, cirrhosis, and alcohol-associated HCC [63]. While typically occurring among those with cirrhosis, alcohol-associated hepatitis may occur at any stage of the spectrum [64]. The finding that not all those chronically exposed to hepatotoxic amounts of alcohol will develop progressive liver disease is a clue to co-factors modulating the risk of ALD.
A retrospective cohort study [65] classified 18,506 CLD-free at enrollment participants in the Mayo Clinic Biobank by self-reported alcohol consumption as: nondrinkers, moderate drinkers (0 to 2 drinks per day), and heavy drinkers (> 2 drinks per day). After a median 5.8 year follow-up among moderate drinkers, the risk of developing hepatic steatosis was high in patients with obesity [adjusted hazard ratio (AHR): 1.31; 95% CI: 1.03 to 1.67], insignificant in the overweight group (AHR: 0.86; 95% CI: 0.58 to 1.26), and decreased in the normal-BMI group (AHR: 0.48; 95% CI: 0.26 to 0.90). Heavy drinkers had an increased risk of hepatic steatosis irrespective of BMI. These data suggest that even moderate alcohol use is associated with the development of hepatic steatosis among individuals with obesity [65].
A prospective cohort study analyzing 414,209 participants from the UK Biobank study found synergistic associations of the PNPLA3 I148M variant, excessive alcohol intake, and obesity with an increased risk of cirrhosis, HCC, and liver disease-related death in the general population [66]. This study suggests that the genetic background is another cofactor associated in the dangerous association linking obesity with progressive ALD. Further studies are needed to examine the factors responsible for ALD in obese individuals, to establish the basis for precision medicine strategies in this area.
Various studies have examined how obesity and central obesity influence the progression of CLD due to hepatitis B virus (HBV) infection (Table 1) [67–75]. Overall, epidemiological data suggest that obesity impacts the entire spectrum of the disease, starting from its prevention through HBV prophylactic vaccination to its most severe complications. For instance, obesity has been linked to reduced response to HBV vaccines [71], higher ALT elevation [68], steato-fibrosis [73], doubled risks of HCC [75], and increased mortality due to liver cancer [74].
HBV and obesity
| Author, year [Ref.] | Method | Findings | Conclusion |
|---|---|---|---|
| Yen et al., 2008 [67] | Cross-sectional study of 8,226 university students | The risk for MetS increases with higher BMI and SUA, while it decreases with vigorous PA and current alcohol consumption. Compared to individuals with seroprotective titers from hepatitis B vaccination [anti-HBs(+) and anti-HBc(–)], those without protective anti-HBs titers after vaccination or without hepatitis B infection [anti-HBs(–) and anti-HBc(–)] had a 34% higher risk for MetS, and those with natural HBV infection [anti-HBc(+)] had a 58% higher risk for MetS. | Hepatitis B vaccination with anti-HBs(+)was associated with a reduced risk of MetS compared to anti-HBs(–). However, HBV infection documented by anti-HBc(+) was associated with a higher risk of MetS. |
| Wang et al.,2010 [68] | Cross-sectional study of 934 patients with HBV infection (25.1% had ALT activity ≥ 40 IU/L). | In a multivariate LRA, BMI and fasting blood glucose levels independently predicted elevated ALT activity, with ORs of 1.73 (95% CI: 1.17 to 2.56) for individuals with a BMI ≥ 25 kg/m2 and 1.88 (95% CI: 1.06 to 3.33) for those with FBG ≥ 126 mg/dL. ALT activity was also associated with BMI among individuals whose ALT activity was within the reference range. | Obesity and fasting hyperglycemia in the diabetic range may worsen liver injury, as assessed by ALT levels. |
| Chung et al., 2012 [69] | Retrospective analysis of 88 treatment-naïve CHB patients treated with ETV | The rate of seroconversion, ALT normalization, and HBV-DNA negativity (< 300 copies/mL) at 3, 6, and 12 months of treatment did not differ significantly between the normal-BMI and high-BMI groups. | Obesity does not impact the outcome of ETV treatment. |
| Chiang et al., 2013 [70] | Cross-sectional analysis of 3,587 HBV-infected participants without liver cirrhosis at study entry | High HBV viral load remained significantly inversely associated with extreme obesity (OR: 0.17; 95% CI: 0.05 to 0.63; P = 0.008) and central obesity (OR: 0.44; 95% CI: 0.25 to 0.78; P = 0.005) in male HBeAg-seropositive participants in sex-stratified analyses. | Extreme obesity and central obesity were linked to a lower prevalence of high HBV viral load in HBeAg seropositive individuals, especially in men. |
| Besharat et al., 2015 [71] | Follow-up study of 304 CHB patients. WC ≥ 102 cm (men) and ≥ 89 cm (women) identified central obesity. LSM ≥ 8 Kpa defined ALS | Nineteen (7.4%) CHB patients developed ALS after 4 years of follow-up. Central obesity and viral load predicted ALS in LRA. | In CHB patients, central obesity is a determinant of ALS. |
| Fan et al., 2016 [72] | Meta-analysis of 16 published studies | Individuals with obesity showed a significant non-response to HBV vaccination (adjusted OR: 2.46; 95% CI: 1.50 to 4.03). | Obesity is significantly associated with a decreased response to HBV vaccines. |
| Sun et al., 2019 [73] | Cross-sectional analysis of 615 CHB patients (287 with abdominal obesity) | In LRA, a high AST value (OR: 2.991; P < 0.001), smoking (OR: 2.002; P = 0.019), and diabetes mellitus (OR: 2.047; P = 0.029) independently predicted liver fibrosis in CHB patients with abdominal obesity. High body weight (OR: 1.113; P < 0.001) and high diastolic blood pressure (OR: 1.079; P = 0.002) were associated with SLD. | Obesity is a significant risk factor for SLD and liver fibrosis among individuals with CHB. |
| Kim et al., 2019 [74] | Study of 13,063 CHB patients, 195 of whom died of liver cancer during a follow-up of 10.6 years | After adjusting for confounders, statin use (HR: 0.17; 95% CI: 0.04 to 0.70) and hypercholesterolemia (HR: 0.46; 95% CI: 0.24 to 0.88 for total cholesterol ≥ 240 mg/dL) were associated with a decreased risk of LCM, while BMI ≥ 30 kg/m2 was associated with an increased risk of LCM (HR: 2.46; 95% CI: 1.20 to 5.06). | Obesity is a risk factor for LCM among patients with CHB. |
| Fan et al., 2021 [75] | Prospective study of 5,754 nucleos(t)ide analogue-treated CHB patients with a 5-year cumulative incidence of HCC of 2.9% | In the overall population, individuals with central obesity (W/H > 0.5) had a significantly higher 5-year incidence of HCC than those without (3.9% vs. 2.1%; HR: 2.06; P = 0.0001). Additionally, in 745 PSM pairs (4.7% vs. 2.3%; HR: 2.04; P = 0.026), central obesity was independently associated with HCC risk (HR: 1.63; P = 0.013). W/H gain within 1 year was associated with a significantly higher HCC risk (AHR: 1.88; 95% CI: 1.12 to 3.13; P = 0.017). | Central obesity is associated with a 2-fold increase in HCC risk among CHB patients receiving antiviral treatment. |
AHR: adjusted hazard ratio; ALS: advanced liver stiffness; ALT: alanine transaminase; anti-HBc: antibody against Hepatis B core antigen; anti-HBs: antibody against hepatitis B surface antigen; AST: aspartate transaminase; BMI: body mass index; CHB: chronic hepatitis B; ETV: entecavir; FBG: fasting plasma glucose; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HR: hazard ratio; LRA: logistic regression analysis; LCM: liver cancer mortality; MetS: metabolic syndrome; OR: odds ratio; PSM: propensity score matched; SLD: steatotic liver disease; SUA: serum uric acid; W/H: waist-to-height ratio; WC: waist circumference
Like HBV infection, the progression of CLD caused by hepatitis C virus (HCV) is also adversely impacted by concurrent visceral obesity (Table 2) [76–87]. BMI serves as an independent predictor of steato-fibrosis [76, 77, 79, 80], cirrhosis, and HCC [84] in individuals with chronic HCV infection. Alarmingly, obesity and excessive alcohol consumption work together to heighten the likelihood of developing liver cancer following sustained virological response achieved through antiviral therapy [87].
HCV and obesity
| Author, year [Ref.] | Method | Findings | Conclusion |
|---|---|---|---|
| Adinolfi et al., 2001 [76] | Cross-sectional study of 180 consecutive patients with biopsy-proven CHC | The grade of steatosis was associated with BMI in HCV genotype 1 infection (r = 0.689; P < 0.001) and with levels of HCV RNA in HCV genotype 3a infection (r = 0.786; P < 0.001). Visceral fat distribution, rather than BMI, was associated with steatosis (P < 0.001). | Visceral obesity and genotype 3a play a role in the development of steatosis. |
| Monto et al., 2002 [77] | Cross-sectional study of 297 consecutive patients with HCV | At LRA BMI (P = 0.0002) and genotype 3a infection (P = 0.02) independently predicted steatosis. After exclusion of subjects with risk factors for NASH, genotype 3a infection remained the only independent predictor of steatosis. Steatosis (P = 0.04) and inflammation (P < 0.0001) scores on liver biopsy were the only independent predictors of fibrosis. | Steatosis in HCV infection is associated with risk factors for NASH, particularly obesity, rather than alcohol consumption. |
| Ortiz et al., 2002 [78] | 114 prospectively recruited subjects who tested positive for HCV-RNA. Rapid disease progression was defined as a rate greater than 0.2 U/year. | At LRA the following variables were associated with progression: advanced age at infection (P = 0.0001), BMI ≥ 25 kg/m2 (P = 0.01), and ALT > 1.5 times ULN (P = 0.01). Among patients with ALT > 1.5 times ULN, these predictors were: age at infection, BMI ≥ 25 kg/m2, diabetes, and transferrin saturation > 45%. Among those with normal ALT levels, only BMI ≥ 30 kg/m2 predicted progression. | Obesity, advanced age at infection, and elevated ALT levels predict rapid fibrosis progression in HCV infection. |
| Friedenberg et al., 2003 [79] | Analysis of 264 consecutive CHC patients | The degree of steatosis and fibrosis both tended to increase with increasing BMI (rho = 0.47, P < 0.001 and rho = 0.13, P = 0.03, respectively). | BMI is an independent predictor of both fibrosis and steatosis in HCV patients. |
| Younossi et al., 2004 [80] | Cross-sectional analysis of 120 CHC subjects who had available liver biopsies | Patients with superimposed NASH had more evidence of obesity (BMI: 30.64 ± 5.23 vs. 29.90 ± 5.35 vs. 27.33 ± 4.07, P = 0.008; W/H: 0.97 ± 0.06 vs. 0.91 ± 0.08 vs. 0.87 ± 0.07, P < 0.001), were more commonly infected with HCV genotype 3 (14% vs. 12% vs. 0%, P = 0.036), and had more advanced fibrosis (95.5% vs. 75.5% vs. 42.9%, P < 0.001). | The extent of steatosis and the type of SLD are associated with BMI, W/H, and HCV genotype 3a. Severe steatosis and superimposed NASH are both associated with advanced hepatic fibrosis. |
| Walsh et al., 2006 [81] | A cohort of 145 individuals with CHC was analyzed to determine host factors associated with non-response to antiviral therapy with either IFN-α or PEG-IFN-α, alone or in combination with ribavirin. | Factors independently associated with NR were viral genotype 1/4 (P < 0.001), cirrhosis on pretreatment biopsy (P = 0.025), and BMI ≥ 30 kg/m2 (P = 0.010). At LRA, SOCS-3 mRNA expression remained independently associated with obesity (P = 0.023). SOCS-3 immunoreactivity was significantly increased in obesity (P = 0.013) and in non-responders compared with responders (P = 0.014). | Obesity reduces the response rate to IFN-alpha via increased expression of inhibitors of interferon signaling in CHC owing to HCV genotype 1. |
