Review
Review

Affiliation:
1Department of Internal Medicine, University of Louisville, Louisville, KY 40202, USA
Affiliation:
1Department of Internal Medicine, University of Louisville, Louisville, KY 40202, USA
Affiliation:
1Department of Internal Medicine, University of Louisville, Louisville, KY 40202, USA
Affiliation:
2Department of Hematology and Oncology, University of Louisville, Louisville, KY 40202, USA
Affiliation:
3Division of Strategic Missions and Technologies, Microsoft Inc., Redmond, WA 98052, USA
Affiliation:
4Louisville Pulmonary Care, Louisville, KY 40207, USA
Email: james.adam.bradley@gmail.com
ORCID: https://orcid.org/0000-0001-5570-4679
Explor Digit Health Technol. 2025;3:101153 DOI: https://doi.org/10.37349/edht.2025.101153
Received: January 30, 2025 Accepted: April 29, 2025 Published: June 11, 2025
Academic Editor: Atanas G. Atanasov, Medical University of Vienna, Austria
Over the last four decades, lung cancer has been the leading cause of death in the United States. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, and historically, treatment consists of surgical resection, chemotherapy, and/or radiotherapy. Over the past decade, targeted immunotherapy has improved overall survival and treatment response. However, immunotherapy is expensive, and only select patients respond to immunotherapy. Recently, there has been much interest in using biomarkers to better identify and predict which patients will respond to therapy. There is much hope that the combined use of artificial intelligence (AI) and omics-based technology will provide enhanced capability to predict response to immunotherapy in patients with NSCLC. We performed a literature review and summarized the various approaches in which AI has been integrated with genomics, radiomics, pathomics, metabolomics, immunogenomics, and breathomics to better understand the tumor immune microenvironment and predict response to immunotherapy.
Lung cancer is the leading cause of cancer-related death and the second most common cancer diagnosed within the United States; the American Cancer Society estimates that there will be a total of 234,580 newly diagnosed patients with lung cancer in the United States in 2024 [1]. Non-small cell lung cancer (NSCLC) accounts for approximately 84% of all lung cancers and has a five-year survival rate of 23% [2]. Classically, for patients with stage I or II NSCLC, treatment is primarily surgical resection with adjuvant therapy, and for stage III or IV disease, treatment consists of chemotherapy and/or radiotherapy. Traditional chemotherapy agents have several limitations though, such as non-specific targeting, low bioavailability, toxicity, and drug-resistance that limit efficacy [3].
The tumor immune microenvironment is a complex and dynamic ecosystem of interactions between tumor cells, immune cells, stromal cells, and various cytokines. Knowledge of these complex interactions led to the development of immunotherapy strategies, which encompass CAR T-cell therapy, monoclonal antibodies, and immune checkpoint inhibitors (ICIs) capable of targeting specific pathways (PD-1/PD-L1, CTLA-4) and promoting immune activation and recognition by T-cells [4]. Notably, ICIs have revolutionized treatment paradigms for NSCLC patients in the neoadjuvant and adjuvant setting, offering an alternative to traditional cytotoxic chemotherapies [5]. Due to significant improvements in overall survival and progression-free survival (PFS), there is much emphasis placed on the role of ICIs in the treatment of NSCLC; however, the sobering reality is that only a subset of patients respond to treatment [6].
Approximately 20–30% of individuals with NSCLC have a durable response with PD-1 or PD-L1 inhibitors [6]. Additionally, the risk of toxicity is well recognized and can lead to a hyperinflammatory state, causing reversible and non-reversible tissue damage and fibrosis (i.e., pneumonitis, colitis, hepatitis) [3]. Of concern, a growing body of evidence also suggests the cost-effectiveness of immunotherapy may be prohibitive without the use of biomarkers capable of selecting patients who benefit from immunotherapy [7].
In general, biomarkers are used to improve the diagnosis, prognostic prediction, management strategies, and outcomes of individuals from different populations; however, the number of biomarkers in medicine that have performed well enough to impact patients’ lives is limited. Classically, biomarkers have been thought of as a laboratory-derived value derived from a biospecimen that is indicative of a disease or disease-state, however the pipeline for conventional biomarker development has significant hurdles and often results in biomarkers of questionable clinical utility [8]. Over the last two decades, technology has evolved to such a degree that the concept of biomarkers in human health and disease has expanded. The omics revolution has resulted in the curation of massive public and private datasets, and the cost and access to imaging technologies such as computed tomography (CT) and magnetic resonance (MR) imaging have improved, resulting in the increased use of imaging modalities. Furthermore, recent advances in the artificial intelligence (AI) community are pioneering an approach in medicine that promises the benefit of systems-level computational analysis on a personalized scale.
As a field, AI can now perform tasks that historically required human-level intelligence, such as image processing, reasoning, and decision making. AI-derived algorithms are adaptive and can learn to process complex tasks. Machine learning (ML, a subset of AI) involves the application of various learning strategies (supervised learning, unsupervised learning, reinforcement learning) and a wide spectrum of computational models (i.e., statistical models, neural networks, model ensembles, multi-modal models, large language models) that arguably represent how reasoning/logic occur. While the performance of each learning strategy and model varies significantly based on the domain and type of data used during training—a common goal for all models is to identify patterns in the data and leverage these patterns for prediction and higher-order reasoning. Specifically, within the fields of pulmonary medicine, radiology, and oncology, a multitude of AI-derived algorithms and prediction models have been developed to assist in the early diagnosis of cancer [9]. For example, algorithms such as Sybil, AdaBoost-SNMV-CNN, and Res-trans have shown great promise in their potential to not only detect malignant pulmonary nodules but also to predict future risk for lung cancer based on CT scans [10–12]. In NSCLC, AI models have shown the ability to assess patterns and interactions among biomarkers to help with earlier diagnosis, cancer type classification, predict response and prognosis, model the complex plasticity of the tumor microenvironment, and detect potential neoantigens [13]. Additionally, improvements in algorithm design and the development of large ground-truth data sets have expanded the potential for AI to facilitate predictive biomarker discovery, helping to identify patients responsive to immunotherapy [14].
