 Review
Review

Affiliation:
1Ospedale Civile di Baggiovara, Department of Internal Medicine, Azienda Ospedaliero-Universitaria di Modena (–2023), 41100 Modena, Italy
†These authors contributed equally to this work.
Email: a.lonardo@libero.it
ORCID: https://orcid.org/0000-0001-9886-0698
Affiliation:
2Institute of Molecular Pathobiochemistry, Experimental Gene Therapy and Clinical Chemistry (IFMPEGKC), RWTH University Hospital Aachen, D-52074 Aachen, Germany
†These authors contributed equally to this work.
Email: rweiskirchen@ukaachen.de
ORCID: https://orcid.org/0000-0003-3888-0931
Explor Dig Dis. 2025;4:100590 DOI: https://doi.org/10.37349/edd.2025.100590
Received: July 22, 2025 Accepted: August 21, 2025 Published: September 02, 2025
Academic Editor: Jose Carlos Fernandez-Checa, Institute of Biomedical Research August Pi i Sunyer (IDIBAPS), Spain
Peroxisome proliferator-activated receptors (PPARs) comprise three isoforms: PPARα, PPARβ/δ, and PPARγ, which regulate the expression of genes involved in fatty acid uptake, β-oxidation, adipogenesis, gluconeogenesis, and insulin sensitivity. Type 2 diabetes (T2D), often accompanied by other features of metabolic syndrome, contributes to vasculopathy, end-stage organ failure, and cancer. Metabolic dysfunction-associated steatotic liver disease (MASLD) refers to steatotic liver disease in the presence of cardiometabolic risk factor(s) and without excessive alcohol consumption. MASLD is prevalent among adults with T2D and carries a high risk of liver fibrosis, metabolic dysfunction-associated steatohepatitis (MASH), cirrhosis and incident T2D. In MASLD, the liver becomes a hub of lipid toxicity, oxidative stress, and fibrotic signalling whenever T2D disrupts hormonal and adipokine signalling, increases free fatty acid flux, and promotes chronic inflammation. MASLD, therefore, results from an impairment of the protection physiologically offered by PPARs through fatty acid oxidation, lipid storage in the adipose tissue, and mitigation of insulin resistance and pro-inflammatory cascades. By examining the molecular mechanisms of PPARα, PPARβ/δ, and PPARγ, as well as their interactions with cofactors like PGC-1α, and their crosstalk with pathways like sterol regulatory element-binding protein (SREBP), NF-κB, AMP-activated protein kinase (AMPK), and adipokines, researchers and clinicians can better understand how T2D-related MASLD can be prevented or treated. Single PPAR agonists, such as fibrates and glitazones, have limited clinical efficacy in achieving hard liver histology endpoints like MASH resolution and fibrosis regression in humans. However, the Pan-PPAR agonist Lanifibranor at the highest doses shows promise in ameliorating these outcomes in subjects with non-cirrhotic MASH. This suggests that activating all three PPAR isoforms together enhances their therapeutic effects on various cells and target organs, restoring insulin resistance, improving gluco-lipidic homeostasis, while inhibiting pro-inflammatory and pro-fibrogenic pathways. Analysis of unresolved issues should dictate the research agenda.
Peroxisome proliferator-activated receptors (PPARs) are a group of ligand-activated transcription factors belonging to the nuclear receptor superfamily, comprising three isoforms: PPARα, PPARβ/δ, and PPARγ [1]. These receptors play fundamental roles in lipid and glucose homeostasis, inflammation, and energy metabolism by regulating the expression of genes involved in fatty acid uptake, β-oxidation, adipogenesis, gluconeogenesis, and insulin sensitivity [2]. Each PPAR isoform has multiple activities in different organs. PPARα orchestrates fatty-acid β-oxidation in liver, heart, and kidney, PPARβ/δ regulates fuel switching and mitochondrial biogenesis in skeletal muscle, endothelium, and adipose tissue, and PPARγ governs adipogenesis, lipid storage, and systemic insulin sensitivity in white and brown fat (Figure 1).
