 Original Article
Original Article

Affiliation:
Griffith Institute for Drug Discovery, Griffith University, Brisbane 4111, Australia
Email: Jessica.smith@griffithuni.edu.au
Affiliation:
Griffith Institute for Drug Discovery, Griffith University, Brisbane 4111, Australia
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-7211-4651
Affiliation:
Griffith Institute for Drug Discovery, Griffith University, Brisbane 4111, Australia
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0001-8981-6652
Explor Med. 2023;4:695–708 DOI: https://doi.org/10.37349/emed.2023.00170
Received: March 31, 2023 Accepted: July 20, 2023 Published: October 25, 2023
Academic Editor: Hua Su, University of California, USA
Aim: Parkinson’s disease (PD) is a complex, chronic neurodegenerative disorder with predominately sporadic etiology. Intricate genetic-environmental interactions lead to the hallmarks of the disease: degeneration of dopaminergic neurons and the deposition of α-synuclein aggregates. The aim of this study was to establish a novel primary patient cell model as an in vitro screen to study α-synuclein processing for drug screening.
Methods: Primary patient olfactory neuroepithelial-derived cells (ONS) were exposed to α-synuclein and examined for cytotoxicity, processing, and solubility over 48 h. Epigallocatechin gallate (EGCG), which is known to destabilise α-synuclein fibrils, was used to investigate the solubilisation of α-synuclein in the model system.
Results: Exposure to 0.1 μmol/L α-synuclein preformed fibrils was not toxic to ONS over 48 h. ONS processing of α-synuclein was observed to be different in PD cells by their increased accumulation in the cytoplasm. Processing deficits in the PD ONS were confirmed by immunoblotting with an increase in sodium dodecyl sulfate (SDS)-insoluble α-synuclein after 48 h.
Conclusions: The data has illustrated the utility of primary patient ONS as a model system to understand the processing of α-synuclein. Considerable differences in α-synuclein processing were identified in PD ONS. Furthermore, the data suggests that primary patient ONS are a viable in vitro drug screening platform for α-synuclein pathology in PD.
Parkinson’s disease (PD) is a common neurodegenerative disorder characterised by the progressive death of dopaminergic neurons in the substantia nigra pars compacta of the midbrain [1–3]. In post-mortem PD brains, these dopaminergic neurons commonly contain proteinaceous inclusions known as Lewy bodies (LBs) [2, 4, 5]. LBs are comprised of accumulated misfolded proteins, primarily α-synuclein and α-synuclein binding proteins. LBs also can contain an abundance of crowded membranous materials including lipids, fragmented vesicles, and lysosomes [6, 7].
α-synuclein was discovered as a causative factor in PD in 1997 when the SNCAA53T variant was associated with autosomal-dominant PD [8]. Duplications and triplications of the gene have also been reported in familial cases of PD, highlighting the importance of α-synuclein in PD pathogenesis [9]. α-synuclein has three structural regions: The N-terminal region (residues 1–60) contains repetitions of highly conserved lysine repeats [10, 11]. The hydrophobic region (residues 61–95) which is known as the non-amyloid β component (NAC) domain is required for amyloid formation and has the propensity to aggregate [12]. Finally, the acidic C-terminal region (residues 96–140) is believed to shield the NAC domain from aggregating spontaneously [13]. Although the primary function of α-synuclein remains elusive in literature, increasing evidence suggests α-synuclein plays a role in synaptic vesicle trafficking and neurotransmitter release by promoting assembly of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex [7, 14, 15].
The pathogenesis of α-synuclein provides direction for drug discovery, as the conformation of the protein and how it evades cellular degradation can alter drug screening techniques. α-synuclein is constantly secreted into the extracellular space and importantly can be taken up by neighbouring neuronal cells, which is an important mechanism for the toxic, prion-like spreading of the protein [16–20]. There are a variety of proposed mechanisms for α-synuclein internalisation, including clathrin-mediated endocytosis, pinocytosis, and cell surface protein-mediated uptake [16–20]. There are a variety of methods of screening α-synuclein inhibitors in cell models. α-synuclein overexpression in B103 neuroblastoma cells saw inhibition of aggregation by the treatment of statins [21]. A recent study utilised trehalose to enhance autolysosomal function in H4 neuroglioma cells and rat hippocampal neurons, demonstrating that indirectly targeting α-synuclein could still enhance clearance and inhibit the toxicity of the protein [22]. A study on SH-SY5Y cells that were pre-treated with α-synuclein oligomers for 40 h demonstrated that curcumin reduced the toxicity of oligomers via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) toxicity assay [23]. However, these cell lines do not have the intricate genetic background and susceptibility to neurodegeneration that a PD patient has and therefore do not provide a robust screening platform to target PD.
