 Case Report
Case Report

Affiliation:
1Faculty of General Medicine, Yerevan State Medical University after Mkhitar Heratsi, Yerevan 0025, Armenia
Email: Fadialtamimi20@gmail.com
ORCID: https://orcid.org/0009-0003-4286-072X
Affiliation:
1Faculty of General Medicine, Yerevan State Medical University after Mkhitar Heratsi, Yerevan 0025, Armenia
ORCID: https://orcid.org/0009-0003-7907-1089
Affiliation:
2Department of Pathophysiology, Yerevan State Medical University after Mkhitar Heratsi, Yerevan 0025, Armenia
ORCID: https://orcid.org/0000-0002-2748-3802
Affiliation:
3Department of Rheumatology, Yerevan State Medical University after Mkhitar Heratsi, Yerevan 0025, Armenia
ORCID: https://orcid.org/0000-0002-8241-8501
Affiliation:
4Faculty of Medicine, The Hashemite University, Zarqa 13133, Jordan
ORCID: https://orcid.org/0000-0001-6855-3718
Explor Med. 2025;6:1001313 DOI: https://doi.org/10.37349/emed.2025.1001313
Received: January 03, 2025 Accepted: March 31, 2025 Published: April 27, 2025
Academic Editor: George N. Goulielmos, University of Crete, Greece
We report a rare case of a female patient with multiple rheumatological conditions. The patient initially presented with periodic, diffuse abdominal pain. This complaint was not fully investigated because polyarthritic symptoms became the predominant ones. This led to the diagnosis of rheumatoid arthritis. Afterward, the patient complained of xerostomia, xerophthalmia, and diffuse rash. After investigations, she was diagnosed with Sjögren’s syndrome. Suspecting a case of methotrexate-induced vasculitis, her initial prescription was changed to azathioprine and then to etanercept. Eventually, her persistent abdominal pain, combined with her Armenian origin, prompted her physician to order a genetic analysis of the MEFV gene, which revealed the V726A/P369S mutation, giving rise to the diagnosis of Familial Mediterranean Fever. In her routine follow-up, the patient was in a stable condition, adherent to the medications, and showed improvement in her symptoms. Therefore, this case shows the importance of early genetic testing in similar cases, which in turn will allow timely diagnosis and treatment.
Deviation of the immune activity from the normal response and the predominance of exaggerated inflammation is the cornerstone of auto-immune and auto-inflammatory diseases. Even though these two types of diseases differ from each other regarding their etiology and pathogenesis, they have overlapping clinical features and treatment methods [1]. Familial Mediterranean Fever (FMF), being an auto-inflammatory disease, is manifested by recurrent attacks of fever and symptoms of serositis [2]. The etiology of FMF can be traced to mutations in the Familial Mediterranean gene (MEFV) located on the short arm of chromosome number 16. It is possible to find cases of the coexistence of FMF with seronegative spondyloarthritis [3], rheumatoid arthritis (RA) [4], or Sjögren’s syndrome [5], however, it is difficult to find publications about the coexistence of FMF with more than one rheumatological condition [6].
A female patient in her early 40s was admitted to the rheumatology department of our university hospital in 2013 with a 3-month history of symmetrical joint pain involving the proximal interphalangeal joints, the metacarpophalangeal joints, the wrist, and the small joints of the feet. The pain in the proximal interphalangeal joint progressed into symmetrical swelling, accompanied by morning stiffness lasting more than an hour. Initial investigations showed a high-positive rheumatoid factor. Consequently, the patient was diagnosed with RA according to the 2010 ACR/EULAR classification criteria [7]. The patient was prescribed methylprednisolone 8 mg once a day, as well as a disease-modifying antirheumatic drug—methotrexate 20 mg once per week. During her regular follow-up appointments from 2013 to 2016, the clinical picture of the patient was under control, and the patient had not mentioned any complaints associated with the treatment plan.