The response to immunotherapy has been assessed by AI through multiple data modalities, including genomics, radiomics, pathomics, metabolomics and lipidomics, immunogenomics, breathomics, and electronic medical record (EMR).
In this manuscript, we provide a general overview of how the architecture of ML models in clinical medicine have evolved over the past decade, and we discuss the potential for AI-derived models to unify disparate data modalities to more comprehensively assess the nuances of the tumor immune microenvironment, allowing for improved models capable of diagnosing NSCLC and predicting immunotherapy treatment response (Figure 1).
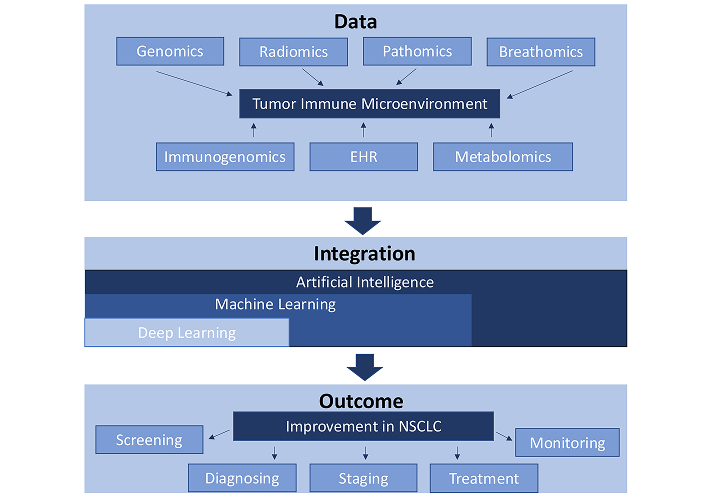
The tumor immune microenvironment refers to the complex and dynamic ecosystem of interactions between tumor cells, immune cells, stromal cells, and various cytokines. This image shows how multi-omics, such as genomics, radiomics, pathomics, breathomics, immunogenomics, electronic health records (EHR), and metabolomics can be used to better understand the tumor immune microenvironment. By using artificial intelligence alongside multi-omics-based technology, there is potential for an enhanced capability to screen, diagnose, stage, treat, and monitor non-small cell lung cancer (NSCLC)
The initial concept of AI was first described in 1950 by Alan Turing, but it wasn’t until the mid-2000s that widespread interest in the use of AI in medicine spurred an impressive number of clinical applications, innovation, and development of custom deep learning (DL) models in the medical domain [15]. Prior to the development of DL models, limitations of AI algorithms were computationally constrained due to existing hardware limitations, the slowing down of Moore’s law, a lack of large annotated datasets, and the public’s general lack of access to high-performance computing resources. The advent of the graphics processing unit and investment in cloud computing solved many of these problems, spawning brand new research efforts into the curation of high-quality labeled datasets (a vital component in the development of modern AI models/systems).
In the 2010s, AI applications in medicine became more pronounced as clinical decision support systems became integrated into EMR systems. Early applications of AI in medicine were limited by small datasets and high risk for model overfitting, but as larger and more robust datasets were curated over the past two decades, these limitations have become more manageable as data science methods have improved. In 2017, following a seminal demonstration of deep neural networks by Krizhevsky et al. [16], the rate of scientific progress in the AI field has taken off, achieving the same amount of discovery in a couple of years that previously took a decade. The leading application for AI in medicine is the classification of medical images. A recent review by Muehlematter et al. [17] found that of the AI-based medical devices approved in the US (n = 220) and Europe (n = 240) between 2015–2020, 58% and 53%, respectively, were approved for radiological diagnostics. CardioAI was the first AI-derived application to receive FDA approval in 2017 for assessment of cardiac MR imaging. In the 2020s, the impact of AI has progressed further to include applications in early detection and diagnosis, patient management, and drug development (Figure 2).
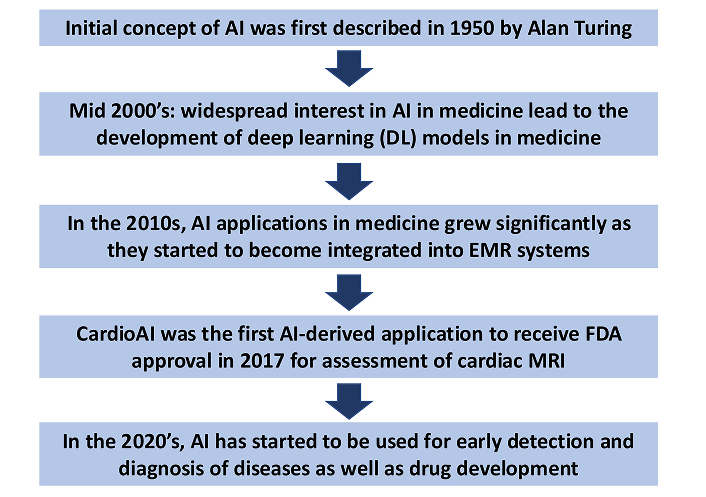
The evolution of artificial intelligence (AI) in medicine. EMR: electronic medical record; MRI: magnetic resonance imaging
Genomics encompasses the various technologies used to identify a particular cell’s genotype. Examples of current techniques include direct deoxyribonucleic acid (DNA) sequencing, DNA allele specific testing, and next generation sequencing (NGS), to name a few. Direct sequencing of the whole genome can be done in a week or less, but is less sensitive than other techniques because the presence of mutations can be hidden if tumor cellularity is low. DNA allele specific testing has replaced direct sequencing because it is faster and more cost effective; however, it can only detect pre-defined mutations. NGS has become a very popular method because it is fast, cost effective, and can analyze multiple genes.