| Nishikawa et al., 2013 [82] | 233 patients with HCV-related HCC who underwent curative SR were recruited. 60 patients (25.8%) were in the obesity group with a BMI greater than 25 kg/m2, while 173 controls had a BMI less than 25 kg/m2. | No differences were found between the obesity group and the control group regarding 1-, 3-, and 5-year cumulative OS (P = 0.818), RFS rates (P = 0.124), nor as regards blood loss during surgery (P = 0.899) and surgery-related serious adverse events (P = 0.813). | Obesity does not affect survival in patients with HCV-related HCC after curative SR. |
| Aguilar et al., 2016 [83] | Analysis of 43,478 new LT waitlist registrants with chronic HCV (21.0% with HCC, 79% without HCC) included in the 2003–2013 UNOS database. | Compared to non-obese patients, those with obesity had lower probability of receiving LT (OR: 0.91; 95% CI: 0.85 to 0.97; P < 0.01) and a lower probability of waitlist mortality (OR: 0.80; 95% CI: 0.72 to 0.89; P < 0.001). | Although it does not affect waitlist survival, obesity is associated with reduced odds of receiving LT. |
| Rao et al., 2017 [84] | Prospective study of two cohorts of HCV patients recruited from the US (n = 1,000) and China (n = 957) | Compared to subjects from China, more US patients had cirrhosis (38.2% vs. 16.0%) and HCC (14.1% vs. 2.7%). AT LRA, significant predictors of advanced disease were age, visceral obesity, diabetes, and prior HCV treatment. | The finding that the US had more advanced liver disease than those from China probably mirrors underlying SLD being a major contributor to this difference. |
| Tsao et al., 2017 [85] | Retrospective cross-sectional analysis of 1,267 medical records | After adjustment for confounders, body fat percentage, FFM/BW and MM/BW were the independent determinants of visceral obesity in HCV-free individuals (P < 0.001) although the trend, still statistically significant (P < 0.05), was not such obvious among HCV-infected individuals with HCV infection and less significant in HCV-infected men. | HCV affects the host’s lipid homeostasis and body fat distribution, with sex differences. |
| Turner et al., 2019 [86] | Analysis of a multiethnic cohort of 748 HCV-infected individuals, with 53% non-Hispanic black, 26% non-Hispanic white, and 22% Hispanic, of whom 23% had advanced liver disease defined as a FIB-4 index score above 3.25. | Among those with obesity (BMI ≥ 30 kg/m2) with a diagnosis of diabetes, the adjusted OR of advanced liver disease for Hispanics vs non-Hispanic black was 7.89 (95% CI: 3.66 to 17.01) and adjusted OR: 12.49 (95% CI: 3.24 to 48.18) for Hispanic vs non-Hispanic white patients (both P < 0.001). | The presence of obesity and diabetes among HCV-infected Hispanics is associated with a far higher risk for advanced liver disease than other ethnic groups. |
| Minami et al., 2021 [87] | Analysis of 2,055 HCV-infected individuals (840 in the IFN group and 1,215 in the DAA group) | At LRA, age, serum albumin, platelet count, AFP, and normal lipidemia, BMI ≥ 25 kg/m2, and alcohol consumption ≥ 60 g/day independently predicted incident HCC, AHR 2.53 (95% CI: 1.51 to 4.25) and 2.56 (95% CI: 1.14 to 5.75), respectively. | Obesity and heavy alcohol consumption are associated with the risk of developing HCC after SVR. |
AFP: alpha-fetoprotein; AHR: adjusted hazard ratio; ALT: alanine transaminase; BMI: body mass index; BW: body weight; CHC: chronic hepatitis C; DAA: direct-acting antivirals; FFM: fat-free mass; FIB-4: fibrosis 4; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; IFN: interferon; LRA: logistic regression analysis; LT: liver transplantation; MM: muscle mass; NASH: nonalcoholic steatohepatitis; OR: odds ratio; OS: overall survival; PEG: pegylated; RFS: recurrence-free survival; SLD: steatotic liver disease; SR: surgical resection; SOCS-3: suppressors of insulin signaling 3; SVR: sustained virological response; ULN: upper limit of normal; UNOS: United Network for Organ Sharing; US: United States
The association of obesity with what is now known as MASLD and MASH dates back to 1867 and 1980, thanks to studies by Flint and Ludwig, respectively [88]. Unhealthy lifestyle habits that contribute to the development of obesity also play a major role in MASLD and MASH by enhancing intrahepatic de novo lipogenesis (DNL), extrahepatic and hepatic IR, dysfunctional gut-liver axis, and metabolic inflammation (metaflammation) [89]. Excess visceral fat leads to an overflow of FFAs to the liver, triggering a harmful cascade including oxidative stress (OS), endoplasmic reticulum (ER) stress, activation of protein kinase C (PKC) pathways, inflammation, excessive intrahepatic steatogenesis, progressive hepatocellular damage, and fibrosing MASH, as discussed in Pathobiology of liver damage in the obese of this article.
A meta-analysis of 21 cohort studies, including 13 prospective and 8 retrospective studies, totaling 381,655 participants, revealed an independent association between obesity and a 3.5-fold increased risk of developing MASLD compared to individuals with normal weight [relative risk (RR) = 3.53, 95% CI: 2.48 to 5.03, P < 0.001]. There was a clear dose-dependent relationship between BMI and MASLD risk with a relative risk of 1.20 per 1-unit increment in BMI (95% CI: 1.14 to 1.26, P < 0.001) [90]. It is essential to note that VAT, being close to the liver, is a significant risk factor for MASLD, even in lean MASLD subjects. Its levels increase in parallel with the stages of liver fibrosis and are closely linked to IR in the liver, muscle, and adipose tissue. This increase in VAT leads to heightened lipolysis and decreased levels of the insulin-sensitizing, anti-steatotic, and anti-inflammatory adipokine adiponectin [91]. These findings help to explain why obesity predicts a worse long-term prognosis in MASLD [92].
More than a quarter of a century ago, Brolin and colleagues [93] reported on 125 individuals with obesity in whom unsuspected cirrhosis was detected during gastric bariatric operations. These authors estimated that cirrhosis may have been caused by severe obesity in 74% of their patients. Although a proportion of these individuals might have had CLD owing to competing etiologies, we know that, in the context of the contemporary epidemic of obesity, these individuals might have progressed from MASLD to fibrosing MASH [94]. Of concern, obesity adversely impacts the natural course of cirrhosis by predisposing to white adipose tissue dysfunction, IR, and portal hypertension through incompletely characterized pathomechanisms [95].
Clinically, patients usually exhibit sarcopenic obesity, which determines higher rates of metabolic and physical dysfunction than either condition alone [96]. The diagnostic criteria for sarcopenic obesity in patients with cirrhosis are scarcely defined, and the most accurate diagnostic tool, cross-sectional imaging, has practical limitations for routine use in clinical practice [96]. Therapeutic approaches aim at improving nutrition, muscular mass, and strength [96, 97].
Future directions in this field comprise consensus definitions for sarcopenia, obesity, and sarcopenic obesity, an improved understanding of the complex pathogenesis of the muscle-liver-adipose tissue axis in cirrhosis [98], and evidence-based management recommendations for nutrition, exercise, and drug treatment [96].
In 2004, El-Serag [99] released the first epidemiological alert, indicating that 15% to 50% of HCC cases lacked evidence of viral or alcoholic risk factors. They also highlighted the emerging risk factor of the MetS for HCC in the USA. Recent epidemiological studies further support the idea that obesity may increase the risk of HCC, with clearer evidence for ALD and more limited evidence for CLD due to HBV, HCV, and MASLD [100]. However, in clinical practice, it is often challenging to separate obesity from more complex metabolic dysfunction. Over the last few decades, CLD due to viral causes, which was previously the most common risk factor for HCC in developing countries, has decreased. This decline has coincided with an increase in MASLD, making HCC relatively more common in Western countries [101]. Sex differences in HCC associated with MASLD and non-MASLD CLD have been discussed in previous studies [102, 103]. Metabolic comorbidities are significant risk factors for HCC and cholangiocarcinoma, the second most common type of primary liver cancer, through various pathomechanisms that have been reviewed elsewhere [101].
Below, we will highlight ALD, chronic viral hepatitis, and hepatic carcinogenesis to illustrate how obesity amplifies pathogenic pathways across a spectrum of distinct hepatic insults. This occurs through metabolic and inflammatory pathways, hastening the progression toward advanced liver disease.
Obesity can amplify both the frequency and severity of ALD by exacerbating OS and promoting a pro-inflammatory milieu in the liver [104]. Under normal circumstances, alcohol metabolism generates ROS [105]. In individuals with obesity, concurrent IR and elevated FFAs can intensify ROS production, compromise hepatic antioxidant defenses, and generate carcinogenic etheno-DNA adducts, thereby leading to the initiation of events leading to HCC [106]. Moreover, dysfunctional adipose tissue in obesity increases the release of pro-inflammatory cytokines, fueling Kupffer cell (KC) activation and hepatic inflammation. This synergy of lipotoxicity and alcohol-induced oxidative damage accelerates fibrogenesis and heightens the risk of cirrhosis and hepatic decompensation.
HBV and HCV infections are exacerbated by untreated obesity, indicating a synergistic effect of metabolic factors and viral hepatitis [107]. Excess adiposity results in an abundance of circulating FFAs, which promote hepatic steatosis, OS, and local immune activation. This environment weakens antiviral responses, and this, in turn, speeds up the progression to fibrosis or cirrhosis. In the case of HCV, fat droplets and increased intrahepatic triglyceride content can boost viral replication [108], while IR further affects interferon expression and signaling [109]. The combined impact of viral infection and obesity-induced lipotoxicity accelerates bridging fibrosis and raises the risk of advanced liver disease.
Obesity predisposes individuals to HCC through chronic inflammation, altered adipokine profiles, and genomic instability in hepatocytes [110]. Persistent lipotoxicity and ROS insult contribute to somatic mutations, DNA damage, and dysregulated cell-cycle control. In cirrhotic livers, irrespective of etiology, local tissue remodeling and compromised immune surveillance allow pre-neoplastic clones to expand. Furthermore, hyperinsulinemia and elevated levels of growth factors in obese individuals can stimulate cellular proliferation and angiogenesis, further promoting cancer development [111]. Therefore, obesity remains a significant factor in the development of HCC, regardless of CLD etiology.
Excess body weight and the accumulation of adipose tissue contribute to the development of MASLD, which begins with simple steatosis and can eventually progress to MASH, fibrosis, cirrhosis, and ultimately HCC in some individuals [16] (Figure 2). The following sections will detail how obesity promotes liver damage through IR, lipotoxicity, systemic inflammation, OS, and alterations in the gut-liver axis, leading to significant and potentially irreversible liver injury.