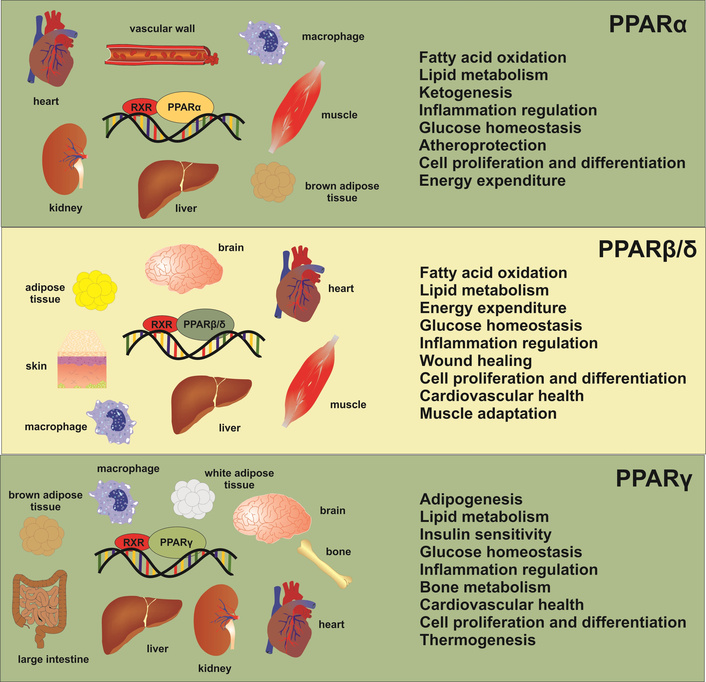
Peroxisome proliferator-activated receptors (PPARs), target tissues, and biological activities. The diagram illustrates the various activities of each PPAR isoform in different organs.
Taken together, this triad forms an inter-organ network that balances glucose-lipid fluxes, modulates inflammation, and even constrains fibrogenic signalling. Consequently, dysregulation of PPAR signalling is intricately linked to the molecular pathogenesis of metabolic disorders, including type 2 diabetes (T2D) mellitus and metabolic dysfunction-associated steatotic liver disease (MASLD), a nomenclature that strongly highlights the metabolic underpinnings of SLD [3]. The pathophysiology of T2D-related MASLD entails a complex interplay between insulin resistance, adipose tissue dysfunction, intestinal dysbiosis, chronic low-grade systemic and hepatic inflammation, culminating in excess lipid accumulation in hepatocytes and subsequent hepatic injury [4]. Understanding how distinct PPAR isoforms modulate metabolic fluxes in key tissues, particularly the liver, adipose tissue, and skeletal muscle, can provide critical insights into the initiation, progression, and potential therapeutic interventions for T2D-related MASLD.
T2D is a chronic dysmetabolic state featuring persistent hyperglycaemia, which, often in concert with concurrent components of the metabolic syndrome, facilitates disabling and life-threatening micro- and macro-vascular complications, end-stage failure of target organs, and certain types of cancers [5–7]. T2D accounts for around 90% of all cases of diabetes and is typically observed in individuals aged over 45 years [5]. However, it is increasingly seen in children, adolescents, and younger individuals due to rising levels of unhealthy lifestyle habits promoting obesity, arterial hypertension, atherogenic dyslipidaemia, and MASLD. MASLD, the most recent nomenclature identifying what had previously been named nonalcoholic fatty liver disease (NAFLD), is defined as SLD in the presence of one or more cardiometabolic risk factor(s) and the absence of harmful alcohol intake [8]. The burden of T2D-related MASLD is substantial [9]. In 2021, MASLD had a prevalence of 1.27 billion people, whereas T2D had a prevalence of 0.51 billion people, with age-standardised prevalence rates of both conditions showing increasing trends over the last decades [10]. In the 21st century, common metabolic diseases pose a significant global health challenge [9], and their global burden is generally higher in males than in females, with the highest age-standardized disability-adjusted life years observed in countries with a low-middle Sociodemographic Index [10].
With this complex backset, our goal is to offer a clear understanding of the bidirectional epidemiological and clinical intersections of MASLD and T2D, as background information useful to better illustrate the role of PPARs in the pathogenesis and drug treatment of MASLD.