A cellular model to screen α-synuclein trafficking inhibitors does not yet exist without the manipulation of α-synuclein, nor has a robust model been developed in primary patient cells. Olfactory neuroepithelial-derived cells (ONS) are primary patient-derived cells from the olfactory mucosa and have PD-specific phenotypes [24–26]. Olfactory function is diminished in PD and is thought to be impacted by α-synuclein pathogenic propagation, making cells derived from olfactory epithelium an ideal candidate for investigating α-synuclein processing [27]. ONS display metabolic and molecular differences in idiopathic PD patient cells when compared with age-matched healthy control cells [24–26].
In the present study, the effect of toxic species of α-synuclein on ONS, which are naive to α-synuclein, was investigated by analysing its processing and solubility. Finally, the utility of ONS as a drug screening system has been investigated for α-synucleinopathies, particularly PD.
All donor tissue and information were obtained with informed and written consent of the participants. All procedures were in accordance with National Health and Medical Research Council Code of Practice for Human Experimentation and approved by the Griffith University Human Experimentation Ethics Committee.
Escherichia coli BL21 (DE3) cells containing pET-22b(+)-α-synuclein were generated previously [28]. Protein expression and purification were based on published protocols with minor modifications [29]. Briefly, isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma Aldrich) induced bacterial pellets were resuspended in resuspension buffer [50 mmol/L Tris (Sigma Aldrich), 10 mmol/L ethylenediaminetetraacetic acid (EDTA, Sigma Aldrich), 150 mmol/L NaCl (ChemSupply Australia), pH 8.0] and placed directly in a dry heat bath at 99℃ for 20 min. Samples were then centrifuged at 4,000 g for 30 min and supernatant was removed to fresh tubes. Next streptomycin sulphate (136 µL of 70 mmol/L solution per mL of supernatant, Sigma Aldrich) and 50% v/v glacial acetic acid (ChemSupply Australia) was added and centrifuged at 4,000 g for 15 min. The supernatant was collected and precipitated with an equal volume of saturated ammonium sulphate (Sigma Aldrich) and centrifuged at 4,000 g for 15 min. The pellet was resuspended in 25 mmol/L ammonium acetate (Sigma Aldrich) in 50% ethanol (ChemSupply Australia) and centrifuged at 4,000 g for 10 min. The final pellet was resuspended in 20 mmol/L Tris (pH 8.0, Sigma Aldrich) and sterile filtered with a 0.22 µm filter (Millipore). An anion exchange column Mono Q (GE Healthcare) was coupled with NGCTM fast protein liquid chromatography (Bio-Rad, NGC Discover 10 Chromatography System) to purify α-synuclein. The sample purity was verified by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Coomassie G-250 staining (Bio-Rad).
Fibrils were generated by incubating 100 μmol/L monomeric α-synuclein in a shaking thermomixer (Eppendorf ThermoMixer®) with 0.02% sodium azide (Sigma Aldrich) at 100 g 37℃ for 7 days. Water bath sonication (45 min, Unisonics Australia FXP10) was performed to prepare the preformed fibrils (PFFs). Fibril and sonicated PFF conformations were confirmed by SDS-PAGE, thioflavin T (ThT) assay, and transmission electron microscopy (TEM, JEOL JEM1011). Sparse fluorescent labelling of PFFs (PFF-488) was performed with an Alexa Fluor® 488 Conjugation Kit-Lightning-Link® (Abcam, #ab236553) as previously described [30].
Stock solution of ThT (1 mmol/L, Sigma Aldrich) was prepared in water and filtered through a 0.22 µm filter (Millipore). α-synuclein monomer, fibril, and sonicated PFFs (equivalent to 100 µmol/L) were incubated with 40 µmol/L ThT for 60 min shaking at 225 r/min, 37℃. ThT fluorescence was measured using SynergyTM 2 multi-mode microplate reader (excitation 450 nm, emission 485 nm, BioTek).