In 2016, the patient visited another hospital complaining of a diffuse and well-expressed petechial rash on her legs, dryness in the mouth, sandy feeling in the eyes, bilateral parotid gland enlargement, and arthralgia involving the small joints of the hand. The latter was attributed to the patient’s incompliance with the prescribed medications. Laboratory investigations are summarized in Table 1. Due to financial limitations, the patient was unable to perform a biopsy or sialography of the enlarged parotid glands. Thus, a diagnosis of secondary Sjögren’s syndrome was established based on the American-European consensus criteria for Sjögren’s syndrome [8]. The presence of high-titer anti-cyclic citrullinated peptide [anti-CCP, (310.9 IU/mL)] antibodies confirms once again her initial diagnosis of RA. Considering the possibility of methotrexate-induced vasculitis, the physician recommended replacing methotrexate with azathioprine 50 mg. Additionally, the dosage for methylprednisolone was increased to 24 mg/day. However, during her routine investigations, the physicians noted an increase in the levels of liver transaminases. After ruling out the potential causes for this elevation, the physician considered it as a side effect of azathioprine, as a result of which azathioprine was replaced by etanercept 50 mg subcutaneous injection once a week. The patient was discharged from the hospital and was given a prescription plan, as mentioned in Figure 1. Her complaints were largely reduced, although periodic abdominal pain was mildly present. The patient refused further investigations that were aimed to understand the cause of this pain.
Results of the laboratory investigations performed upon admission to the hospital
| Marker | 2016 | 2024 |
|---|---|---|
| RF | 600 IU/mL | 173.8 IU/mL |
| ANA | 7.6 IU/mL | 5.244 IU/mL |
| Anti-SS-A | 387.5 IU/mL | > 200 IU/mL |
RF: rheumatoid factor; ANA: antinuclear antibodies; anti-SS-A: anti Sjögren’s syndrome related antigen A
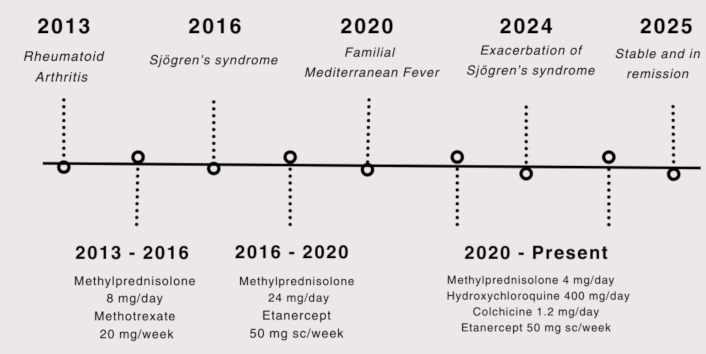
Timeline showing the patient’s diagnoses (above the horizontal line) and management plans (below the horizontal line)
In 2020, the patient visited our university hospital complaining of exacerbated abdominal pain and thoracic pain lasting 4 to 5 days. Even though these pains were not accompanied by fever, they were being repeated on average every 2 months. At this point, the patient mentioned that she had been complaining of mild periodic abdominal pain for more than 10 years but had never thought of investigating the etiology of this pain. Because of the character of the pain, her Armenian origin, and her family history of FMF diagnosis (maternal uncle), the physician ordered a genetic test for FMF. The latter revealed V726A/P369S compound heterozygous mutations in the MEFV gene, as well as α/β mutation of the serum amyloid A1 (SAA-1) gene. Thus, the patient was diagnosed with FMF based on these results, combined with the diagnostic criteria suggested by Livneh et al. [9].