Through the identification of driver mutations, genomics is already being used to select targeted therapy for patients and has been shown to improve outcomes in NSCLC [18–20]. For example, the detection of mutated epidermal growth factor receptor (EGFR) allows for the selection of receptor tyrosine kinase-specific inhibitors against EGFR [21]. Whole genome sequencing of malignancies has also shown clinical relevance. In 2017, Jamal-Hanjani et al. [22] demonstrated the relationship between tumor cell variation and the likelihood of resistance to treatment and a tendency to evolve.
Given the large volume of data inherent to the field of genetics, DL models are particularly suited to analyze genomic data to assess relationships between genotype and clinical endpoints. DeepVariant-AF, created by Google Health, has been applied to large data sets and has been shown to have considerable reliability in identifying gene variants [23]. Aradhya et al. [24] trained DL models using Invitae’s Evidence Modeling Platform to predict protein structure and function based on sequence data. AI has also been used in laboratory settings to identify chromosomal abnormalities such as translocations, deletions, and duplications [25].
The use of AI to evaluate genomic data for predicting treatment response in patients with NSCLC is not currently utilized in routine clinical practice, but several groups are developing models that have this capability. For example, the DL model DrugCell, developed on data from the Cancer Therapeutics Response Portal v2 and the Genomics of Drug Sensitivity in Cancer databases, can predict the in vitro response of tumor cell lines to various drugs. Interestingly, the performance of their interpretable model of drug response was not significantly different from conventional black-box models [26]. In the clinical setting, Sammut et al. [27] created a predictive model using combined sequence and digital pathology data to predict response to neo-adjuvant therapy in breast cancer patients. The predictive features were processed by univariable selection followed by an unweighted ensemble classifier. The resultant predictive function of this model was derived from the average of three underlying algorithms and was validated by applying them to an external cohort of 75 patients. The authors showed that their integrated model could accurately derive predictors of response with good discrimination power [area under the curve (AUC) 0.87] [27].
Genomics is being used to guide the selection of targeted therapy, but AI is not routinely used to assess the likelihood of response for a given therapy based on the specific genotype. While its abilities have been demonstrated in research settings, its lack of validation and questions regarding the explainability of these models have limited its routine use in clinical practice. As the use of these models becomes more mainstream in other areas, their reliability, accuracy, and reproducibility will likely improve. More work is needed to train these DL algorithms to analyze database information. Multidisciplinary collaboration within the fields of oncology, bioinformatics, and pathology will be crucial for the accurate validation and improved predictive analytics of AI-based algorithms and databases.
Digital pathology refers to the electronic digitization of pathological specimens. In the late 1990s, this technology expanded to include whole-slide imaging. As the name suggests, lab instruments can now read and piece together the entirety of the slide for subsequent storage and analysis by pathologists.
In oncology, digital pathology occupies a central role because tissue analysis remains the foundation of accurate diagnosis. Microscopic features of tissue and individual cells have both diagnostic and prognostic value. Advancements in the digitization of pathology specimens have allowed improvements in the storage and sharing of large amounts of data. In oncology, immunohistochemistry (IHC) staining techniques are commonly used to identify the expression of oncoproteins. This information often guides the choice of therapy. For example, the FLEX study demonstrated that the presence of EGFR can be used to select patients for cetuximab and platinum-based chemotherapy in the setting of advanced NSCLC [28]. Since digital pathology data collection has quickly become a technological requirement in modern clinical practice, the integration of AI can positively impact the pathologist’s workflow by aiding in the identification of new biomarkers of disease, improving quality assurance, and consolidating clinical and pathological information.
DL models have been used to analyze whole-slide digital images. Specifically, convolutional neural networks (CNNs), which are a standard in DL-based approaches to computer vision and image processing, can process complex 2D images by utilizing convolutional and pooling layers to maximize the processing power and analysis of digital path images. DL models that incorporate segmentation of specific cellular and tissue features, cellular and mitosis detection, and tissue classification have demonstrated their diagnostic abilities as well as their ability to predict genotype. For example, the pretrained OverFeat model has been shown to accurately predict the Gleason score using region-level tissue classification [29].
Additionally, the use of AI in digital pathology has shown the potential to predict treatment response. Hoang et al. [30] developed a two-step model, ENLIGHT-DeepPT, which can predict genome-wide tumor mRNA expression and treatment response to targeted and immune therapies based on digital pathology images of hematoxylin and eosin-stained (H&E) tumor slides. This model successfully predicted responders in five independent cohorts of patients, which included six different primary malignancies (including NSCLC) and four different treatments [30]. In 2020, Hu et al. [31] showed that a CNN model could predict response to immune-checkpoint blockade based on the analysis of H&E slides alone. This is particularly interesting because it highlights the possibility that AI may be able to predict response to therapy based on morphologic features of tissue specimens alone, without the additional use of specialized stains and reagents [31].
A 2023 analysis of 380 patients with small cell lung cancer showed that PathoSig, an unsupervised DL model with a contrastive clustering computational framework (DL-CC), could predict response to chemotherapy by identifying phenotypic clusters from H&E digital images. The group observed that patients categorized as high-risk by PathoSig had a statistically significant shorter disease-free survival compared to patients with low and intermediate risk labels. Additionally, the high-risk patients had higher rates of recurrence. Furthermore, PathoSig maintained its independent prognostic efficacy even after adjusting for various clinical features using multivariable Cox regression analysis [32].