Five microlitres of monomer, fibril, and sonicated PFFs were loaded onto formvar-coated, carbon-stabilised copper grids (400 mesh, ProSciTech) for 5 min. Grids were blotted with filter paper and negatively stained with 1% uranyl acetate (ProSciTech) for 2 min. Grids were washed and blotted with filter paper and then examined using a JEM 1011 TEM (JEOL).
ONS derived from patients with PD are referred to as PD cells (2803, 2810, 2505, 2818, and 2820). Cells derived from healthy control subjects are referred to as control cells (1901, 1905, 1803, 1811, and 11401). Cell lines from patient and healthy control donors were established as previously described [24, 25]. Frozen aliquots of patient-derived and control-derived cells were thawed and cultured in Dulbecco’s modified eagle medium (DMEM)/F12 (ThermoFisher) supplemented with 10% fetal bovine serum (FBS, ThermoFisher) at 37℃, 5% CO2. All assays were performed with cells cultured for similar periods after nasal biopsy (under 15 passages from the initial plating). For analysis of internalisation, cells were pre-treated with dynog-4a (D4a, 25 µmol/L, Sigma Aldrich) or wortmannin (Wort, 1 nmol/L, Sigma Aldrich) for 30 min prior to addition of α-synuclein PFFs.
Cells were seeded and incubated in 6 well plates (NuncTM). After 48 h incubation with 0.1 μmol/L α-synuclein, cells were washed with phosphate buffered saline (PBS, Sigma Aldrich) and incubated with 200 μL lysis buffer [40 mmol/L Tris pH 7.5 (Sigma Aldrich), 150 mmol/L NaCl (ChemSupply Australia), 1 mmol/L EDTA (Sigma Aldrich), 1% SDS (Sigma Aldrich)] containing complete protease inhibitor cocktail (Roche Molecular Biochemicals) for 15 min. For Epigallocatechin gallate (EGCG) treatment, cells were treated with EGCG (+EGCG), 30 min prior to addition of 0.1 μmol/L α-synuclein. Samples were then sonicated (20% duty cycle for 20 sec, Branson 450 Sonifier®) and centrifuged at 14,000 g for 30 min to separate SDS-soluble (supernatant) and SDS-insoluble (pellet) fractions. Samples (equivalent proportions of SDS-soluble and -insoluble) were electrophoresed on discontinuous 12% polyacrylamide gels and proteins were transferred onto nitrocellulose membranes (GE Healthcare). Membranes were blocked in 5% non-fat dry milk (Coles) in PBS for 1 h at room temperature and then incubated with mouse anti-α-synuclein (1:3,000 dilution, BD Biosciences, #610786) and mouse anti-α-tubulin (1:3,000 dilution, Sigma Aldrich, #T5168) at 4℃ overnight. The membranes were washed five times with PBS containing 0.1% Tween 20 (PBS-T, Bio-Rad) and then incubated with secondary antibody (goat-anti-mouse-680, 1:10,000 dilution, LI-COR) for 60 min at room temperature. Membranes were washed five times in PBS-T and bands were observed on an Odyssey® XF imager system (LI-COR).
ONS were seeded in black-walled clear bottom 96 well plates (Greiner, #655087) and incubated with α-synuclein sonicated PFFs (0.1 μmol/L) for 48 h. Cells were washed with PBS and stained with 1 μg/mL propidium iodide (PI, ThermoFisher) and 50 ng/mL Hoechst (ThermoFisher) for 15 min. Plates were imaged immediately on an Operetta® CLSTM system (PerkinElmer) with a 20×/0.4 NA objective.