In the first period following the diagnosis of FMF, the patient was in remission, with little to no complaints. However, in 2024, the patient was once again admitted to our university hospital. Her complaints were diffuse joint pain and exacerbation of symptoms of Sjögren’s syndrome. On physical examination, the patient had gained a Cushingoid appearance as a consequence of prolonged glucocorticoid usage, livedo reticularis was noted on her arms and legs, as well as an erythematous rash on the face and sun-exposed areas of the chest. The patient mentioned that, in general, she had poor compliance with the medications due to the side effects that had developed and because of the financial burden that these medications and frequent hospital visits had caused. The diagnosis and management plan are mentioned in Figure 1. After getting her condition stabilized, the patient was discharged from the hospital.
After being discharged, the patient continued visiting the hospital regularly for her follow-up appointments. She is in stable remission and has not developed any relapse. She adheres to the prescribed medications (mentioned in Figure 1) and is able to perform her daily activities without major limitations.
Based on the literature review, the coexistence of RA, Sjögren’s syndrome, and FMF has not been reported yet. Due to the similarity in their clinical manifestation, a physician may face diagnostic challenges and misdiagnose or have a late diagnosis of one of the diseases. Since the patient had already been diagnosed with two diseases, the FMF-associated symptoms might have been ignored or masked [10]. Consequently, the diagnosis of FMF at this age is not a case of late-onset but simply a late diagnosis.
The coexistence of FMF with more than one rheumatological condition has been reported by Dörtbaş et al. [6]. The authors describe the case of a 34-year-old female patient whose FMF diagnosis was confirmed by a genetic test revealing M694V and R202Q homozygous mutations in MEFV gene. She later developed Sjögren’s syndrome and ankylosing spondylitis.
RA is a chronic auto-immune condition targeting primarily the synovial membrane of the small joints of the hand, feet, and wrist. The exact etiology of RA is yet to be defined, however, genetics and environmental factors have their share in the development of this condition [11]. The diagnosis of RA in our patient was made according to the 2010 ACR/EULAR classification criteria for RA, with a total score of 8 (3 points for the involvement of 4–10 small joints, 3 points for having a high-positive rheumatoid factor (RF) value, 1 point for having abnormal ESR value and 1 point for the duration of symptoms being more than 6 weeks) [7].
Sjögren’s syndrome is another auto-immune condition characterized by xerostomia, xerophthalmia, enlarged parotid glands, pruritus, dry cough, symmetric arthritis, and esophageal mucus atrophy, among others [12]. The etiology and pathogenesis are unknown, though females have a higher risk than males [12]. For the diagnosis of Sjögren’s syndrome, the American-European Consensus Criteria for Sjögren’s syndrome was taken into consideration. According to these criteria, our patient fulfilled 4 (ocular signs and symptoms, oral symptoms, and autoantibodies) out of the 6 items mentioned in the American-European Consensus Criteria for Sjögren’s syndrome [8].
Genetic mutation in the MEFV gene is the contributing factor for the development of FMF, which is a hereditary autoinflammatory disease [2]. FMF usually presents with periodic attacks of fever, abdominal pain, and symptoms of serositis [2]. The MEFV is located on chromosome 16p13.3 and consists of 10 exons. Different mutations have been recorded, and numerous mutation combinations have been detected. A cohort study conducted by Kriegshäuser et al. [13] among Armenian patients with FMF reveals most of these mutations. According to the results, 12.02% of the patients had M694V heterozygous mutation (M694V/-), 20.22% of the patients had compound heterozygous mutation (M694V/V726A), and 11.12% had M694V homozygous mutation (M694V/M694V) [13]. Furthermore, five patients (0.05%) out of a total of 10,370 patients had the V726A/P369S compound heterozygous mutation, which is the same as our patient’s mutation. The type of mutation has an influence on the clinical manifestation of the disease. For example, M694V, M694I, M680I, and V726A mutations have been associated with a more severe course of the disease, where patients can develop amyloidosis [14]. On the other hand, E148Q and P369S mutations are associated with a milder course of the disease [14]. Genetic testing in our patient revealed the presence of a rare combination of V726A/P369S mutation. Thus the diagnosis of FMF was confirmed, in addition to the diagnostic criteria suggested by Livneh et al. [9] where our patient satisfied two of the major criteria (monoarthritis and incomplete abdominal attack) and two of the minor criteria (incomplete attack involving the joints and a favorable response upon initiation of 1.2 mg/day colchicine treatment).