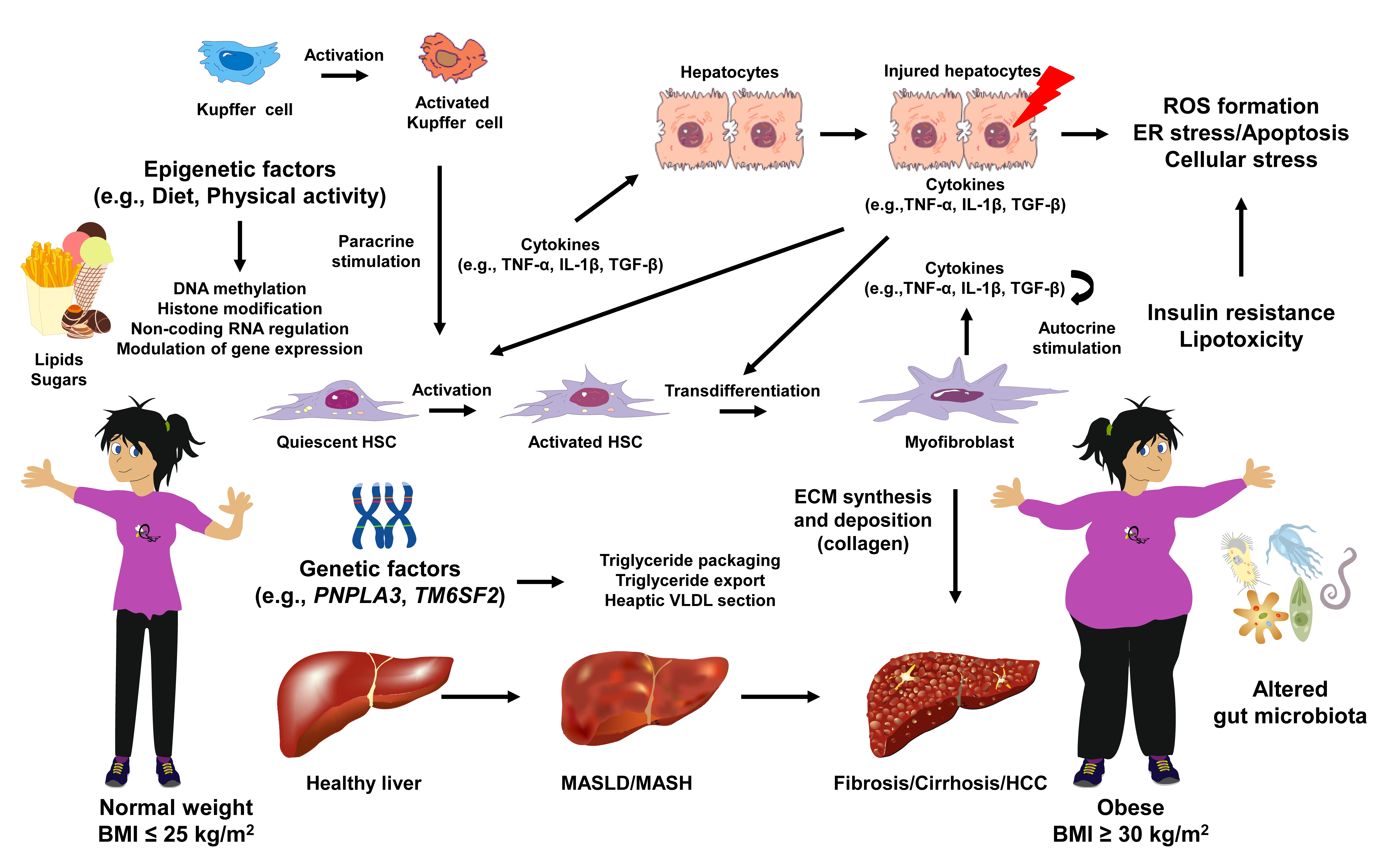
Pathobiology of liver damage in obesity. This diagram illustrates how interconnected metabolic and inflammatory pathways collaborate to generate the spectrum of obesity-linked CLD. Gene variants and epigenetic factors, such as unhealthy diets and sedentary behavior, promote the development of MASLD and MASH, which can progress to fibrosis, cirrhosis, and HCC. Excessive FFAs can stimulate KCs to produce increased levels of pro-inflammatory and pro-fibrotic cytokines. Additionally, the development of IR can further contribute to hepatic steatosis and cause lipotoxic injury to hepatocytes. OS and ER stress can also damage hepatocytes and worsen inflammation. In response to these signals, quiescent HSCs can transform into matrix-producing myofibroblasts, leading to ECM synthesis and hepatic fibrogenesis. Accumulated ECM disrupts the normal hepatic architecture, ultimately resulting in advanced fibrosis and cirrhosis. In more advanced stages, the chronically injured, inflamed, and fibrotic liver has an increased risk of HCC. CLD: chronic liver disease; ECM: extracellular matrix; ER: endoplasmic reticulum; FFAs: free fatty acids; HSCs: hepatic stellate cells; HCC: hepatocellular carcinoma; IR: insulin resistance; MASH: metabolic dysfunction-associated steatohepatitis; MASLD: metabolic dysfunction-associated steatotic liver disease; OS: oxidative stress; ROS: reactive oxygen species
In obesity, elevated levels of FFAs and pro-inflammatory cytokines can interfere with insulin signaling pathways, impairing the ability of insulin to adequately regulate blood glucose levels and suppress hepatic glucose output, leading to metabolic dysfunction [112]. This state of IR has profound effects on glucose and lipid homeostasis. As peripheral tissues, such as adipocytes, become increasingly resistant to the effects of insulin, they release more FFAs into circulation. The liver is then exposed to these circulating FFAs, which are taken up by hepatocytes via plasma-membrane-associated proteins and are either oxidized or stored as triglycerides within the liver [113]. With prolonged excess supply, the hepatic lipid storage capacity of the liver can become overwhelmed and maladaptive, leading to steatosis, which lays the groundwork for potential progression to steatohepatitis and ultimately more advanced liver disease.
Steatosis initiates a series of cellular stress responses collectively known as lipotoxicity. Various forms of lipids, particularly certain FFAs and their derivatives (e.g., diacylglycerols, ceramides), can be toxic to hepatocytes and induce lipid-induced hepatic IR [114]. When hepatic lipid species accumulate, they impair mitochondrial function, disrupt ER homeostasis, and generate ROS [115]. Excessive fatty acid load can overwhelm and damage mitochondria, leading to abundant ROS production that further compromises mitochondrial integrity and causes oxidative damage to cellular lipids, proteins, and DNA [115]. Additionally, ER stress can trigger the unfolded protein response (UPR), which aims to restore proper protein folding. If the UPR fails to relieve ER stress, it can result in hepatocyte apoptosis or necrosis. Therefore, in obesity, lipid overload and lipotoxicity generate a cycle of progressive liver damage.
A key feature of obesity is the presence of chronic low-grade inflammation. Adipose tissue, particularly VAT, serves not only as a passive energy storage depot but also as an active endocrine organ that secretes numerous hormones and cytokines, such as TNF-α, interleukins (e.g., IL-6, IL-1β), and adipokines (e.g., leptin, adiponectin). In obesity, the balance of these secreted factors may shift towards a pro-inflammatory profile, further worsening IR and promoting local and systemic inflammation. Within the liver, KCs, which are resident macrophages, play a crucial role in orchestrating the inflammatory response [116]. Under normal conditions, KCs help clear pathogens and cellular debris, maintain tissue homeostasis, and limit tissue injury [117, 118]. However, in obesity, exposure to elevated levels of fatty acids, endotoxins, and inflammatory mediators activates KCs, leading to the production of various cytokines, including TNF-α, IL-1β, and IL-6, which perpetuate inflammatory signaling within the liver [116]. This increased inflammatory environment attracts circulating monocytes and other immune cells, contributing to a continuous inflammatory cycle. Over time, persistent inflammation triggers fibrogenic responses, paving the way for more advanced liver damage [119].
The progression from simple steatosis to MASH may be accompanied by liver fibrosis, primarily driven by hepatic stellate cells (HSCs). Normally quiescent in the space of Disse, they store vitamin A and other retinoids [120]. However, when exposed to inflammatory mediators, ROS, and signals from injured hepatocytes, HSCs transform into activated myofibroblast-like cells. This activation is characterized by a significant increase in extracellular matrix (ECM) production, including collagens I and III, loss of normal stellate cell functions [121], and secretion of pro-inflammatory and pro-fibrotic cytokines. In advanced disease stages, excessive ECM deposition distorts liver architecture and leads to cirrhosis, which gradually reduces liver function and can result in portal hypertension, variceal bleeding, liver failure, and increased risk of HCC. Due to their crucial role in fibrogenesis, HSCs have become a focal target of anti-fibrotic therapies [121].
OS plays a central role in obesity-related CLD [122]. When the capacity of mitochondria for effective oxidative phosphorylation is exceeded or partially reduced, ROS accumulate [14, 15]. These ROS, such as superoxide anions, hydrogen peroxide, and hydroxyl radicals, are highly reactive molecules that can damage cellular structures. In addition to mitochondria, other sources of ROS include peroxisomal and microsomal oxidation of FAs, as well as activated immune cells in the liver. Some of the main downstream consequences of elevated ROS levels in hepatocytes include (i) lipid peroxidation—ROS attack polyunsaturated fatty acids in cell membranes, producing breakdown products that further propagate oxidative damage, (ii) protein oxidation—oxidative changes to proteins can impede their function and lead to their degradation, affecting structural proteins and enzymes crucial for cellular functionality, (iii) DNA damage—ROS can cause mutations and strand breaks in DNA, contributing to genomic instability that could increase the risk of malignant transformation, (iv) OS also interacts with inflammatory pathways, promoting the activation of NF-κB and other transcription factors that increase the production of cytokines and promote carcinogenesis [123]. Therefore, oxidative damage directly harms hepatocytes and contributes to fueling harmful cycle of inflammation and cellular stress.
Adipose tissue secretes a complex array of liver-impacting adipokines, including leptin, adiponectin, resistin, visfatin, and others [124]. In obesity, the profile of these adipokines can be altered in ways that promote both metabolic dysregulation and inflammation.
Leptin levels, which physiologically regulate appetite and energy expenditure, tend to rise in obesity due to leptin resistance. High levels of leptin can stimulate KCs, which further drive the production of pro-inflammatory mediators and directly influence HSCs to promote fibrosis [125]. Serdyukov and colleagues [126], based on analysis of young and middle-aged men, have proposed the definition of “metabolically neutral obesity” based on the presence of MHO and leptinemia < 3.5 ng/mL. These men with “metabolically neutral obesity” had a low prevalence of dyslipidemia, prediabetes, and carotid atherosclerosis compared to MHO subjects. However, no details have been published regarding liver health in these subjects, and additional studies comprising female subjects, older individuals, and longitudinal hard cardiometabolic outcomes are expected.
Adiponectin, which normally activates glucose transport and FFA oxidation and downregulates inflammation through the PPARγ pathway, is typically reduced in obesity [127]. Adiponectin exerts insulin-sensitizing, anti-inflammatory, and anti-fibrotic effects, in part by improving FA oxidation in hepatocytes and attenuating the production of pro-inflammatory cytokines [128]. Reduced adiponectin levels in obesity thus remove an important protective mechanism against liver injury. The imbalance in these and other adipokines highlights the complex endocrine crosstalk between adipose tissue and the liver in driving the pathogenesis of MASLD.
Accumulating evidence emphasizes the importance of the gut-liver axis in the pathobiology of obesity-related liver damage [129]. The gut microbiota, consisting of trillions of microorganisms, plays a central role in metabolism, bile acid turnover, and immunity [129]. In obesity, dysbiosis can lead to an increased permeability of the intestinal barrier, often referred to as “leaky gut”, allowing bacterial endotoxins, particularly lipopolysaccharides (LPS), to translocate from the intestinal lumen into the portal circulation [130]. These endotoxins stimulate KCs and other immune cells in the liver to secrete pro-inflammatory cytokines, worsening liver inflammation and IR [131]. Additionally, changes in bile acid profiles modulated by gut bacteria can also impact metabolic processes in the liver, further exacerbating steatosis and inflammation [129].
The ER is responsible for the proper folding and processing of proteins before their transport to other cellular compartments or to the extracellular space [132]. When hepatocytes are exposed to nutrient overload, pro-inflammatory signals, and toxic lipid metabolites, the protein-folding capacity of the ER can be overwhelmed, leading to an accumulation of misfolded or unfolded proteins [133]. This activates an adaptive program known as the UPR aimed at restoring homeostasis by reducing protein translation, promoting proper protein folding by up-regulating chaperones, and enhancing protein degradation pathways [132, 133]. If these compensatory mechanisms fail, the cell may activate apoptotic pathways, leading to hepatocyte cell death. Chronic ER stress and subsequent apoptosis contribute to ongoing liver injury and inflammation, perpetuating MASLD and MASH. Importantly, signaling molecules such as C/EBP homologous protein (CHOP) and JNK (c-Jun N-terminal kinase) have been implicated in ER stress-induced apoptosis, providing potential molecular targets for therapeutic intervention [134].
Polymorphisms in genes involved in lipid metabolism, insulin signaling, and inflammatory pathways may increase the risk of MASLD and accelerate its progression to fibrosis. For example, variants in the patatin-like phospholipase domain-containing protein 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), and membrane-bound O-acetyltransferase domain-containing protein 7 (MBOAT7) genes have been strongly associated with MASLD and MASH by affecting triglyceride packaging and export, hepatic VLDL secretion, and predisposing to more severe disease phenotypes. In addition, epigenetic modifications can alter gene expression patterns influenced by various environmental factors [135–137]. This complex interplay of genetics and environment may account for the large inter-individual disease variability and heterogeneity, underscoring the importance of personalized approaches to prevention and treatment.
Although many obese individuals with MASLD remain stable or have only mild liver disease, a proportion may progress to MASH, cirrhosis, end-stage liver failure, and MASH-HCC [138, 139]. Therefore, early identification and monitoring of MASH patients at high risk of cirrhosis and HCC are of paramount importance.
As discussed in Principles of treatment of obesity-associated CLD, lifestyle interventions remain the initial cornerstone of therapy for MASLD and MASH [140]. However, adherence to long-term lifestyle changes can be challenging for many individuals. Novel agents targeting specific pathogenic mechanisms, such as IR, de novo lipogenesis, and fibrogenesis, are currently under investigation. These drugs modify bile acid signaling (e.g., obeticholic acid), reduce lipotoxic intermediates (e.g., acetyl-CoA carboxylase inhibitors), activate the thyroid hormone receptor, or target inflammatory cascades (e.g., IL-1β or TNF-α inhibitors), representing promising avenues for the treatment of MASLD/MASH (Table 3) [119, 141–156].