Cells were seeded onto 384 well plates (CellCarrier Ultra microplates, PerkinElmer, #6057302) and α-synuclein sonicated PFFs (0.1 μmol/L) were added at varying timepoints. At the end of the experiment, cells were washed once with PBS and fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 15 min at room temperature. Fixed cells were blocked and permeabilised in 1% bovine serum albumin (BSA, Sigma Aldrich), 0.1% Triton X-100 (ThermoFisher) for 60 min at room temperature and were then incubated with mouse anti-α-synuclein (1:500 dilution, BD Biosciences, #610786) for 90 min. Unbound antibody was removed by 3 washes in PBS and primary antibodies were detected using donkey-anti-mouse-Alexa FluorTM 555 (1:1,000 dilution, ThermoFisher). For F-actin staining cells were incubated with phalloidin-Atto 565 (1:2,000 dilution, Sigma Aldrich, #94072) for 15 min. Imaging was performed on an Operetta® CLSTM system (PerkinElmer) with either a 20×/0.4 NA or 40×/0.6 NA objective.
Cytotoxicity cell counts were measured using Harmony® software (version 4.9, PerkinElmer) whereby Hoechst-stained (Hoechst+) nuclei (all cells) and the number of PI-stained (PI+) nuclei (dead/dying cells) were quantified. Fiji (http://fiji.sc) was used to process immunofluorescence images and perform immunoblot densitometry analysis. PFF puncta counts were performed on 100 cells per cell line per treatment. F-actin staining was used to exclude cell boundaries to quantify intracellular puncta. Statistical analysis was performed using Prism (version 8, GraphPad Software). Statistical tests performed were either unpaired two-tailed t-test or one-way ANOVA with Sidak’s multiple comparisons. Significance levels were defined as follows, *: P < 0.05; **: P < 0.01; ***: P < 0.001.
In order to establish if the ONS were a feasible model for the trafficking of α-synuclein, it was first determined whether α-synuclein sonicated fibrils were cytotoxic, as ONS are naive to α-synuclein [26]. α-synuclein sonicated fibrils were generated and the species were charactertised by immunoblot, ThT assay, and TEM (Figure S1).
As the physiological concentration of α-synuclein at the neuronal synapse is equivalent to 0.1 μmol/L sonicated PFFs [31], this concentration was utilised throughout the experiments. To determine if 0.1 μmol/L α-synuclein sonicated PFFs were cytotoxic Hoechst/PI live cell staining analysis was performed. As PI can only enter cells with a compromised plasma membrane (apoptotic, necrotic, or compromised cells), the percentage of viable cells can be calculated as a fraction of the total number of the membrane-permeable Hoechst+ nuclei. Two control cell lines (1901 and 1905), and two PD lines (2803 and 2810) were initially selected, as full genetic analyses have previously been conducted on these lines (unpublished data, Sykes AM and Mellick GD). Cells were treated with or without 0.1 μmol/L α-synuclein sonicated PFFs for 48 h and cell viability was assessed. There was no significant difference in viability between untreated control cells (95% ± 2%) and PD cells (93% ± 1%). Furthermore, after incubation with α-synuclein, no significant change in viability in control cells (96% ± 2%) nor PD cells (94% ± 1%) was observed indicating that 0.1 μmol/L α-synuclein PFFs were not toxic to ONS over 48 h (Figure 1).
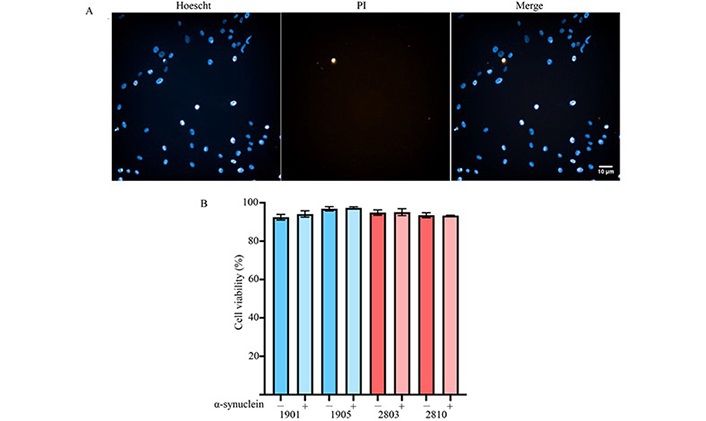
Cytotoxicity of α-synuclein sonicated PFFs on ONS. Control and PD cells were exposed to 0.1 μmol/L sonicated PFFs for 48 h. (A) Representative live imaging of ONS stained with Hoechst and PI; (B) the fraction of Hoechst+/PI+ cells was calculated and no significant difference was observed after PFF treatment. Data presented as mean ± SD, n = 3 independent experiments. –: without PFF treatment; +: with PFF treatment; SD: standard deviation. Scale bar = 10 μm
To investigate the mechanism of α-synuclein uptake in ONS, the endocytosis inhibitors D4a and Wort were next assessed. D4a is a potent dynamin inhibitor, dynamin is a guanosine triphosphatases (GTPase) enzyme that cleaves membrane-bound clathrin-coated vesicles for entry into the cell [32]. D4a inhibits all dynamin-mediated endocytosis.