A comprehensive literature search was conducted from January 1, 2014, to July 11, 2025, to identify studies examining the role of PPARs in the molecular pathogenesis of T2D-related MASLD. PubMed was searched using the query: (PPARs[Title/Abstract]) AND (type 2 diabetes[Title/Abstract]) AND (liver[Title/Abstract]), resulting in an initial set of 101 articles. To ensure the inclusion of all relevant sources, the reference lists of these articles were also reviewed and cross-checked for additional pertinent studies. Publications were evaluated based on their relevance to PPAR biology, T2D pathophysiology, and liver-related metabolic dysfunctions. Additional bibliographic resources, including Google Scholar, Scopus, and Web of Science, were consulted for a thorough assessment of the topic. Priority was given to the most recent publications, while “historical” citations were maintained when useful for comparison with the most updated studies. To ensure an unbiased synthesis of literature, only articles written in languages other than English were preliminarily discarded. Detailed information on the specific research strategy adopted for each research question is indicated in the footnotes of the tables included in this manuscript.
Data summarized in Table S1 indicate that the prevalence of steatosis/NAFLD/MASLD [11–20] among adults with T2D ranges from 59% [19] to 87% [15]. These variations in the estimated prevalence may be explained by geographical/ethnic differences, T2D patient selection (primary care vs. referral clinics), study design (retrospective vs. prospective), as well as variable sensitivity of the tools used to diagnose MASLD [conventional ultrasonography, FibroScan, magnetic resonance imaging (MRI)]. Of concern, any type of liver fibrosis is found in 28% of cases [14, 20], and “at-risk MASH” (metabolic dysfunction-associated steatohepatitis)—defined as fibrosing liver disease with prominent signs of histological inflammatory activity—is found in 13.6% of cases, with cirrhosis identified in 6.8% of cases [13]. Collectively, these findings support the notion that early diagnosis of MASLD should be part of the holistic assessment among T2D patients [12, 18]. Certain predictors of progressively fibrosing liver disease may further guide the selection of subjects for more in-depth hepatological assessment for aggressive anti-fibrotic drug schedules [13]. These predictors include anthropometric indices suggesting severe (visceral and overall) obesity [12, 14, 18], female sex [20], low educational level [12], hypertransaminasemia [18], and hyperferritinaemia in non-hemochromatosis individuals [16].
Table S2 provides a more comprehensive assessment of the bidirectional association linking NAFLD/MASLD and T2D [21–29].
On one hand, pre-existing NAFLD exposes individuals to a doubled risk of incident T2D over a 5-year follow-up [21, 22]. The risk of incident T2D is also observed in the so-called “lean NAFLD” [26], suggesting that it is not caused by obesity but is rather mediated by the severity of NAFLD/MASLD [23, 26]. On the other hand, adults with T2D have a prevalence of MASLD ranging from 55.5% [24] to 59.67% [22], 65.04% [27], and 65.33% [28] in various studies. The risk appears to be higher in Europe than elsewhere [28], probably mirroring the variable methodology of European studies or a genuinely higher risk of T2D associated with European ethnicities or lifestyle habits.
In parallel with the development of NAFLD/MASLD, T2D also provides a biological milieu that facilitates the progression of liver disease. Accordingly, the global prevalence of NASH and advanced fibrosis among T2D subjects reportedly ranges from 37.3% and 17.0% [24], to 31.55% and 14.95% [27], to 40.78% and 15.49% [28], respectively. Collectively, these data reinforce a holistic approach aimed at triaging NAFLD/MASLD and fibrosing NASH/MASH among individuals with T2D. To this end, it may be important to consider that, in T2D patients, body mass index, age, male sex, the cut-off of vibration-controlled transient elastography and Asian ethnicity are associated with elevated liver stiffness, a surrogate, non-invasive biomarker of liver fibrosis [25].
MASLD, as a multisystem disorder, not only increases the risk of adverse liver outcomes but also has major extra-hepatic complications [30], such as cardiovascular disease (CVD), chronic kidney disease (CKD), and certain extra-hepatic cancers.
Compared to the extensive literature summarized in Table S2, studies assessing the prevalence of T2D among those with NAFLD are significantly limited in number, yet crucial for evaluating the global clinical and economic impact of T2D in patients with NAFLD or MAFLD.