Representative pharmacotherapies currently under investigation for the treatment of MASLD/MASH
| Mechanism | Selected compounds | Structure | Remarks |
|---|---|---|---|
| FXR agonism | Obeticholic acid | 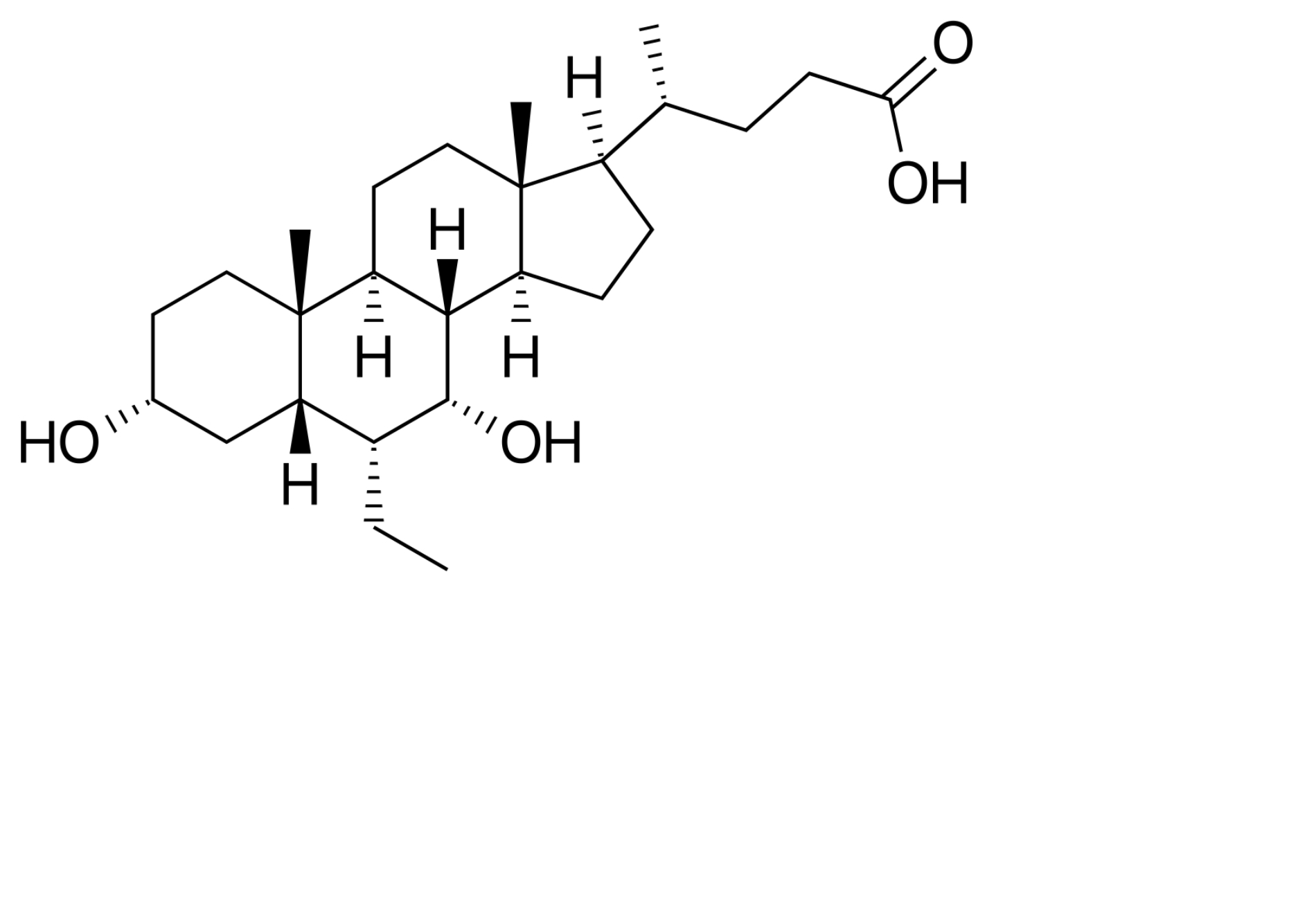 | Obeticholic acid is a semi-synthetic bile acid data that is FDA approved for primary biliary cholangitis. It ameliorates obesity and hepatic steatosis by activating brown fat [141]. |
| Tropifexor | 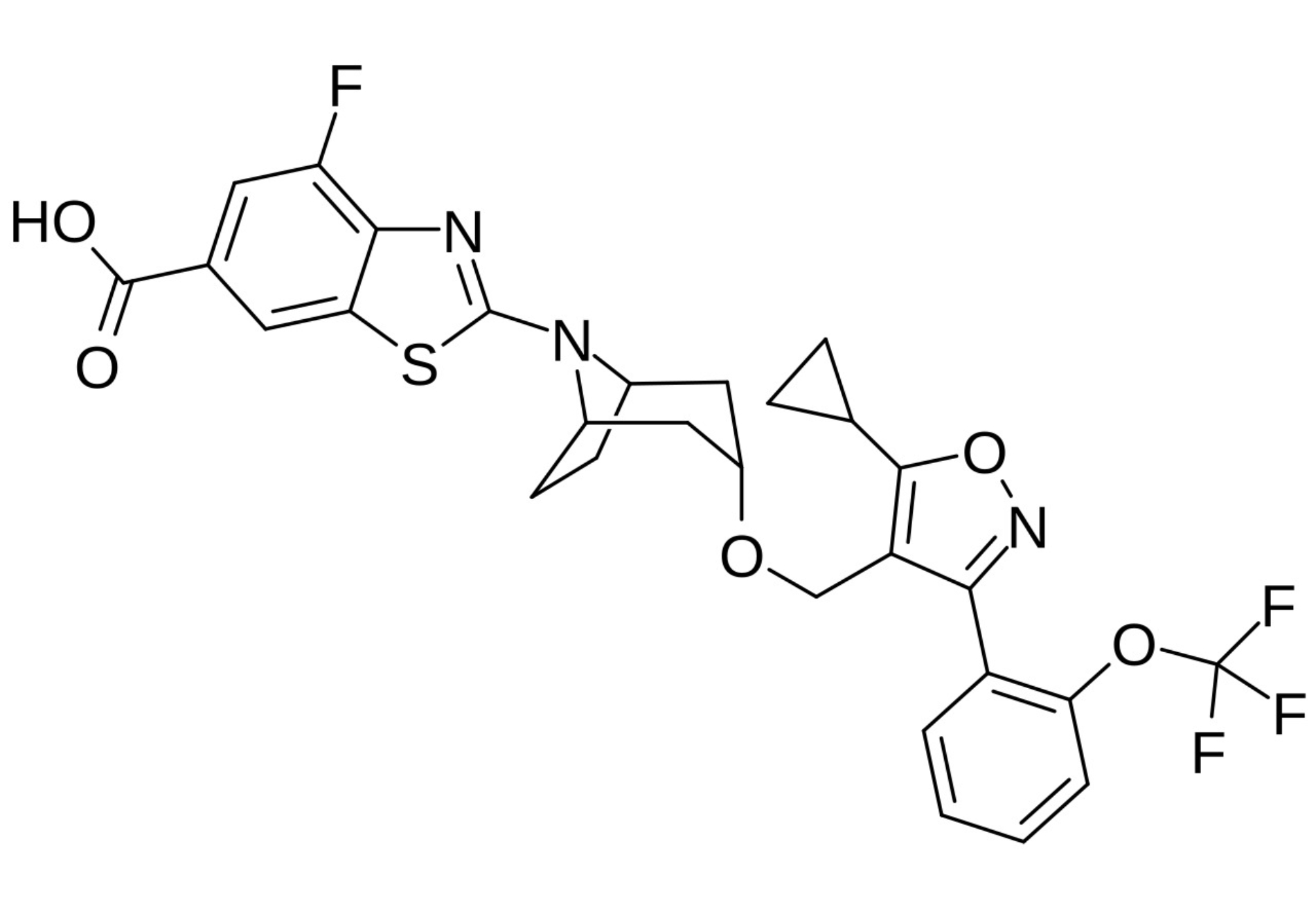 | Tropifexor was developed for the treatment of cholestatic liver diseases and MASH [142]. | |
| Cilofexor | 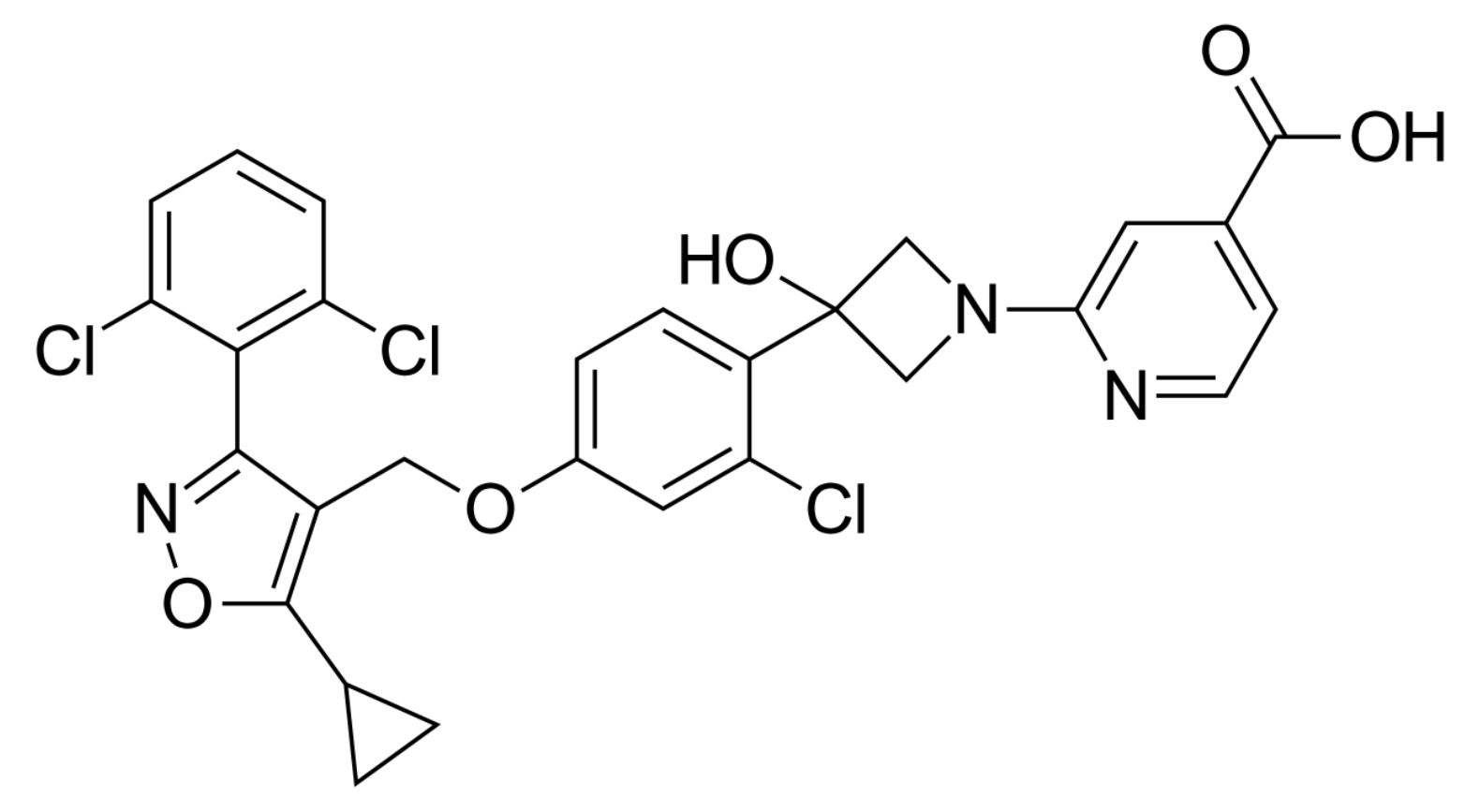 | Cilofexor is a nonsteroidal FXR agonist for the treatment of MASLD, MASH, and primary sclerosing cholangitis [143]. | |
| PPAR agonism | Elafibranor | 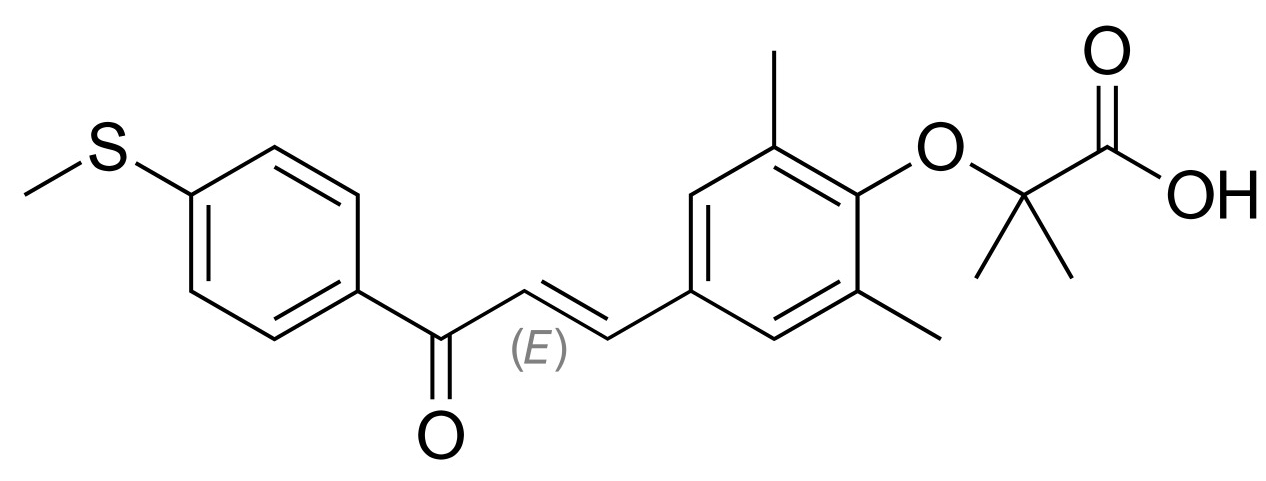 | Elafibranor is a dual PPAR α/δ agonist that is approved for the treatment of primary biliary cholangitis, showing beneficial effects in MASH [144]. |
| Saroglitazar | 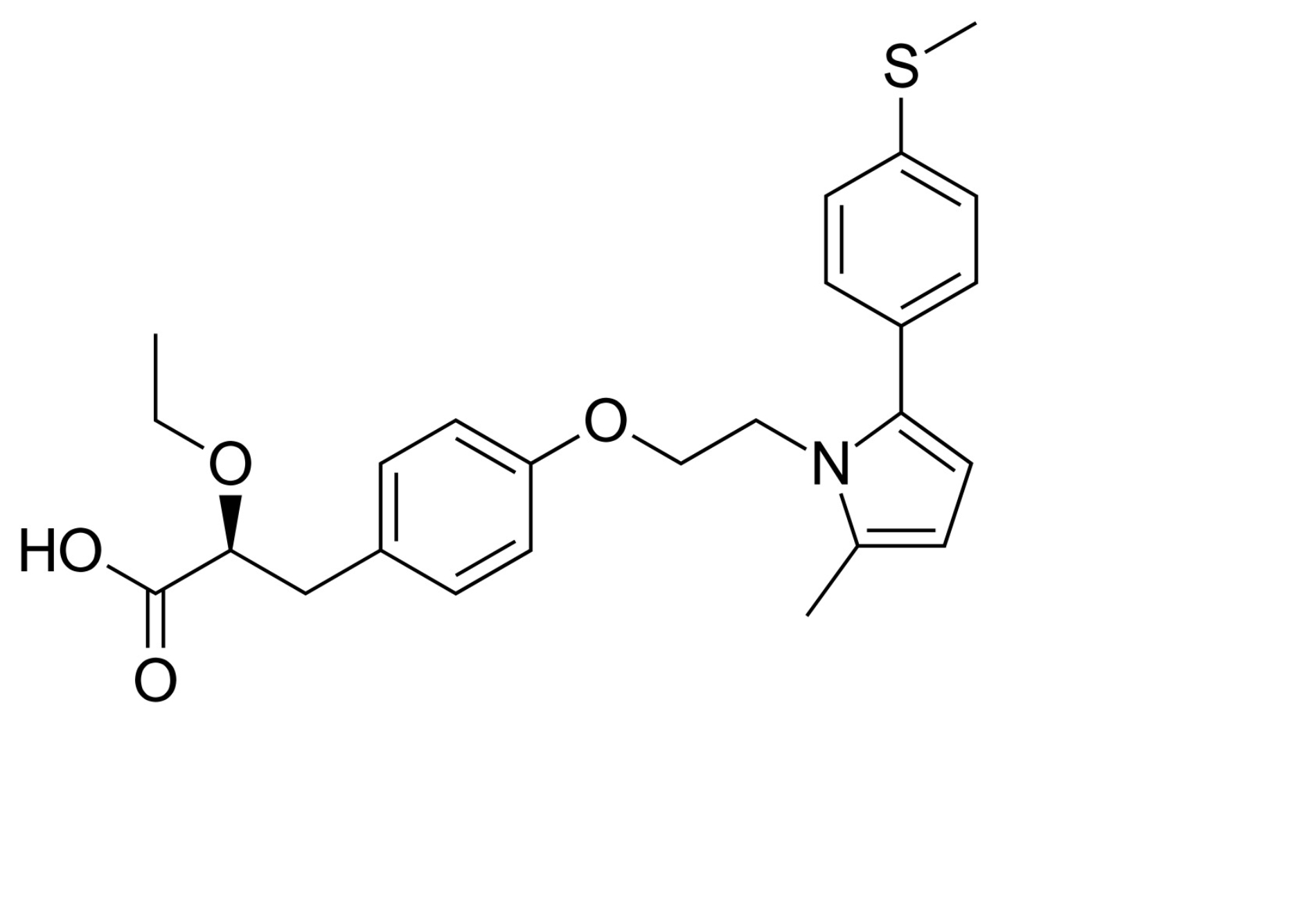 | Saraglitazar is a dual PPARα/γ agonist acting as an insulin sensitizer and has beneficial effects on adipose tissue and extracellular matrix deposition in obesity. Therefore, it is a drug for the treatment of T2DM, dyslipidemia, MASLD, and MASH [145]. | |
| Lanifibranor | 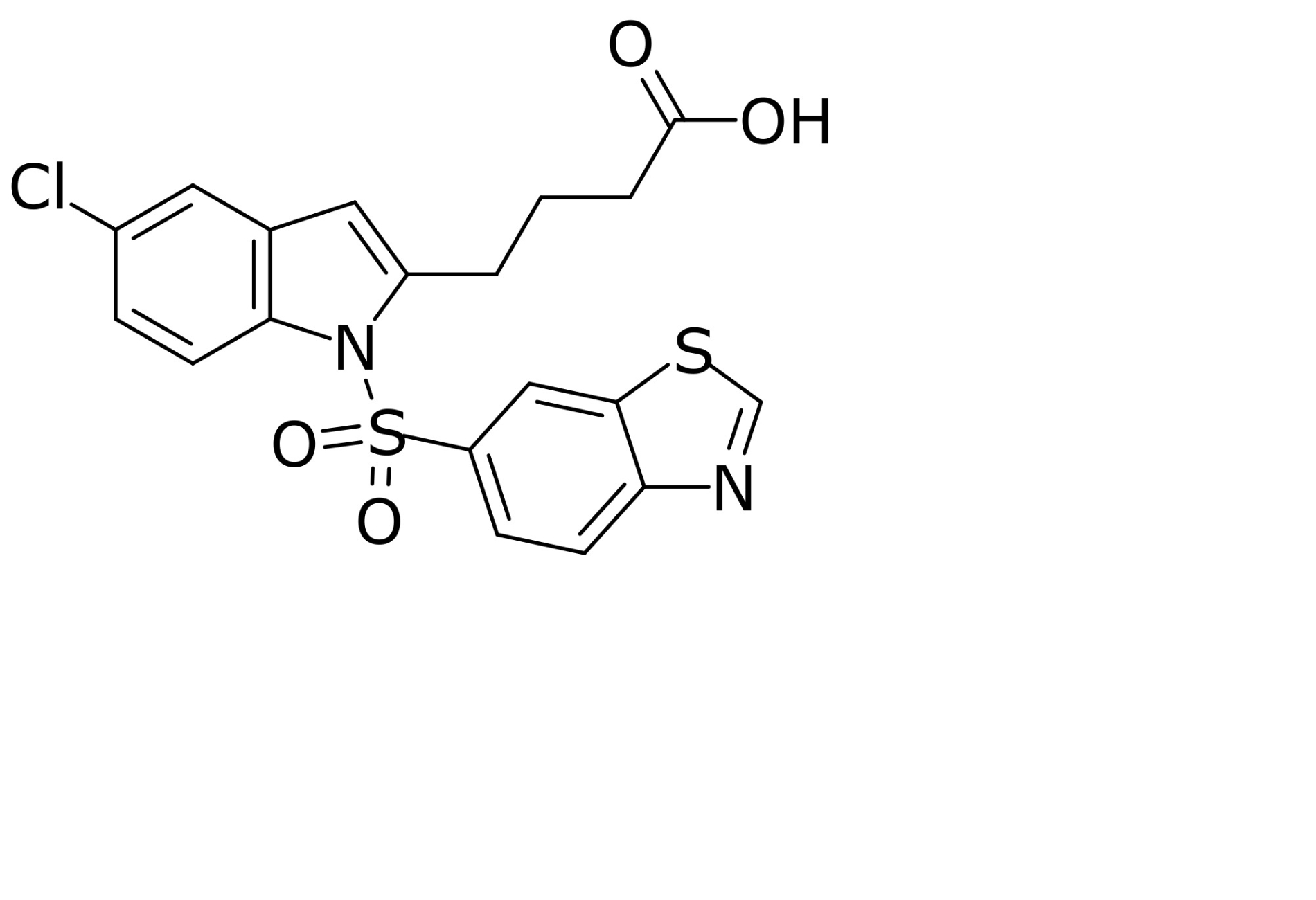 | Lanifibranor is a pan-specific PPAR agonist with balanced α, β/δ, and γ activity. It improves liver health, IR, adipose tissue function, and provokes weight gain in MASH patients [146, 147]. | |