Wort is a phosphatidylinositol 3-kinase (PI3K) inhibitor, where PI3K is involved in early endocytic activity including endosome fusion [33]. ONS were treated with D4a or Wort 30 min prior to the addition of 0.1 µmol/L PFF-488 for 4 h (Figure 2A–C). Internalisation of PFF-488 was significantly decreased in control cell lines by D4a (53% inhibition, P < 0.01) and Wort (52%, P < 0.01; Figure 2D). Uptake was also diminished in PD cell lines by D4a (50%, P < 0.001) and Wort (47%, P < 0.001; Figure 2D). Inhibition by Wort suggests that PFFs are entering the cell via endocytosis. Furthermore, the reduction of PFF uptake by D4a suggests that PFFs are partially endocytosed via dynamin-mediated endocytosis. Interestingly, there were significantly more PFF-positive puncta in PD cell lines when compared to controls (P < 0.05) indicating that PD cell lines internalise more α-synuclein (Figure 2D).
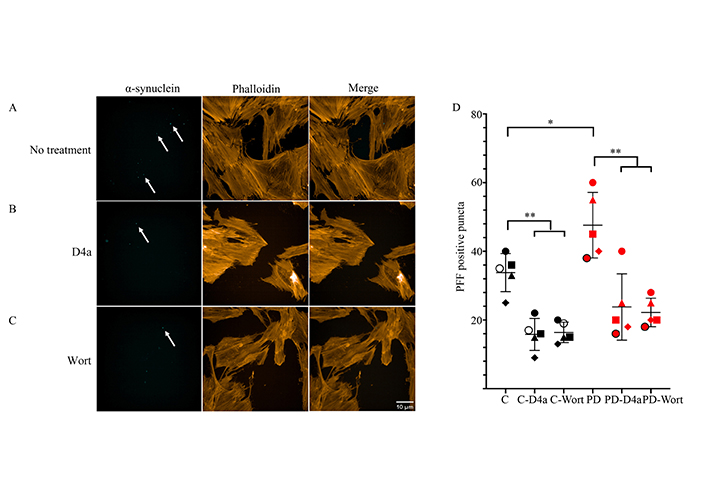
Endocytic uptake of α-synuclein PFFs is mediated by dynamin in ONS. ONS were pre-treated with 25 μmol/L D4a or 1 nmol/L Wort before incubation with 0.1 μmol/L α-synuclein PFF-488 for 4 h. (A) PFF-488 only; (B) D4a treatment; (C) Wort treatment; (D) scatterplot of intracellular PFF-488 puncta counts of 100 cells, n = 5 cell lines indicated by different shapes. Scale bar = 10 μm. Black/white shapes represent control ONS: circle = 1901, square = 1905, triangle = 1803, diamond = 1811, white circle with black border = 11401; Red shapes represent PD ONS: circle = 2803, square = 2810, triangle = 2505, diamond = 2818, circle with black border = 2820. Arrows to PFF-488-positive puncta. C: control. * P < 0.05; ** P < 0.01; *** P < 0.001
To further investigate the trafficking of α-synuclein in ONS, α-synuclein immunocytostaining analysis on ONS exposed to PFFs over 48 h was performed. Firstly, it was observed that α-synuclein PFFs were detectable in all ONS (Figure 3A–D). In agreement with the accumulation of cytoplasmic α-synuclein, all ONS had more detectable α-synuclein at 48 h, when compared to the 1 h and 12 h timepoints (Figure 3A–D). It was next investigated if there were any differences in α-synuclein accumulation between control (Figure 3A–B) and PD (Figure 3C–D) ONS. No significant differences in the relative fluorescent intensity of α-synuclein were detected between the control lines, nor between the PD lines (Figure 3E).