In their meta-analysis of 395 published studies totalling 6,878,568 participants with NAFLD and 1,172,637 participants with MAFLD, Younossi et al. [31] reported that approximately 23% of patients with NAFLD also have T2D. Similarly, a more recent meta-analysis of 395 studies totalling 6,878,568 participants with NAFLD and 1,172,637 participants with MAFLD from 40 countries found that the pooled prevalence of T2D among NAFLD or MAFLD patients was 28.3% [95% confidence interval (CI) 25.2–31.6%] and 26.2% (95% CI 23.9–28.6%) globally [32].
Finally, a study involving 30,633 participants found that NAFLD increased the risk of pre-diabetes and T2D [33]. Conversely, pre-diabetes and T2D were also associated with an increased risk of NAFLD. In cross-lag path analysis, NAFLD was found to significantly affect the incidence of pre-diabetes (β = 0.285, p < 0.001), while the effect on T2D was not statistically significant. Additionally, pre-diabetes and T2D had a weak effect on the risk of NAFLD. In conclusion, this study demonstrates a bidirectional causal association between NAFLD and T2D through progression from NAFLD to pre-diabetes and then to T2D.
The pathways leading from MASLD to pre-diabetes and overt T2D involve the upregulation of hepatokines due to hypercaloric diets and hyperglycaemia, as well as peripheral and hepatic insulin resistance. Additionally, de novo lipogenesis is sustained by the liver being overwhelmed by hyperglycaemia and free fatty acids, leading to increased oxidative stress, inflammation, and fibrosis associated with intrahepatic oxidative phosphorylation [34]. These complex pathogenic connections explain why antidiabetic agents can reduce intrahepatic fat content [35] and effectively cure MASH in randomized controlled trials [34].
The presence of comorbid T2D worsens the natural course of MASLD in many hepatic and extra-hepatic outcomes. In this complex context, PPARs play a major role in the natural history of MASLD due to their characteristics as master regulators of metabolism in various cells and tissues, as well as owing to their sex-specificity [36, 37].
It is universally agreed that T2D significantly increases the risk of developing advanced fibrosis and cirrhosis in patients with MASLD, resulting from the combined effects of insulin resistance, inflammation, and lipotoxicity inherent in both conditions [38]. In a series of 501 individuals with T2D aged 50 years and older, showing a high prevalence of non-invasively assessed liver fibrosis and cirrhosis, Ajmera et al. [11] found that obesity and insulin treatment were associated with increased risks of advanced fibrosis (OR 2.50; 95% CI 1.38–4.54; p = 0.003 and OR 2.71; 95% CI 1.33–5.50; p = 0.006, respectively). In the same study, 2 out of 29 patients with cirrhosis were found to have hepatocellular carcinoma (HCC), and one subject had gallbladder adenocarcinoma [11]. This study clearly illustrates the severe outcomes of the interaction between MASLD and T2D, which also include a higher risk of liver-related mortality and the need for liver transplantation [39].
The pathomechanisms of MASLD-related hepatocarcinogenesis among those with T2D have been reviewed elsewhere [40, 41]. In short, they involve increased reactive oxygen species (ROS) production, endothelial damage, release of proinflammatory cytokines, and activation of the insulin-like growth factor (IGF) pathway. Additionally, sex and liver fibrosis play significant roles as modifiers for the risk of HCC compared to extra-hepatic cancers [42].
In addition to the increased risks of fibrosis, cirrhosis, primary liver cancer, and end-stage liver disease, MASLD has significant extra-hepatic implications [30], such as CVD, CKD, and certain extra‐hepatic cancers [43–45]. Emerging data suggest a progressively rising risk of CVD that is linked to the severity of MASLD. Incorporating liver-specific variables allows for improved risk stratification and targeted interventions to reduce the impact of CVD in this high-risk patient population [46]. In terms of CKD and end-stage renal disease, which are influenced by both T2D and MASLD [47], there appears to be a synergistic and deleterious interaction between these two conditions. In a national population-based retrospective cohort study, Park et al. [48], by analyzing 816,857 individuals over a median follow-up of 7.7 years, found that the presence of NAFLD was associated with a higher risk of adverse clinical outcomes in individuals with CKD.