| ACC inhibition | Firsocostat (GS-0976) | 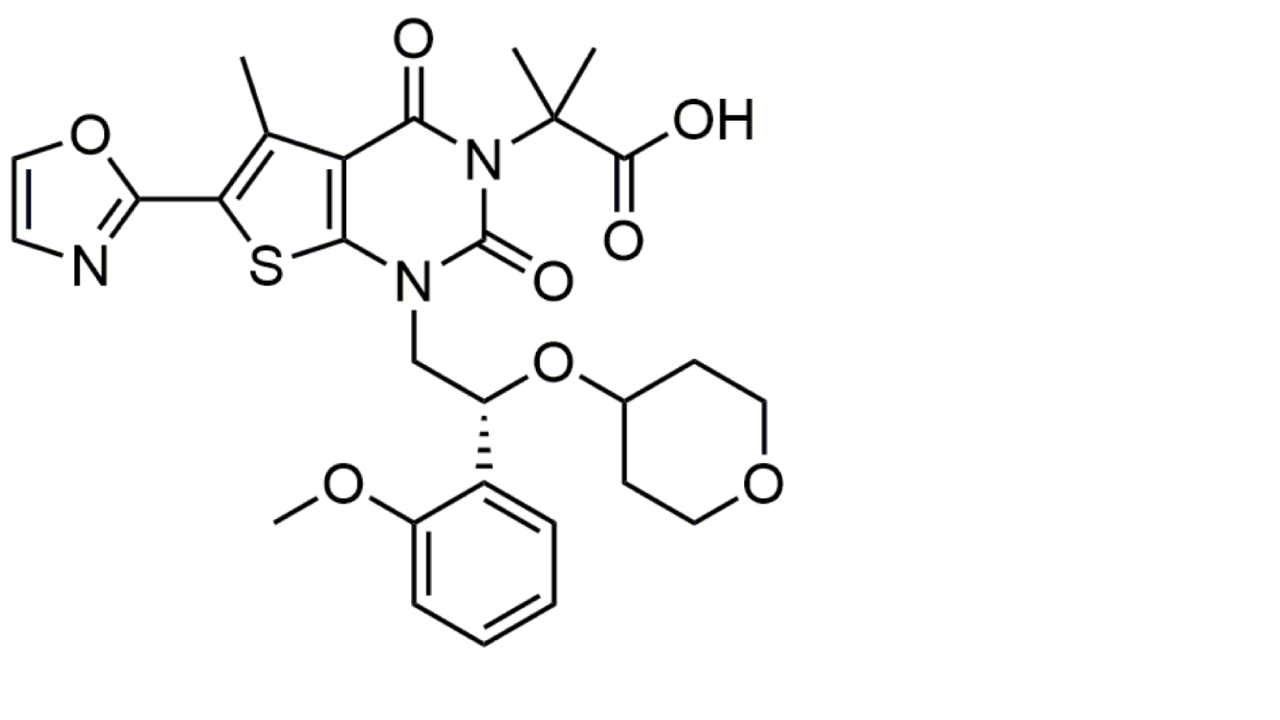 | Firsocostat is a liver-targeted ACC inhibitor that inhibits hepatic de novo lipogenesis and decreases the deleterious effects of lipotoxicity. It reduces hepatic steatosis that, in combination with other drugs, is therapeutically effective in MASH [148]. |
| PF-05221304 (Clesacostat) | 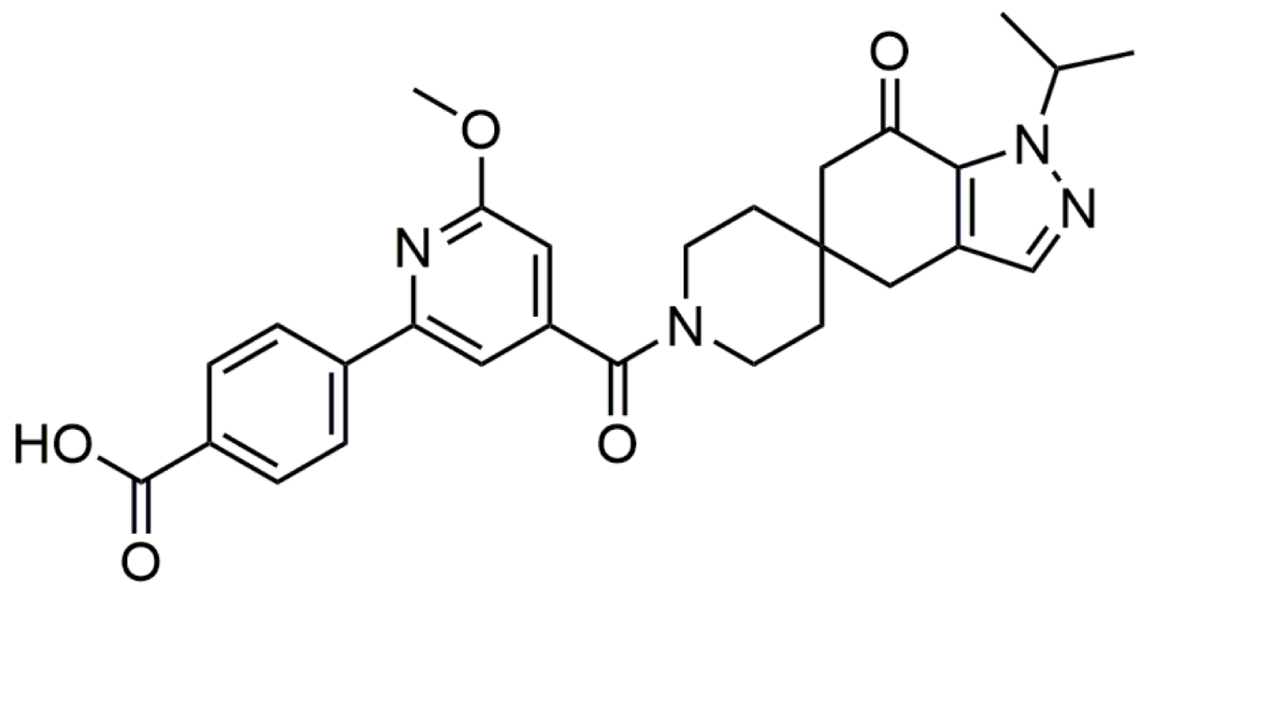 | PF-05221304 is an orally bioavailable, liver-targeted inhibitor of ACC. It inhibits de novo lipogenesis and is therefore under close investigation for treatment of MASLD/MASH [149]. | |
| THRβ agonism | Resmetirom | 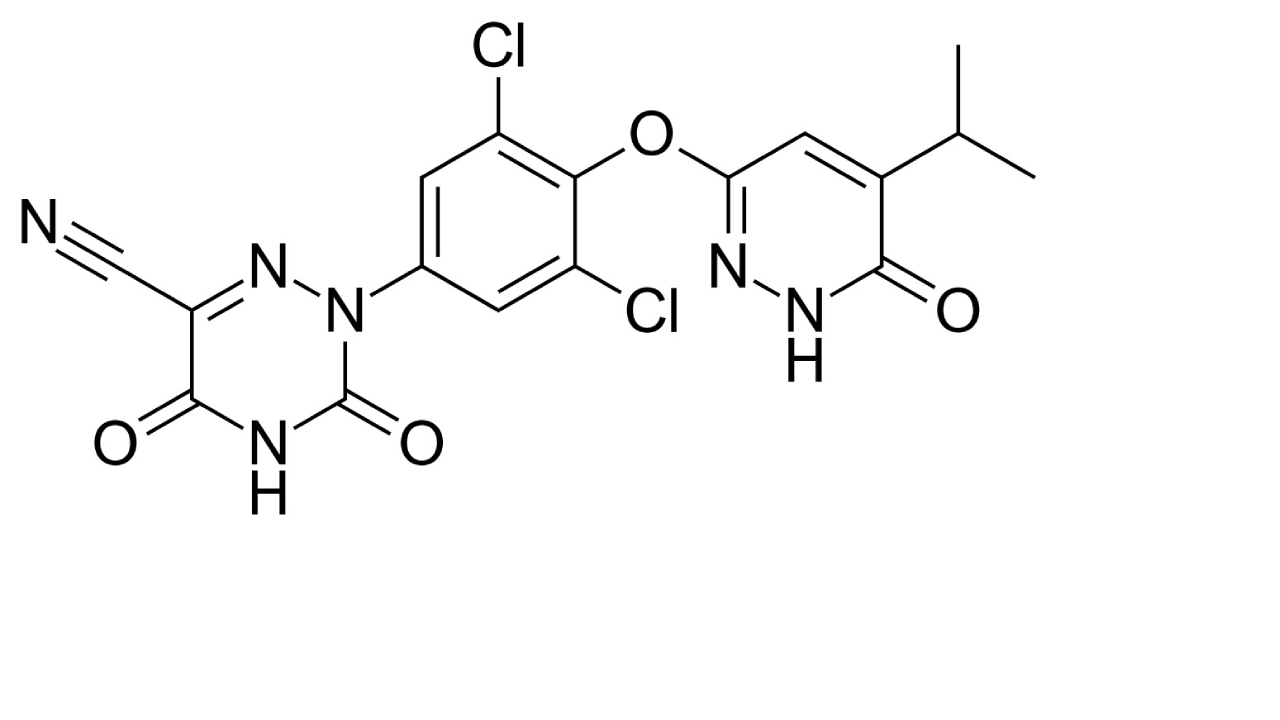 | Resmetirom is a FDA-approved drug used for treatment of non-cirrhotic MASH with moderate to advanced liver fibrosis [150]. |
| FGF17 mimetics | Aldafermin | 5-194 FGF19 (5-Methionine, 6-Arginine, 8-Serine, 9-Serine, 11-Leucine); calculated molecular weight of 21,300 Da. | Aldafermin is an engineered analog of the human hormone FGF19 that improves liver histology in patients with non-cirrhotic MASH and patients with compensated MASH cirrhosis [151]. |
| FGF21 mimetics | Pegbelfermin (BMS-986036) | (109-(4-(1-((2-((ω-Methoxypoly(oxyethylene))formamido)ethoxy)imino)ethyl)-L-Phenylalanine))homo sapiens FGF21; calculated average molecular of 50,000 Da. | Pegbelfermin is a polyethylene glycol-conjugated analog of human FGF21 that impacts energy metabolism and reduces hepatic fat fraction in patients with MASH [152, 153]. |