A common challenge in the clinical interpretation of pathological imaging is the degree of positivity of staining seen on IHC. Recent development of DL models to address this issue has shown promise. In 2022, Cheng et al. [33] developed a DL model to assess the immunohistochemical expression of PD-L1 in lung cancer patients. Their study included 1,288 patients with lung cancer, and they evaluated the performance of three AI models (M1, M2, and M3) to quantitatively score PD-L1 expression. Even in samples with low PD-L1 expression (< 1%), which can be problematic for pathologists to accurately assess, their AI models (particularly M2 and M3) demonstrated high accuracy (96.4%) and specificity (96.8%) [33]. Given the degree of discordance that can occur when evaluating PD-L1 expression between pathologists, an AI model developed by Choi et al. [34] demonstrated the ability to improve consensus of reads between pathologists as well as predict response to treatment in patients with NSCLC.
In 2024, Ligero et al. [35] developed a Retrieval with Clustering-guided Contrastive Learning RetCCL model to quantify the degree of positivity of PD-L1 on IHC slides, which was then used to predict the response to ICIs by estimating PFS [35]. They created a two-step process: feature extraction was performed with the RetCCL model, followed by aggregation and classification by a weakly supervised attention-based multiple instance learning model. Cox proportional-hazards and Kaplan–Meier curve analysis demonstrate an association between PD-L1 positivity and PFS. Their model demonstrated the proof of concept that a weakly supervised DL model is capable of distinguishing different histologic patterns in IHC and quantifying PD-L1 expression.
Currently, clinical use of AI in the field of digital pathology is limited. In 2021, van der Laak et al. [36] considered that the lack of generalizability of DL algorithms is likely the biggest barrier for use in clinical practice, in addition to the inherent ambiguity of “black-box” algorithms [36, 37]. The lack of generalizability was attributed to the lack of variation of data sources in the research setting compared to data encountered in clinical practice [38]. Clinically, it has been recognized that there are limitations in data resources due to a lack of population diversity, and this can result in decreased performance of algorithms when they are applied to large external datasets [39]. In the future, the development of large, heterogeneous databases that have been carefully curated will drive the generalizability of AI models in clinical practice.
Radiomics involves extracting a diverse array of features from medical images, including shape, size, texture, and intensity. These features can then be quantified and analyzed by advanced algorithms and computational methods. This quantitative approach offers a more objective measure compared to traditional visual assessments. The extracted radiomic features can be input into ML models to create algorithms, which can help in diagnosing disease, predicting treatment response, and evaluating prognosis based on imaging data [40, 41].
In a study by Mu et al. [42], a small residual convolutional network was employed to analyze PET/CT images and clinical data from NSCLC patients, to develop a DL score that predicted PD-L1 expression. Achieving an AUC of 0.82 in two external validation cohorts, this model was able to distinguish between PD-L1 positive and negative patients. This study presents an attractive alternative to traditional IHC for the assessment of PD-L1 expression [42].
Tumor mutation burden (TMB) is a key predictor of the efficacy of ICIs. He et al. [43] developed a novel non-invasive biomarker by integrating DL technology with CT characteristics. Their approach was successful in differentiating between NSCLC patients with high-TMB and low-TMB tumors as well as predicting outcomes and treatment efficacy.
Several studies have explored the use of radiomics to predict response to immunotherapy in lung cancer. A systematic review by Dercle et al. [44] evaluated 87 studies on the integration of AI-based models and radiomics in the care of patients receiving immunotherapy. Of the 87 studies reviewed, the primary goal of the algorithms that were developed was prognostication (33%), prediction of treatment response (27%), and characterization of tumor phenotype (16%) or immune environment (15%). Although many of the studies demonstrated proof of concept, the median radiomics quality score was 12 (max score of 36), which suggests that the field of radiomics needs to establish more robust and innovative methodologies behind model development before AI-based algorithms can be expected to have a widespread clinical impact [44, 45].
In a study by Trebeschi et al. [46], 1,055 primary and metastatic lesions from 203 patients with advanced melanoma and NSCLC undergoing anti-PD1 therapy were analyzed. The authors utilized AI to characterize each lesion based on pretreatment contrast-enhanced CT imaging data to develop a noninvasive ML biomarker that would distinguish between immunotherapy responders and non-responders. Their biomarker demonstrated good performance in patients with NSCLC lesions, with AUC 0.83 (P < 0.001), and a borderline significant result for melanoma lymph nodes, AUC 0.64 (P = 0.05). When these lesion-level predictions were aggregated at the patient level, the biomarker achieved an AUC of 0.76 for both cancer types (P < 0.001) and indicated a 1-year survival difference of 24% (P = 0.02) [46].
Mu et al. [47] demonstrated that radiomic features from baseline pre-treatment 18F-FDG-PET/CT scans can predict clinical outcomes for NSCLC patients undergoing checkpoint blockade immunotherapy. Their study included 194 patients with histologically confirmed stage IIIB-IV NSCLC who had pre-treatment PET/CT images. Radiomic features were extracted from PET, CT, and PET + CT fusion images using minimum Kullback–Leibler divergence criteria. Their model successfully predicted patients who experienced durable clinical benefit, achieving an AUC of 0.86 (95% CI: 0.79–0.94) in the training cohort, 0.83 (95% CI: 0.71–0.94) in the retrospective test cohort, and 0.81 (95% CI: 0.68–0.92) in the prospective test cohort [47].