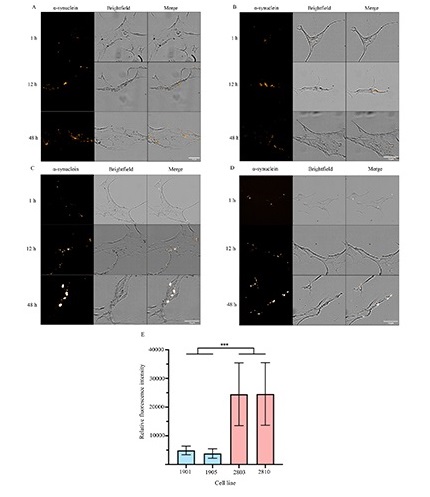
PD cells have impaired processing of α-synuclein and accumulation can be seen over time. ONS were incubated with 0.1 μmol/L α-synuclein for 1 h, 12 h, or 48 h and immunostained for α-synuclein. (A) 1901 cell line; (B) 1905 cell line; (C) 2803 cell line; (D) 2810 cell line; (E) quantification of the relative fluorescent intensity of α-synuclein per cell after 48 h. Scale bar = 10 μm. Data presented as mean ± SD, n = 20 cells. * P < 0.05; ** P < 0.01; *** P < 0.001
However, there was a significant increase in α-synuclein fluorescence intensity between control and PD cell lines. 1901 vs. 2803 (P < 0.0001), 1901 vs. 2810 (P < 0.0001), 1905 vs. 2803 (P < 0.0001), and 1905 vs. 2810 (P < 0.0001, Figure 3E). This data supports the α-synuclein processing differences between control and PD lines over 48 h in culture. Whilst α-synuclein positive puncta was observed intracellularly, there was also evident accumulation of α-synuclein on the cell boundaries, which could suggest evidence of pathogenic propagation of α-synuclein over time.
Genetic studies have revealed defects in endolysosomal processing are common amongst a subset of PD patients and α-synuclein toxicity can be a result of impaired trafficking [7, 34]. To further examine if there were processing deficits in PD ONS and to determine if the increased accumulation of α-synuclein over 48 h was pathogenic (Figure 3C–D). As a known characteristic of toxic α-synuclein is SDS-insolubility in PD brains and in vitro models of α-synuclein [31], the SDS-solubility of α-synuclein in ONS was investigated. To investigate the formation of SDS-insoluble α-synuclein species, ONS were treated with 0.1 µmol/L PFFs for 48 h, washed the cells twice in PBS to remove residual α-synuclein, lysed in 1% SDS, and sonicated to resolve all bands as a single 15 kDa band for analysis (Figure S2). The efficiency of SDS-solubility was first assessed by investigating α-tubulin, which is soluble in 1% SDS. As expected, a single, 50 kDa band was observed in the SDS-soluble fraction (Figure 4A) of all four cell lines and was absent in the SDS-insoluble fraction (Figure 4A). Furthermore, α-synuclein was also detected as an SDS-soluble, 15 kDa band in all ONS, indicative of non-toxic α-synuclein species (Figure 4A). Next, the SDS-insoluble fraction of the control cells was investigated and discovered that only a small proportion of the α-synuclein was SDS-insoluble (4.5% ± 1.1%, Figure 4B). Strikingly, there was significantly more SDS-insoluble α-synuclein (17.8% ± 3.1%, Figure 4B) detected in the PD ONS, likely to be toxic species of α-synuclein [29]. This phenotype was ONS-specific, as no SDS-insoluble species could be detected in SH-SY5Y cells, HEK293T cells, or HeLa cells (Figure S3). In support that there was increased α-synuclein in PD cells, there was significantly more total α-synuclein in PD cells in comparison to control cells (P < 0.05, Figure 4C).