| SCD1 inhibition | Aramchol | 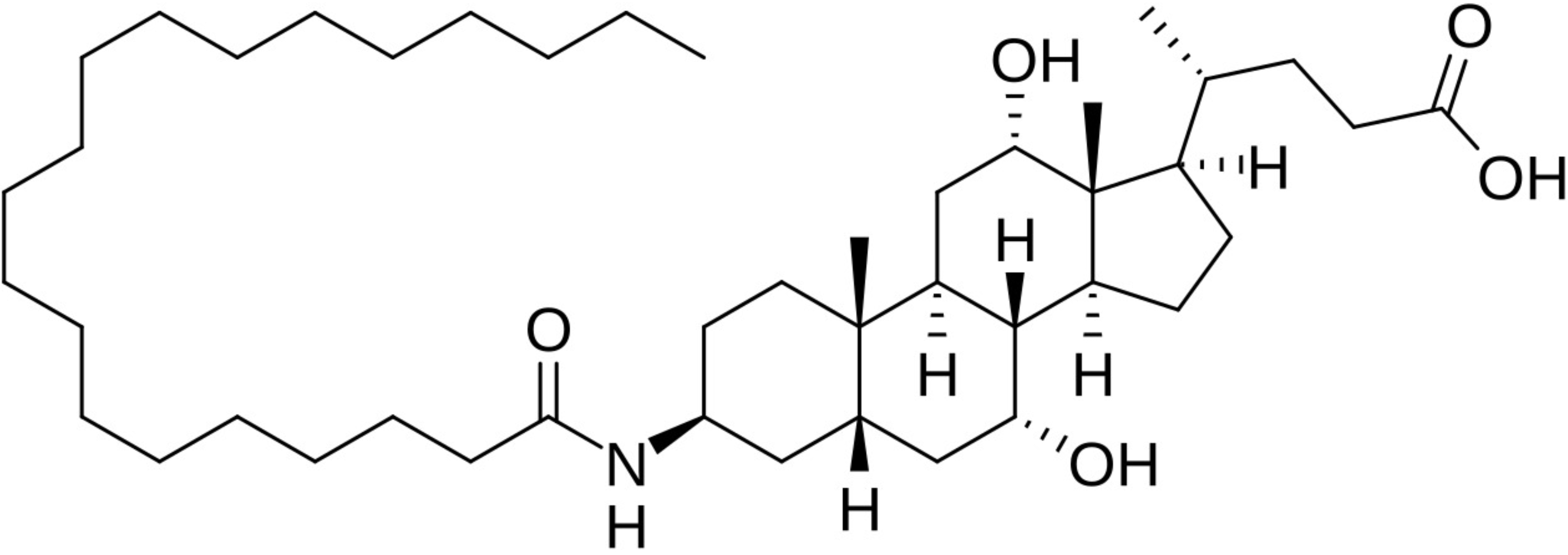 | Aramchol is a conjugate of cholic acid and arachidic acid that can be orally administered for treatment of MASLD/MASH. It affects liver fat metabolism and reduces liver fat content by inhibiting the activity of SCD1 in the liver [154]. |
| CCR2/CCR5 inhibition | Cenicriviroc | 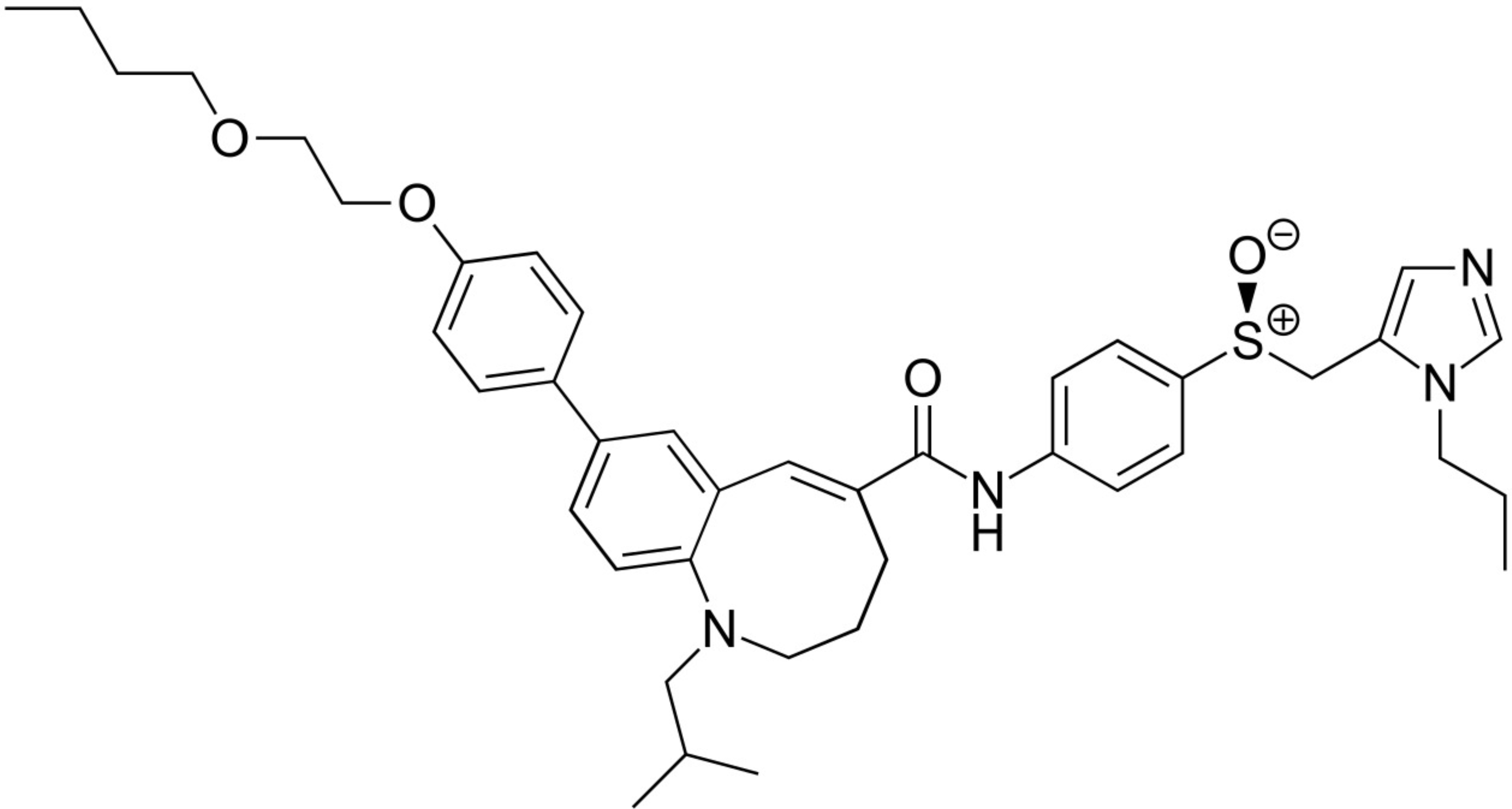 | Initially developed for the treatment of HIV infections, this oral dual CCR2 and CCR5 antagonist reversed liver fibrosis in patients with MASH [155]. However, novel data relativize this finding in MASH [156]. |
ACC: acetyl-CoA carboxylase; CCR: chemokine CC motif receptor; FGF: fibroblast growth factor; FXR: farnesoid X-activated receptor; PPAR: peroxisome proliferator-activated receptor; SCD1: stearoyl-CoA desaturase 1; THRβ: thyroid hormone receptor-β
Moreover, the emerging focus on the gut-liver axis has prompted research into probiotics, prebiotics, and drugs aimed at restoring a healthy microbiome [157]. In selected cases, bariatric surgery may be considered to reduce body weight and improve liver histology [22]. However, larger-scale studies are needed to elucidate the long-term effects of these interventions on cirrhosis and complications such as HCC. In addition to refining therapeutic modalities, further research into genetic and epigenetic factors will be crucial for risk stratification and personalized treatment approaches. As the prevalence of obesity continues to rise, effective disease prevention, diagnosis, and management strategies are becoming increasingly urgent for healthcare systems worldwide.