Hyperprogression is an unusual response pattern to immune checkpoint inhibition observed in NSCLC. In a study by Vaidya et al. [48], image-based radiomic markers extracted from baseline CT scans of advanced NSCLC patients treated with PD-1/PD-L1 inhibitors were investigated for their potential to identify patients at risk of hyperprogression. Using a random forest classifier with key features associated with hyperprogression, the model achieved an AUC of 0.85 ± 0.06 in the training set (D1 = 30) and 0.96 in the validation set, effectively distinguishing hyperprogression from other radiographic response patterns [48].
Li et al. [49] developed a CT-based radiomics model that accurately predicted hyperprogression and pseudoprogression in NSCLC patients undergoing immunotherapy. They retrospectively analyzed 105 NSCLC patients from three institutions who were treated with ICIs, dividing them into training and independent testing sets. The logistic regression model demonstrated excellent performance in distinguishing between pseudoprogression and hyperprogression, achieving an AUC of 0.95 [95% confidence interval (CI): 0.91–0.99] in the training set and 0.88 (95% CI: 0.66–1.00) in the test set [49].
Metabolomics encompasses the study of the metabolome, which includes the composition and complex interactions of small-molecule metabolites. Metabolites of interest include sugars, amino acids, lipids, and other organic compounds. Metabolomics has been used to evaluate the functional capacity and cellular activity of both single-cell processes and whole biological systems in various stages of disease. This data can then be integrated with various omics domains to provide a more comprehensive, holistic assessment of disease status.
Two of the most common analytical techniques for studying metabolomics are nuclear MR spectroscopy and mass spectrometry (MS). Metabolomics results in large data sets, containing thousands of data points representative of known and unknown metabolites with complex relationships [50]. Targeted metabolomics measures the concentration of metabolites determined a priori, while untargeted metabolomics represents a semi-quantitative and unbiased measurement of thousands of metabolites. In the 1990s, Curry et al. [51] developed an artificial neural network to help classify MS spectrometry. Subsequently, in 2002, Ball et al. [52] illustrated the use of ML approaches to analyze MS data and identify metabolic signatures that could differentiate patients with low versus high-grade astrocytoma [51, 52].
AI algorithms that process raw MR and MS data have been developed to identify metabolites and metabolic signatures as novel biomarkers of disease, as further discussed by Barberis et al. [53]. A 2016 study by O’Shea et al. [54] coupled an artificial neural network model with sputum metabolomics to identify six metabolites (phenylacetic acid, L-fucose, caprylic acid, acetic acid, propionic acid, and glycine) that were elevated in patients with small cell lung cancer compared to NSCLC. As a proof of concept, Xie et al. [55] used multiple different ML techniques to identify six metabolites (proline, L-kynurenine, spermidine, amino-hippuric acid, palmitoyl-L-carnitine, and taurine) that distinguished stage 1 lung cancer patients from healthy controls.
Many of the metabolomic studies that have been completed thus far are focused on identifying specific metabolites and metabolic profiles that could be used as diagnostic biomarkers for diagnosis, but very few have been assessed for prognostic significance. For example, metabolomic studies have identified metabolites that could be used to predict treatment response in rheumatoid arthritis [56]. There are a few proof of concept studies that have been performed that illustrate the potential for changes in the metabolome to predict response to treatment, but none have been performed in patients with NSCLC to our knowledge. In patients with HER2+ breast cancer, metabolomic and transcriptomic analyses were used to identify a metabolic signature and develop a prediction model of the treatment response to neoadjuvant chemotherapy [57]. A separate study involving patients with breast cancer showed some changes in serum metabolites and lipids in response to neoadjuvant chemotherapy [58]. Additionally, similar studies that assess metabolic changes in response to treatment have been performed in patients with colorectal cancer, bladder cancer, and melanoma [59–61].
Lipidomics is considered a subfield within metabolomics. Lipids are essential in energy storage, signal transduction, and cell membrane formation, and disruption of lipid homeostasis has a role in tumorigenesis; thus, there has been interest in using lipidomics to monitor treatment response. As discussed in the prior section, AI algorithms have also been used to identify and evaluate changes in various lipid species [62].
In patients with NSCLC, Jiang et al. [63] used lipidomic profiling to identify six key lipids and develop a predictive model capable of predicting treatment response to chemo-immunotherapy with AUC of 0.87. Utilizing lipidomics, Yu et al. [64] identified nine distinct lipids that were able to predict immune related adverse events in NSCLC patients undergoing treatment with ICIs.
Immunogenomics is an interdisciplinary field that integrates genomics and immunology to understand the genetic basis of immune responses and their implications in various diseases, particularly cancer. This field leverages the power of high-throughput sequencing technologies and computational analyses to analyze the interaction of both tumor and immune cells within the tumor immune microenvironment. For example, techniques such as T-cell receptor sequencing and immune microenvironment deconvolution have been utilized to reveal important insights into the tumor-immune microenvironment [65, 66]. In a prospective cohort study of patients with pulmonary nodules, Chen et al. [67] showed that ground-glass associated lung cancers were less metabolically active and had a less active immune microenvironment compared to patients with solid lung nodules. A separate study by Sun et al. [68], which utilized IHC and RNA-sequencing data, suggests that NSCLC patients who lack either PD-L1 expression or immune infiltration may not benefit from immunotherapy.