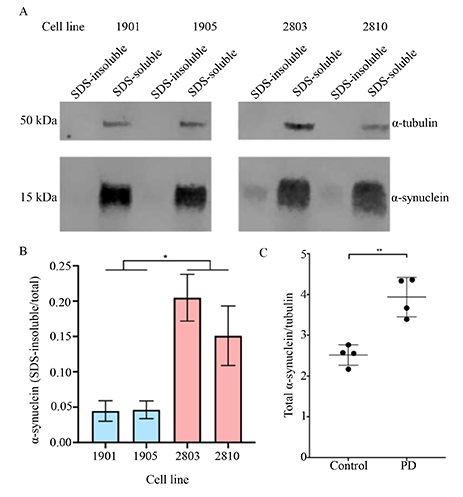
SDS-solubility of α-synuclein is decreased in PD ONS. ONS were incubated with 0.1 μmol/L α-synuclein sonicated fibrils for 48 h and its solubility in SDS was analysed. (A) Immunoblot SDS-solubility analysis of α-synuclein; (B) quantification of SDS-insoluble/total α-synuclein. Control lines had significantly less SDS-insoluble α-synuclein than PD ONS. Data presented as mean ± SD, n = 3 independent experiments; (C) total α-synuclein was calculated by adding SDS-insoluble fraction and SDS-insoluble fraction and normalised to tubulin. PD lines generated significantly more α-synuclein per cell. Data presented as mean ± SD, n = 4 independent cell lines. * P < 0.05; ** P < 0.01; *** P < 0.001
EGCG is a known small molecule inhibitor of α-synuclein PFFs [35, 36], therefore it was hypothesised that pre-treatment of the PD line 2803 with EGCG would result in a reduction of the α-synuclein SDS-insoluble species. To investigate the viability of ONS as an in vitro α-synuclein drug screening model, the PD line 2803 was treated with α-synuclein in the presence or absence of EGCG. As EGCG can remodel mature α-synuclein fibrils and prevent aggregation of new fibrillar structures at an equimolar ratio [35–37], cells were incubated with 0.1 μmol/L EGCG for 30 min prior to the addition of 0.1 μmol/L sonicated fibrils. As previously observed (Figure 4A), exposure to α-synuclein resulted in the generation of SDS-insoluble species in the absence of EGCG treatment (20.9% ± 3.0%, –EGCG, Figure 5). Treatment with EGCG significantly reduced the formation of the SDS-insoluble α-synuclein species as only 7.4% (± 0.02%) SDS-insoluble was detected (+EGCG, Figure 5) comparable to the control ONS (Figure 4). To further investigate the reduction of accumulated, insoluble α-synuclein in ONS by EGCG, α-synuclein immunocytostaining analysis was performed on ONS exposed to PFFs over 48 h, with or without 30 min pre-treatment of EGCG. There was no reduction in intracellular PFF puncta between –EGCG and +EGCG (Figure 5C). However, there was less accumulation of PFFs on the cell boundaries, likely to be pathogenic species of α-synuclein in agreement with immunoblot data (Figure 5A).
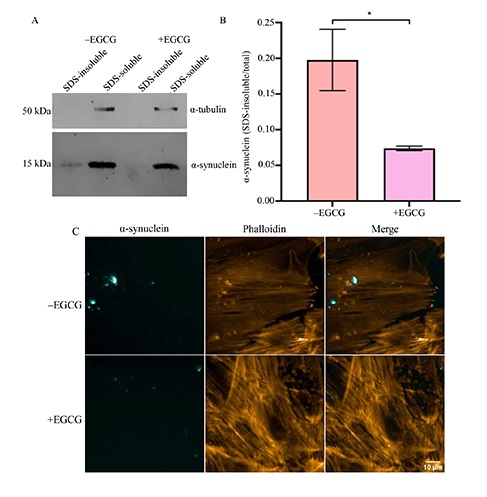
Incubation with EGCG results in a reduction of SDS-insoluble α-synuclein species. (A) Immunoblot of PD ONS 2803 –EGCG or +EGCG treatment prior to incubation with 0.1 μmol/L α-synuclein PFF for 48 h; (B) quantification of SDS-insoluble/total α-synuclein. Data presented as mean ± SD, n = 3 independent experiments; (C) immunohistochemistry of ONS exposed to 0.1 μmol/L α-synuclein PFF –EGCG or +EGCG. * P < 0.05; ** P < 0.01; *** P < 0.001
Prior research has demonstrated that PD ONS have alterations in gene and protein expression that display a disease-relevant phenotype that can be utilised to model PD [24–26].