Moving forward, a comprehensive research agenda should prioritize:
Development of precise phenotyping strategies incorporating metabolic markers, imaging technologies, and genomics to accurately classify obesity subtypes.
Conducting prospective studies to clarify the role of the gut-liver axis and identify optimal interventions to reverse gut dysbiosis.
Investigating the long-term effects of new pharmacotherapies and surgical approaches on the natural history of liver disease, including the risk of cirrhosis and hepatic and extra-hepatic malignancy.
Exploring patient-centered outcomes and cost-effective treatment models that can be incorporated into guidelines and real-world clinical practice.
Strengthening patient education initiatives and public health policies that address dietary habits, physical activity, and wider socio-economic determinants of obesity based on current scientific evidence, rather than unproven assumptions that blame individual responsibility for the development of obesity [9].
Offer subjects with “clinical obesity” prompt, evidence-based treatment, aiming to induce improvement (or remission, whenever possible) of clinical signs and symptoms of obesity, thereby halting the progression to target-organs impairment and failure [9].
For the implementation of these goals, several activities are required that must be addressed by patients, clinicians, researchers, and food companies (Table 4). Implementing these priorities will refine the classification of obesity, clarify the role of the gut-liver axis in disease progression, and support the safe long-term application of emerging therapies and surgical approaches. In parallel, patient education, cost-effective treatment models, and public health policies that address socio-economic determinants will foster more effective and equitable strategies for obesity prevention and care worldwide.
Key research and implementation priorities for obesity-related liver disease
| Implementation priority | Action required |
|---|---|
| Refining obesity phenotyping | There is a clear need for robust classification methods that integrate metabolic, anthropometric, and genomic information to better distinguish obesity subtypes and predict liver disease risk. The incorporation of advanced imaging techniques, such as MRI-based proton density fat fraction (PDFF) measurement, may further refine these stratifications. |
| Clarifying the gut-liver axis | Research is needed to understand how dysbiosis, increased intestinal permeability, and altered bile acid signaling contribute to liver inflammation and fibrosis. Future investigations should explore whether interventions like probiotics, prebiotics, and fecal microbiota transplantation can confer durable benefits. |
| Long-term evaluations of emerging therapies | Numerous drugs targeting pathways from insulin resistance (IR) to fibrogenesis are under development. Longitudinal studies examining how these agents influence outcomes, especially cirrhosis and hepatocellular carcinoma, are vital, as are evaluations of combination therapies that may yield synergy without excessive toxicity. |
| Cost-effective, patient-centered models of care | Integrating novel diagnostics and treatments into clinical practice must be balanced against cost considerations. Research is necessary to explore how best to incorporate telemedicine, digital health tools, and multidisciplinary teams in ways that optimize patient adherence and outcomes while minimizing healthcare expenditures. |
| Preventive and public health strategies | Effective management of obesity-related liver disease also requires upstream interventions that address environmental and socio-economic factors underpinning obesity itself. Enhanced public policies, improved nutritional labeling, and community-based programs have the potential to reduce the overall disease burden if implemented broadly and supported through sustained funding. |
A wide range of options is available to treat obesity, including lifestyle changes, pharmacological treatments, endoscopic procedures, and surgical techniques. One initial question is whether all individuals with obesity should be treated, including those with MHO. It is important to consider that individuals with MHO are still at a higher risk of developing health issues, so they should be encouraged to maintain or adopt a healthy lifestyle to avoid the negative effects of obesity [158]. Supporting this intervention, weight loss has been shown to trigger anti-inflammatory responses in individuals with MHO, highlighting the significance of weight reduction in this group [159]. Additionally, individuals with “clinical obesity”, as previously defined, should always receive prompt treatment to manage the clinical symptoms of obesity and prevent the progression of organ damage to end-stage complications [9].
A systematic review with meta-analysis of 15 RCTs or cohort studies has shown that adopting a low-calorie diet (LCD) is effective in reducing body weight [–9.1 kg vs. the controls (95% CI: –12.4 to –5.8)] but not serum ALT (–5.9 IU/L, 95% CI: –13.9 to 2.0). Total Dietary Replacement reduced intrahepatic fat content (IFC) by –9.1% vs. the control (95% CI: –15.6% to –2.6%). The Mediterranean-LCD for ≥ 12 months reduced ALT (–4.1 IU/L, 95% CI: –7.6 to –0.5) and for 24 months reduced liver stiffness versus other LCDs. The Green-Mediterranean-LCD reduced intrahepatic lipid content, irrespective of body weight [160]. Therefore, additional RCTs should be conducted, using standardized methods to determine IFC and liver fibrosis among those of non-Caucasian ethnicity.
In the context of sex-specific approaches, it is also worth noting that all-cause mortality in MUO cohorts is associated with different macronutrient consumption subtypes, their quality, meal timing, and sex-specific effects between men and women [161]. Studies suggest that high phenolic extra virgin olive oil effectively contributes to the prevention and therapeutic dietary approach to cardiometabolic diseases among those with MUO [162]. Finally, improvement of intrinsic motivation should be pursued [163]. Collectively, these findings strongly support precision medicine approaches in the implementation of diets among people with obesity.
A systematic review and meta-analysis of 10 randomized trials totaling 737 nonalcoholic fatty liver disease (NAFLD) adults [164] has demonstrated that Mediterranean diet was associated with a decreased liver stiffness (kPa) by –0.42 (95% CI: –0.92 to 0.09) (P = 0.10) and significantly reduced total cholesterol by –0.46 mg/dL (95% CI: –0.55 to –0.38) (P = 0.001) without any documented effects on liver enzymes and WC. According to the European MASLD guidelines, all MASLD individuals should follow a Mediterranean Diet, minimizing processed meat, ultra-processed foods, and sweetened drinks and increasing the proportion of either unprocessed or minimally processed food [165]. However, the guidelines also pinpoint that more longitudinal investigation is necessary to define the long-term efficacy of weight loss obtained with diet therapy on clinical liver-related outcomes and liver-related mortality.
A meta-analysis of 116 RCTs totaling 6,880 adults with overweight or obesity has shown that engaging in 30 minutes of aerobic exercise weekly was associated with modest reductions in body weight, WC, and body fat measures. Additionally, aerobic training exceeding 150 minutes weekly at moderate intensity or greater is needed to achieve clinically meaningful reductions in body weight [166].
It is important to note that the benefits of cardiometabolic health in adults with overweight or obesity, even in the presence of comorbid conditions, go beyond the effects on BMI. A systematic review of 54 published articles has demonstrated that exercise was associated with improved insulin sensitivity HOMA-IR [standardized mean difference (SMD) = –0.34 (95% CI: –0.49 to –0.18), P < 0.0001], particularly among patients with T2DM [SMD = –0.50 (95% CI: –0.83 to –0.17), P = 0.003]. Moreover, exercise was also associated with significantly reduced IFC [SMD = –0.59 (95% CI: –0.78 to –0.41), P < 0.00001, I2 = 0%], with a larger effect size after high-intensity interval training [167]. Long-term improvement in Cardiorespiratory Fitness is a desirable objective to improve insulin sensitivity among men with visceral obesity [168].
According to the European guidelines [168], physical activity and exercise of more than 50 minutes per week of moderate intensity or 75 minutes per week of vigorous intensity are strongly recommended. This should be adapted according to the individual’s preference and capacity to reduce steatosis in adults with MASLD. However, the guidelines also emphasize with strong consensus that, despite the well-documented cardiometabolic benefits, the benefits of physical activity and exercise on liver histology and liver outcomes are less well documented.
Successful weight loss requires more aggressive interventions than minimal care (MC) and usual care (UC), and this can be achieved and accomplished through behavioral therapy (BT) and cognitive BT (CBT). A recent systematic review with pairwise meta-analysis and network meta-analysis [169] found that, compared to no treatment as a common comparator, CBT was the most effective intervention for achieving body weight loss, followed by BT, UC, and MC. Similar findings have been reported in another meta-analytic review conducted in patients with obesity and comorbid T2DM [170]. Additionally, a pilot study conducted in 85 MASLD individuals with visceral obesity showed that a 6-month course of CBT led to a significant decrease in the atherogenic coefficient and arterial hypertension [171].
Fecal microbiota transplantation (FMT) involves transferring fecal matter from a donor into the intestinal tract of a recipient with the aim of modifying the recipient’s gut microbiota to provide health benefits [172]. A systematic review and meta-analysis of studies on individuals with obesity and related metabolic issues demonstrated significant improvements in body weight (WMD = –4.77, 95% CI: –7.40 to –2.14), BMI values (WMD = –1.59, 95% CI: –2.21 to –0.97), HOMA-IR (WMD = –0.79, 95% CI: –1.57 to –0.00), and HbA1c (WMD = –0.65, 95% CI: –0.75 to –0.55) after FMT treatment compared to pre-FMT baseline values [173]. The success of FMT depends on the microbial diversity and composition of the donor feces [174], underscoring the necessity for further research to identify the characteristics of an ideal donor.
While data from published studies are promising, additional investigation is necessary before FMT is utilized for CLD. The effectiveness and safety of microbiome-based treatments must be evaluated through rigorous pharmacological studies and larger RCTs in individuals with MASLD [165]. Key areas requesting further research include the optimal FMT regimen, documenting efficacy and safety with large RCTs, and gaining pathophysiological insights into the reasons underlying failure of FMT in the individual patient [175].