Immunogenomic technology and methods have been used to assess the repertoire of T-cell and B-cell receptors in lung cancer patients, and identify infiltrating immune cells and neoantigens that influence the efficacy of ICIs [65, 69]. By integrating genomic data with immunological insights, immunogenomics provides a foundation for precision medicine approaches in oncology, enabling the development of personalized therapeutic strategies based on the unique genetic and immunological landscape of each patient’s tumor. Genetic polymorphisms in immune-related genes can influence the efficacy and survival outcomes of patients undergoing PD-1/PD-L1 blockade therapy. For instance, polymorphisms in genes such as ATG7, CD274, and TLR4 have been identified as predictors of response to PD-1/PD-L1 blockade, with certain alleles associated with increased risk of tumor progression and poorer PFS [66]. In lung cancer, germline polymorphisms in immune-related genes, such as HLA-DRB5, KIR3DL1, and KIR3DL2, have been shown to negatively impact the response to EGFR tyrosine kinase inhibitors, resulting in decreased PFS and overall survival [70]. This data illustrates that polymorphisms expressed in genes expressed by immune cells can influence the response to immunotherapy.
Recently, Liu et al. [71] combined data from scanned histology slides and RNA-sequencing to develop an AI-based immunoscore model capable of predicting survival outcomes in patients with NSCLC who had received chemoimmunotherapy [71]. Additionally, recent work by Kong et al. [72] illustrated an AI framework that uses network-based analyses to identify biomarkers that were highly predictive of ICI treatment response in patients with melanoma, gastric cancer, and bladder cancer [72]. Integration of immunogenomic technology with AI-based algorithm design processes is still in its infancy and will continue to elicit profound insights into the tumor immune microenvironment.
Breathomics refers to the molecular analysis of exhaled breath. In 1971, Pauling et al. [73] used gas chromatography methods to show the presence of volatile compounds in human breath and urine [74]. Since then, there have been more than 3,000 volatile organic compounds (VOCs) that have been discovered [74]. Gas chromatography-MS (GC-MS) is a method in which samples of exhaled breath are collected and stored in containers [75]. Helium subsequently pushes the sample of breath through a column, and the VOCs undergo separation [75]. In 1985, Gordon et al. [76] used GC-MS to investigate breath samples from 12 individuals with lung cancer and 17 individuals without disease. Of the individuals in that study, 22 VOCs were identified that showed the largest difference between those with or without lung cancer [76]. Three VOCs (acetone, methyl ethyl, and n-proponal) were further assessed due to increased occurrence and peak. Using the three VOCs, Gordon et al. [76] were able to accurately classify 93% of the samples. In 1999, Phillips et al. [77] used GC-MS to investigate breath samples from 108 individuals who had abnormal chest imaging that were scheduled to have a bronchoscopy. They were able to confirm lung cancer in 60 of the patients. Out of the breath samples, 22 VOCs had 100% sensitivity and 81.3% specificity [77]. In a 2010 study by Poli et al. [78], the total amount of exhaled aldehydes was increased in individuals with NSCLC compared to individuals without disease.
Some previous studies show that GC-MS analysis of VOCs could prove to be beneficial in diagnosing lung cancer; however, there have been notable discrepancies in the literature. In a 2010 study by Kischkel et al. [79] that compared individuals with lung cancer to healthy controls and smokers, they found that age, sex, smoking history, and inspired substance concentrations were confounding factors that impacted exhalation profiles. Other confounding factors that can affect the exhalation profile when using GC-MS is how the breath collection is obtained, the stability of different compounds, and the storage container used for sample collection [75]. Overall, GC-MS is a method that shows potential to help with screening and diagnosing cancer, but there are weaknesses with the method that need to be further investigated prior to routine clinical use.
Nanoarray analysis, such as the electronic nose (e-nose), is another promising technology that may prove to be beneficial in the future for screening and diagnosing lung cancer. The e-nose works similarly to a human nose, however, it possesses the ability to have breath samples filtered into several different sensors and further analyzed by AI-derived algorithms to determine a “breath print” comprised of VOC’s [74, 75]. In 2003, Di Natale et al. [80] used an e-nose (composed of eight quartz microbalance gas sensors) combined with partial least squares discriminant analysis (an AI method for classification tasks of high-dimensional data) to correctly classify 100% of lung cancer patients (n = 35). In 2008, Dragonieri et al. [81] used e-nose technology to separate 85% of individuals with NSCLC from people with COPD and 90% of individuals with NSCLC from healthy controls.
Other types of sensors and nanoarray analysis, such as colorimetric sensors, are being investigated for their potential to diagnose lung cancer. In 2007, Mazzone et al. [82] used colorimetric sensors to accurately predict individuals with lung cancer versus individuals with other lung diseases. Overall, the e-nose technology is very promising; however, it is early in development, and the clinical impact of this technology in real-world settings remains unclear at this time. Although there may be a role for the use of breathomics to monitor treatment response to immunotherapy in the future, current limitations of the methodology and issues with generalizability prevent its routine use in clinical oncology.
Over the past few decades, electronic health records (EHRs) have revolutionized the health care industry. Important information included in EHRs are patient’s biographical information, past medical history, symptoms, immunization history, medication history, laboratory results, pathological reports, as well as any imaging that was completed [83]. Recently, research has started to investigate EHR based interventions to help with identifying patients at higher risk for lung cancer and/or poor prognosis.
In 2024, Marmarelis et al. [84] used a nudge-based intervention with an EMR to investigate whether it led to increased molecular testing and better guideline-concordant care. Using a nonrandomized prospective trial design at the University of Pennsylvania hospitals, and molecular genotyping with tissue- and/or plasma-based NGS methods, the authors included 533 patients with NSCLC; 376 in the preintervention and 157 in the post-intervention. Following the intervention with the EMR-based nudge, more patients successfully underwent comprehensive molecular genotyping in the postintervention period.