PD has been difficult to model, particularly within drug discovery, as it is a complex neurodegenerative disease that progresses generally due to an intricate interplay between genetic and environmental insults over a person’s life [38]. ONS display susceptibility to stress in a disease-like manner and have varying genetic and metabolomic differences [25, 26]. Furthering this work, the results have demonstrated that the PD-specific stressor, α-synuclein, was not toxic to patient-derived ONS, however, there were significant protein processing differences identified between control and PD cell lines. Imaging analyses illustrated that over 48 h, all ONS accumulated α-synuclein. However, the PD lines had significantly higher amounts of α-synuclein fluorescence, which could demonstrate an increased concentration of α-synuclein [39]. Processing differences were further demonstrated by immunoblot analysis of SDS-solubility of α-synuclein species. The PD ONS generated significantly more SDS-insoluble α-synuclein after 48 h incubation in comparison to the control ONS, indicating that the PD patients have a general deficit in the clearance of α-synuclein and produce more toxic α-synuclein species. Many genetic risk factors for PD have been elucidated and suggest protein trafficking as a vital cellular pathway for PD pathogenesis, and therefore avenues for pharmacological intervention. Insults to endocytic processing, retromer formation, and autophagolysosomal fusion and function have all been linked to the pathogenesis of PD, where deficits in these vital pathways can result in the formation of toxic α-synuclein species [7, 40–42].
Recent proteomics analyses on the patient-derived ONS identified a significant decrease in endolysosomal processing proteins in systems including clathrin vesicle budding, trans-Golgi network vesicle budding and biogenesis, and in retromer complex formation [26]. This reduction in the proteins involved in the endolysosomal system [40, 43] was not previously known to have functional consequences, however, this study has shown that there was a significant difference in the cellular processing of α-synuclein that resulted in increased SDS-insoluble (and potentially pathogenic) species. Endolysosomal disruptions have been previously linked to the loss of neurons in PD, manifesting as the accumulation of proteinaceous LBs [35].
Here, ONS were identified as a suitable in vitro model to screen inhibitors of α-synuclein pathology, using EGCG to reverse the PD ONS phenotype. As EGCG is easily oxidised and therefore has a low bioavailability, it was not suitable for clinical use [44]. However, as EGCG decreases SDS-insoluble α-synuclein to levels comparable to healthy control cells in this in vitro model, it is a good assay control for future drug screening projects. This data enables the proposal of a new, disease-relevant model for screening inhibitors of α-synuclein pathology.
Overall, the data has validated the use of the ONS in understanding PD, particularly in the in vitro processing of α-synuclein. Considerable differences were observed in protein processing which is consistent with previous genetic, proteomic, and metabolomic data. Functional consequences of these previous studies were identified, whereby α-synuclein processing is diminished in PD cell lines, evident by imaging and immunoblot analysis. Furthermore, the data suggests the ONS model can be a viable drug screening platform for α-synuclein pathology in PD. Future directions include screening small molecule inhibitors against α-synuclein pathology in ONS lines. There have been a number of small molecule inhibitors of α-synuclein pathology identified in recent years by in situ assays that would be vital to screen in a disease-relevant in vitro model.
+EGCG: cells were treated with epigallocatechin gallate
D4a: dynog-4a
EGCG: epigallocatechin gallate
–EGCG: the absence of epigallocatechin gallate treatment
Hoechst+: Hoechst-stained
LBs: Lewy bodies
ONS: olfactory neuroepithelial-derived cells
PBS: phosphate buffered saline
PD: Parkinson’s disease
PFFs: preformed fibrils
PI: propidium iodide
PI+: propidium iodide-stained
SDS: sodium dodecyl sulfate
TEM: transmission electron microscopy
ThT: thioflavin T
Wort: wortmannin
The supplementary figures for this article are available at: https://www.explorationpub.com/uploads/Article/file/1001170_sup_1.pdf. Other supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/1001170_sup_2.pdf.
JKS: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. GDM: Conceptualization, Writing—review & editing, Supervision. AMS: Conceptualization, Investigation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
All procedures were in accordance with National Health and Medical Research Council Code of Practice for Human Experimentation and approved by the Griffith University Human Experimentation Ethics Committee.
All donor tissue and information were obtained with informed and written consent of the participants.
Not applicable.
Not applicable.
JKS was supported by the
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