Confronted with the often-disappointing results of lifestyle changes alone, pharmacological treatment of obesity remains one of the key options. Recent advances have revolutionized present treatment schedules and promise even more substantial innovations soon [176]. According to guidelines, those eligible for drug treatment of obesity are subjects who still have a BMI ≥ 30 kg/m2 (or ≥ 27 kg/m2 with an obesity-related comorbidity) despite attempting lifestyle changes [177]. While analysis of rare obesity syndromes with metreleptin and setmelanotide is beyond the scope of this review, the principal drugs approved for chronic use in non-syndromic obesity are orlistat, phentermine/topiramate, naltrexone/bupropion, liraglutide, semaglutide, and tirzepatide [177].
Semaglutide is one of the most studied and widely used anti-obesity medications. It is available as a weekly subcutaneous injection or a daily oral medication and has been shown to effectively promote an average weight loss of 11.62 kg and reduce WC by up to 9.4 cm compared to placebo [178]. In addition, semaglutide has been found to reduce blood pressure, fasting blood glucose, C-reactive protein, and improve lipid profiles, offering cardiovascular benefits to patients with established atherosclerotic CVD. It also reduces the risk of renal outcomes and cardiovascular-related mortality and may improve metabolic dysfunction in some patients [178]. The use of semaglutide has been associated with improved mental health and quality of life. However, weight regain often occurs after discontinuing semaglutide, necessitating long-term maintenance strategies [178]. Some potential side effects of semaglutide include mild-to-moderate gastrointestinal complaints and the risk of hypoglycemia [178].
Meta-analytic evidence from three RCTs involving 458 individuals supports the effectiveness and tolerability of semaglutide as treatment for MASLD. Zhu et al. [179] have demonstrated that semaglutide treatment increases the likelihood of resolving MASH (OR: 3.18, 95% CI: 1.70 to 5.95; P < 0.001), reduces steatosis (OR: 2.83, 95% CI: 1.19 to 6.71; P = 0.03), lobular inflammation (OR: 1.81, 95% CI: 1.11 to 2.96; P = 0.02), and hepatocellular ballooning (OR: 2.92, 95% CI: 1.83 to 4.65; P < 0.001). However, it does not significantly impact fibrosis stage (OR: 0.71, 95% CI: 0.15 to 3.41; P = 0.67). Nevertheless, imaging techniques revealed that liver stiffness (SMD: –0.48, 95% CI: –0.86 to –0.11; P = 0.01) and steatosis [mean difference (MD): –4.96%, 95% CI: –9.92 to 0.01; P = 0.05] were reduced. Moreover, it also significantly reduces transaminases, HbA1c, and body weight [179]. Future studies should investigate the effects of this drug on fibrosis regression in individuals with MASLD.
Tirzepatide is the most recently approved anti-obesity drug. Meta-analytic evidence from 12 RCTs totaling 11,758 patients demonstrates that it effectively reduces BMI, WC, and body weight, albeit with some gastrointestinal side effects that require vigilance [180]. A published study of two cases of Alström syndrome, a rare obesity syndrome, offers promising perspectives on the reduction of liver fat content (LFC) assessed noninvasively [181]. Importantly, Loomba et al. [182] in their industry-funded, phase 2, dose-finding RCT have shown that a 52-week treatment with tirzepatide was more effective than placebo as regards MASH resolution without worsening of fibrosis. The safety profile of tirzepatide was encouraging since the most common adverse events in treated subjects were mild-to-moderate gastrointestinal complaints. This pioneering study calls for more extensive, independent assessment of tirzepatide in MASH.
In conclusion, the best choice of anti-obesity medication should ideally be tailored to the patient’s clinical and biochemical profile, co-morbidities, drug contra-indications and side effects, as well as the expected weight loss and improvements in hepato-cardio-nephro-metabolic risk profile [176]. Further research is needed in the field of precision medicine approaches for individuals living with obesity.
Traditionally, certain endoscopic techniques, such as intragastric balloon (IGB), have been considered as a bridge to bariatric surgery for individuals at high surgical risk. Interestingly, a recent meta-analysis of 19 published studies totaling 911 patients has shown that IGB may play an important role in addressing the treatment gap in NAFLD management. This is achieved by improving not only body weight, BMI, glycated hemoglobin and HOMA-IR but also histological parameters including NAFLD activity score (NAS): MD: –3.0 (95% CI: –2.41 to –3.59), ALT: MD: –10.40 U/L (95% CI: –7.31 to –13.49), liver volume: MD: –397.9 (95% CI: –212.78 to 1,008.58), and liver steatosis: MD: –37.76 dB/m (95% CI: –21.59 to –53.92) [183].
More recently, endoscopic duodenal mucosa hydrothermal ablation of the superficial duodenal mucosal layers, followed by resurfacing of the mucosal interface, shows promise in resetting and correcting any abnormal signaling from the duodenal mucosa. This procedure eventually results in improved pancreatic endocrine function and glucose tolerance due to restored normal mucosal surface [184]. The procedure is safe and well-tolerated and has demonstrated metabolic benefits and reduced LFC primarily in diabetic patients. Its effectiveness in non-diabetic obesity remains under evaluation.
According to the European Guidelines [165], metabolic/bariatric endoscopic procedures need more validation as therapy for MASH and are not currently recommended for treating MASH.
Adults with a BMI ≥ 40 kg/m2 (or ≥ 35 with obesity-related comorbidities) are candidates for bariatric and metabolic surgery [185]. These surgeries restrict food intake, reduce nutrient absorption, or alter gut hormones to achieve weight loss and improve metabolic health through surgical manipulation of the proximal GI tract [186].
A meta-analytic review of thirty published studies totaling 3,134 patients has shown that bariatric surgery significantly reduced BMI (ratio of means, 0.79), induced a 72% reduction of LFC (assessed with MRI-PDFF) a reduction of NAS by 60% at 3–6 months and by 50% at 36–60 months; a reduction of the fraction of patients with steatosis; a decrease of lobular inflammation, ballooning degeneration and significant fibrosis (≥ F2) [187]. Of interest, Roux-en-Y gastric bypass induces the reversal of metabolic dysfunction among individuals with MUO [188].
Finally, a somewhat neglected topic of great clinical and scientific significance is the outcome of cirrhotic individuals with portal hypertension and severe obesity [189]. Additional studies are therefore required to provide evidence-based recommendations.
The European guidelines recommend considering bariatric surgery in adult individuals who have non-cirrhotic MASLD and an approved indication, as these surgeries can have long-term beneficial hepatic effects in addition to inducing T2DM remission and improving the profile of cardiometabolic risk [165]. Similarly, bariatric surgery can also be considered for adults with MASLD-related compensated advanced CLD/compensated cirrhosis who have an approved indication. However, a careful assessment of indications, surgical technique, and the presence of clinically significant portal hypertension should be conducted by an expert multidisciplinary team [165].
In recent decades, the global increase in obesity has led to a sharp rise in the prevalence of obesity-related liver disease. This review has provided insights into the definitions, the relative burden, and multiple pathways involved in the pathobiology of liver injury in individuals with obesity. The interplay between metabolic changes, inflammatory cascades, and adipose tissue dysfunction promotes a spectrum of liver abnormalities ranging from simple steatosis to end-stage cirrhosis and HCC. Effective management requires a multifaceted approach including lifestyle interventions, pharmacological regimens, and, in selected cases, endoscopic or surgical modalities. BMI, the anthropometric measure most widely used to identify obesity, has major limitations [190].
Lifestyle-based approaches, as well as BMI-centric approaches (which involve encouraging individuals to eat less and move more), are not sustainable for long-term weight loss and maintenance for most individuals. This outdated approach may have caused more harm than good [190]. Therefore, the focus should be on actionable lifestyle-related risk factors that can improve the overall quality of diet while increasing levels of physical activity among the general population [190]. Intensive lifestyle changes and anti-obesity interventions aim to encourage individuals to adopt healthy eating and physical activity behaviors. Although they are initially effective, lifestyle changes are hard to maintain in the long term, especially when faced with environmental factors that promote unhealthy behaviors [191]. To ensure the long-term success of lifestyle modifications, individual responsibility must be complemented by collective and institutional responsibility to create a healthier food and activity environment for the general population, not just those living with obesity [191].
Despite remarkable advances in understanding the pathophysiology, there are still gaps remaining regarding genetics, early disease biomarkers, subtle mechanisms that sustain disease development and progression, and the influence of discrete obesity phenotypes on hepatic outcomes. Candidate biomarkers, whether genetic, epigenetic, or proteomic, need further validation in large, diverse cohorts to refine risk stratification and optimize patient selection for intervention. Additionally, the dynamic nature of obesity requires longitudinal studies that capture the interplay between weight fluctuations, ectopic fat storage, and subclinical inflammation. In a rapidly changing therapeutic landscape, pharmacological interventions must transcend weight reduction and target specific pathomechanisms such as hepatic inflammation, IR, and fibrogenesis. Research into novel drugs that modulate bile acid metabolism, gut microbiota composition, and immune signaling pathways is promising. Individualizing therapy based on metabolic phenotypes and genetic risk scores may ultimately lead to better clinical outcomes and fewer adverse effects. Moreover, the question of which patients benefit most from bariatric surgery or endoscopic interventions remains incompletely answered, highlighting the need for comparative effectiveness research.
In conclusion, obesity-related liver injury represents a significant and evolving public health challenge that requires a coordinated, evidence-based response. Ongoing work to refine the classification of obesity phenotypes, standardize diagnostic algorithms, and develop targeted therapies will be critical in reducing the burden of liver disease. By integrating molecular insights, advanced imaging, and translational research, we can pave the way for improved prevention and treatment strategies, ultimately contributing to better liver health worldwide. Moreover, the use of big data analytics in combination with multicenter registries can enable the identification of patient subgroups most likely to respond to specific interventions, including lifestyle programs, medical therapies, or surgical procedures. Integrative science, bringing together clinicians, basic researchers, and public health experts, will be crucial in developing a coherent, holistic framework to address, with a precision medicine approach, the rising tide of obesity-related liver disease on a global scale.
AHR: adjusted hazard ratio
ALD: alcohol-related liver disease
BAT: brown adipose tissue
BMI: body mass index
BT: behavioral therapy
CBT: cognitive behavioral therapy
CLD: chronic liver disease
CVD: cardiovascular disease
DNL: de novo lipogenesis
ECM: extracellular matrix
ER: endoplasmic reticulum
FFAs: free fatty acids
FMT: fecal microbiota transplantation
HBV: hepatitis B virus
HCC: hepatocellular carcinoma
HCV: hepatitis C virus
HSCs: hepatic stellate cells
IFC: intrahepatic fat content
IGB: intragastric balloon
IR: insulin resistance
KC: Kupffer cell
LCD: low-calorie diet
LFC: liver fat content
MASH: metabolic dysfunction-associated steatohepatitis
MASLD: metabolic dysfunction-associated steatotic liver disease
MC: minimal care
MD: mean difference
MetS: metabolic syndrome
MHO: metabolically healthy obesity
MUO: metabolically unhealthy obesity
NAFLD: nonalcoholic fatty liver disease
NAS: nonalcoholic fatty liver disease activity score
NPY: neuropeptide Y
OR: odds ratio
OS: oxidative stress
RCT: randomized controlled trial
ROS: reactive oxygen species
SMD: standardized mean difference
T2DM: type 2 diabetes mellitus
TNF-α: tumor necrosis factor-alpha
UC: usual care
UPR: unfolded protein response
VAT: visceral adipose tissue
WC: waist circumference
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/1001334_sup_1.pdf.
The graphical abstract was generated using Servier Medical Art, provided by Servier. The authors would like to express their gratitude to Sabine Weiskirchen from the University Hospital Aachen for preparing Figure 2 of this review. Additionally, we would like to extend our sincere thanks to the World Obesity Federation for assisting and granting permission to publish Figure 1B and the Supplementary material.
AI and AI-assisted Technologies: The authors of this article used the Large Language Model RWTHgpt by RWTH Aachen University exclusively for minor translations and grammatical corrections in this work. All sentences revised by RWTHgpt were reviewed and verified by the authors.
AL and RW: Conceptualization, Writing—original draft, Writing—review & editing. Both authors read and approved the submitted version.
Amedeo Lonardo who is the Associate Editor of Exploration of Medicine had no involvement in the decision-making or the review process of this manuscript. Another author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