In 2021, Yuan et al. [85] assembled a large cancer cohort from an EHR. They used a ML algorithm, incorporating extraction strategies for both unstructured and structured data. In their study, they identified patients with at least one lung cancer diagnostic code in an EHR in the Massachusetts General Brigham health system from July 1988 to October 2018. Among 42,069 people with lung cancer, they were able to extract structured data and notes by creating a customized natural language processing tool using the EHR. The positive predictive value of this study was 94.4% [85]. The authors then went on to develop a prognostic model for NSCLC using the EHR cohort. Their findings suggest that a prognostic model based on data commonly found in the EHR may facilitate the prediction of NSCLC survival [85]. Overall, there has not been a significant amount of work utilizing AI-based methods that further assess data already contained within EHR’s though. As generative AI and large language models, such as ChatGPT, become more advanced, this type of data may be further integrated in a multi-omics approach toward further understanding and predicting lung cancer diagnosis and treatment response.
Over the past two decades, AI has had a profound impact on the ability of multiomics to diagnose NSCLC and identify biomarkers capable of predicting treatment response. While many of the current AI applications in lung cancer primarily utilize imaging characteristics or digital biomarkers to diagnose, assess prognosis, and predict treatment response, there has been widening interest in the use of AI to combine analyses of other data modalities, such as genomics, metabolomics, proteomics, histopathology imaging, and immunogenomics to improve diagnostic accuracy and provide a more holistic understanding of the tumor immune microenvironment (Table 1). This unique approach promises more robust models capable of implementing the nuances of personalized medicine into predictive analytics and patient care.
Studies illustrating the potential of artificial intelligence in multiomics
| Study | Genomics |
|---|---|
| Chen et al. [23] | A DL model, DeepVariant-AF, created by Google Health is applied to large data sets and reliably identifies gene variants. |
| Aradhya et al. [24] | DL models were trained using Invitae’s Evidence Modeling Platform to predict protein structure and function based on sequence data. |
| Kuenzi et al. [26] | A DL model, DrugCell, predicts the in vitro response of tumor cell lines to various drugs. |
| Sammut et al. [27] | A predictive model using combined sequence and digital pathology data predicts response to neo-adjuvant therapy in breast cancer patients. |
| Pathomics | |
| Källén et al. [29] | The DL model, OverFeat, accurately predicts the Gleason score using region-level tissue classification. |
| Hoang et al. [30] | A two-step model, ENLIGHT-DeepPT, predicts genome-wide tumor mRNA expression and treatment response to targeted and immune therapies based on digital pathology images of hematoxylin and eosin-stained (H&E) tumor slides. |
| Hu et al. [31] | CNN model predicts response to immune-checkpoint blockade based on the analysis of H&E slides alone. |
| Zhang et al. [32] | The DL model, PathoSig, predicts response to chemotherapy by identifying phenotypic clusters from H&E digital images. |
| Cheng et al. [33] | A DL model and three AI models quantitatively score PD-L1 expression. |
| Choi et al. [34] | A DL model improves the consensus of reads between pathologists and predicts response to treatment in patients with NSCLC. |
| Ligero et al. [35] | A Retrieval with Clustering-guided Contrastive Learning (RetCCL) model quantifies the degree of positivity of PD-L1 on IHC slides and predicts response to ICIs by estimating progression-free survival. |
| Radiomics | |
| Mu et al. [42] | A small residual convolutional network was employed to analyze PET/CT images and clinical data from NSCLC patients in order to develop a DL score that predicted PD-L1 expression. |
| He et al. [43] | Developed a novel non-invasive biomarker by integrating DL technology with CT characteristics to differentiate between NSCLC patients with high-TMB and low-TMB tumors and predict treatment efficacy. |
| Mu et al. [47] | Radiomic features from baseline pre-treatment 18F-FDG-PET/CT scans can predict clinical outcomes for NSCLC patients undergoing checkpoint blockade immunotherapy. |
| Vaidya et al. [48] | Radiomic markers extracted from baseline CT scans of advanced NSCLC patients treated with PD-1/PD-L1 inhibitors identifies patients at risk of hyperprogression. |
| Li et al. [49] | A CT-based radiomics model accurately predicts hyperprogression and pseudoprogression in NSCLC patients undergoing immunotherapy. |
| Metabolomics | |
| Curry et al. [51] | Developed an artificial neural network to help classify MS spectrometry. |
| Ball et al. [52] | Machine learning approaches to analyze MS data and identify metabolic signatures can differentiate patients with low versus high grade astrocytoma. |
| O’Shea et al. [54] | Use of an artificial neural network model with sputum metabolomics identifies six metabolites that were elevated in patients with small cell lung cancer compared to NSCLC. |
| Xie et al. [55] | Machine learning techniques identify six metabolites that distinguish stage 1 lung cancer patients from healthy controls. |
| Lipidomics | |
| Jiang et al. [63] | Lipidomic profiling identifies six key lipids, used to develop a predictive model for treatment response to chemo-immunotherapy. |
| Yu et al. [64] | Nine distinct lipids were used to predict immune related adverse events in NSCLC patients undergoing treatment with ICIs. |
| Immunogenomics | |
| Chen et al. [67] | Ground-glass associated lung cancers were less metabolically active and had a less active immune microenvironment compared to patients with solid lung nodules. |