The MEFV gene encodes the protein pyrin, which is part of the pyrin inflammasome that regulates the production of pro-inflammatory cytokines. This mutation will result in the hyperactivity of the pyrin inflammasome and recruit caspase-1. The latter, in turn, will cleave IL-1 into IL-1β, which is the most potent cytokine and is responsible for most FMF manifestations [2].
One of the potential links between FMF and RA is IL-1β. This cytokine can influence the activity of the NF-κB signaling pathway, increasing the production of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6, among others [15]. TNF-α, the prominent cytokine involved in the pathogenesis of RA, is another activator of the NF-κB pathway [11]. The continuous activation of the NF-κB pathway results in the formation of a vicious cycle, which constitutes the basis of chronic inflammation and contributes to the progression of the diseases [16]. IL-18, a pro-inflammatory cytokine, is greatly increased during the acute phase of FMF. The latest research suggests considering IL-18 as a biomarker to distinguish patients in remission from those in an acute phase of the disease [17]. Furthermore, in a research study led by Eriksson et al. [18], researchers have found a positive correlation between serum IL-18 and the development of RA and Sjögren’s syndrome. They hypothesize that IL-18 might be responsible for the increased amount of synthesized immunoglobulins observed in these conditions, as IL-18 can amplify the ongoing inflammation [18].
Though not proven definitively, FMF may, indirectly, lead to the development of auto-immune diseases through chronic inflammation and cytokine dysregulation, especially if the patient has genetic susceptibility to such diseases [19]. For example, an exaggerated innate immune response as in FMF can serve as a source of modified neoantigens that activate the adaptive immune system, bridging auto-inflammatory diseases with autoimmune aggression. One such example is neutrophil-derived intranuclear antigens after NETosis, seen in Systemic Lupus Erythematosus (SLE) (anti-dsDNA, anti-histone, anti-Smith antibodies). In SLE patients, even specific neutrophil populations (so-called low-density granulocytes) have been identified, which are more prone to undergo NETosis and are more actively producing cytokines than other neutrophils. The complex intracellular changes occurring during NETosis, including arginine citrullination in histones by peptidyl arginine deiminase 4 (PAD4), are linked to the formation of anti-cyclic citrullinated antibodies (anti-CCAs), which are known to be the most specific autoantibodies in RA [20, 21].
Indeed, auto-inflammatory and auto-immune diseases differ from each other from a classical pathophysiological point of view, but these two groups of diseases can share a similar clinical picture. Consequently, the presence of multiple conditions simultaneously can be a diagnostic challenge, especially for newly developing symptoms. Therefore, a detailed history and proper investigation of each symptom are crucial in revealing the conditions present in a patient. In cases similar to our patient, especially in the endemic regions of FMF, early genetic testing should be considered, as such rare cases of coexistence are possible.
Further research should focus on understanding whether or not these conditions share a common underlying mechanism whereby one can trigger the development of the other. This, in turn, will help conduct early investigations and prevent or delay the development of the other expected conditions as well as identify and diagnose a present condition without any delays.
FMF: Familial Mediterranean Fever
MEFV: Familial Mediterranean gene
RA: rheumatoid arthritis
SLE: Systemic Lupus Erythematosus
FA, RA: Conceptualization, Investigation, Writing—original draft. AS: Conceptualization, Writing—review & editing. KG: Investigation, Supervision, Writing—review & editing. YA: Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
According to the regulations of YSMU Ethics Committee, this study is exempt from ethical approval as a case report.
Informed consent to participate in the study was obtained from the participant.
Not required.
The datasets that support the findings of this study are available from the corresponding author upon reasonable request.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 4240
Download: 25
Times Cited: 0