| Sun et al. [68] | Immunohistochemistry and RNA-sequencing data show that NSCLC patients who lack either PD-L1 expression or immune infiltration may not benefit from immunotherapy. |
| Liu et al. [71] | Combined data from scanned histology slides and RNA-sequencing to develop an AI-based immunoscore model capable of predicting survival outcomes in patients with NSCLC who had received chemoimmunotherapy. |
| Breathomics | |
| Gordon et al. [76] | Gas chromatography-mass spectrometry (GC-MS) analysis of breath samples classifies 93% of patients with vs. without lung cancer. |
| Philipps et al. [77] | GC-MS analysis of 108 individuals confirms lung cancer in 60 patients. |
| Di Natale et al. [80] | Use of the electronic nose combined with partial least squares discriminant analysis correctly classifies 100% of lung cancer patients. |
| Mazzone et al. [82] | Colorimetric sensors accurately predict individuals with lung cancer versus individuals with other lung diseases. |
| Electronic health record | |
| Marmarelis et al. [84] | A nudge-based intervention with an EMR increases molecular testing and better guideline-concordant care. |
| Yuan et al. [85] | Developed a machine-learning-based prognostic model for NSCLC by the extraction of unstructured and structured data. |
DL: deep learning; CNN: convolutional neural network; NSCLC: non-small cell lung cancer; EMR: electronic medical record
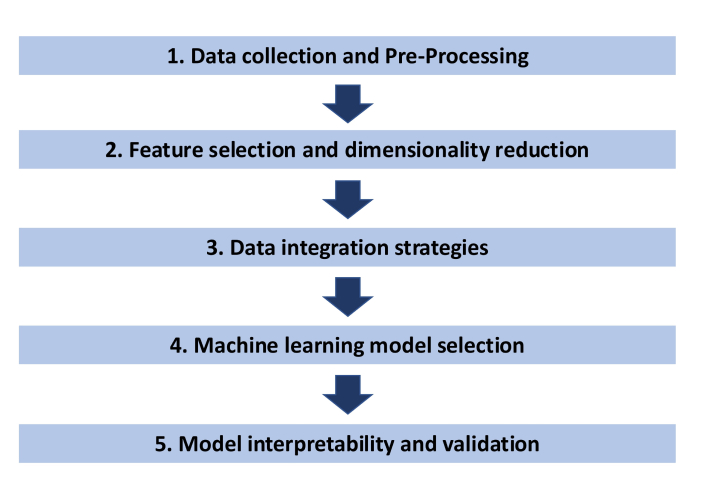
To integrate multiomics data with AI, there are several key steps that must be considered: 1) data collection and pre-processing, 2) feature selection and dimensionality reduction, 3) data integration strategies, 4) ML model selection, and 5) model interpretability and validation (Figure 3). With the cost of genomic technology plummeting and widespread use of NGS in the care of lung cancer patients, there are ever-increasing databases of genomic and laboratory data such as the Lung Cancer Gene database, Lung Cancer Explorer, and the All of US database hosted by the National Institute of Health [86–88]. As the work toward multiomics integration occurs, the importance of accurately curated and shared data is paramount to the success and generalizability of future models; resources such as the Omics Discovery Index, which aim to link publicly available omics datasets, will prove invaluable [89].
Feature selection and dimensionality reduction are particularly important in complex, heterogeneous disease states as found in NSCLC and have been discussed in several recent reviews [90]. The most common linear dimensionality reduction technique used in medical research is principal component analysis, but recent evidence by Khadirnaikar et al. [91] and Alanis-Lobato et al. [92] suggests that non-linear dimensionality reduction techniques exhibit superior performance when assessing biological data. For example, several groups have illustrated the effectiveness of an autoencoder (a non-linear DL-based technique) for dimensionality reduction in multiomics analysis [93–95].
Various data integration strategies to analyze noisy and highly dimensional datasets have been developed, each with pros and cons. These can be categorized as early, mixed, intermediate, late, or hierarchical and have been discussed in depth in prior publications [90]. Although many of the currently used AI models were designed for single-omics analysis, new multiomics integration techniques are continuously being proposed. DL architectures are quite flexible, allowing for the integration of multi-layer datasets, highlighting their unique potential for NSCLC research and patient care. However, compared with radiology datasets, DL algorithms have been used less frequently in lung cancer clinical studies that incorporate omics data. These specific types of models can be difficult to train and often result in overfitting; thus, newer techniques such as transfer learning (used in image recognition) are currently being investigated and applied in multiomics research.
Although there have been many promising results over the past 5–10 years in the field of AI and medicine, rapid improvement in multiomics data integration strategies is still needed prior to routine use in clinical care. As previously discussed, many of the AI models that have been developed are at high risk of model overfitting and not generalizable across different patient populations. Advancements in computational methodologies throughout the previous steps highlighted in this section and across the different omics fields highlight a robust potential for AI-derived algorithms to assist in the diagnosis of patients with NSCLC and the prediction of treatment response to immunotherapy. Furthermore, multidisciplinary collaboration between clinicians, bioinformaticians, data scientists, and basic science researchers will be needed to develop robust models that undergo multisite external validation to illustrate true generalizability and clinical efficacy.
AI: artificial intelligence
AUC: area under the curve
CNN: convolutional neural network
CT: computed tomography
DL: deep learning
DNA: deoxyribonucleic acid
EGFR: epidermal growth factor receptor
EHR: electronic health record
EMR: electronic medical record
e-nose: electronic nose
GC-MS: gas chromatography-mass spectrometry
H&E: hematoxylin and eosin-stained
ICI: immune checkpoint inhibitor
IHC: immunohistochemistry
ML: machine learning
MR: magnetic resonance
MS: mass spectrometry
NGS: next generation sequencing
NSCLC: non-small cell lung cancer
PFS: progression-free survival
RetCCL: Retrieval with Clustering-guided Contrastive Learning
TMB: tumor mutation burden
VOC: volatile organic compounds
BW: Conceptualization, Writing—original draft, Writing—review & editing. EH: Writing—review & editing. BK: Writing—review & editing. PP: Writing—review & editing. Joshua B: Writing—review & editing. James B: Conceptualization, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.