The spectrum of extra-hepatic neoplasms associated with MASLD includes cancers of the (extra-hepatic) digestive system and urinary tract [45, 49]. Similarly, a meta-analysis of published observational studies assessing the associations between T2D and cancer incidence or mortality, with 151 cohorts globally totalling over 32 million people, 1.1 million cancer cases, and 150,000 cancer deaths [50] documented that T2D was associated with increased risk of nearly 100% for liver, pancreatic, and endometrial cancer, 86% for gallbladder cancer, 67% for kidney cancer, 64% for colon cancer, 62% for colorectal cancer, and less than 50% of other cancer incidences. Additionally, there was a 92% increased risk for pancreatic cancer mortality in individuals with T2D [50]. These findings support a causal association between T2D and liver, pancreatic, and endometrial cancer incidence, as well as pancreatic cancer mortality. The potential pathomechanism connecting MASLD, T2D, and the risks of extra-hepatic cancers has been previously discussed in this journal [51] and will not be reiterated here. The key points regarding the association between MASLD and T2D may be summarized as follows:
The prevalence of MASLD in T2D adults ranges from 59% to 87%.
Various factors can modify this prevalence, including geographical and ethnic differences, the selection of T2D patients, study design, and diagnostic techniques used to identify MASLD.
Liver fibrosis, at-risk MASH, and cirrhosis are present in 28%, 13.6%, and 6.8% of cases, respectively.
Predictors of more advanced forms of MASLD in T2D include severe obesity, sex, ethnicity, educational level, hypertransaminasaemia, and hyperferritinaemia.
Having pre-existing NAFLD doubles the risk of developing incident T2D over a 5-year period, and this risk is also observed in individuals with lean NAFLD.
Between 23% to 28.3% of patients with NAFLD/MAFLD also have T2D.
Having comorbid T2D exacerbates the risks of hepatic and extra-hepatic outcomes in MASLD.
The PNPLA3 I148M variant is a significant genetic risk factor for MASLD, leading to the accumulation of liver inflammation and progression to steatohepatitis, cirrhosis, and HCC, even in individuals without other metabolic risk factors [52]. This gene variant affects the severity of liver disease and fibrotic progression by potentially influencing PPAR signaling. For example, PPARα activation may decrease PNPLA3 expression, while I148M may disrupt PPAR pathways through unclear mechanisms [53]. Moreover, the I148M variant could exacerbate inflammation by interfering with the anti-inflammatory effects of PPARγ. PNPLA3 and PPARs also interact with pathways like liver X receptor-sterol regulatory element-binding protein-1c (LXR-SREBP-1c) involved in liver metabolism and inflammation [53].
PPARα, primarily expressed in organs with high oxidative capacity such as the liver, heart, and muscles, regulates fatty acid oxidation and energy expenditure [54]. By increasing the expression of genes involved in mitochondrial and peroxisomal β-oxidation, PPARα helps preserve the normality of lipid homeostasis. However, in the insulin-resistant state characteristic of T2D-related MASLD, lipid homeostasis can be overwhelmed by excessive free fatty acid influx and dysregulated by inflammatory signalling [55]. PPARβ/δ, which is widely distributed, supports fatty acid catabolism, insulin sensitivity, and mitochondrial function. However, its protective functions can be weakened in the presence of chronic systemic metabolic stress. PPARγ, initially identified as a factor induced during adipocyte differentiation, is mainly found in adipose tissue but is also present in the liver [56]. It plays a crucial role in adipocyte differentiation, lipid storage, and insulin sensitization. Nevertheless, in conditions closely related to T2D, such as adipocyte hypertrophy and inflammation, its activity may be disrupted, leading to a decreased ability to buffer lipids in adipose tissue and increased ectopic accumulation of intrahepatic lipids [56].
In T2D-related MASLD, insulin resistance in the adipose tissue leads to the release of free fatty acids into the bloodstream, overwhelming the liver’s capacity to oxidize them. Under physiological conditions, hepatic PPARα is activated to increase the expression of genes that facilitate β-oxidation, aiding in the efficient clearance of free fatty acids and thereby preventing the harmful build-up of triglycerides [57]. However, in individuals with T2D, hyperinsulinemia, coupled with hyperglycaemia and elevated levels of lipotoxic substrates, such as diacylglycerols and ceramides, can disrupt the normal function of PPARα. The persistently elevated levels of insulin stimulate the production of SREBP-1c, a potent factor that promotes steatogenesis if it is not balanced by PPARα-mediated fatty acid oxidation [58, 59]. Moreover, a pro-inflammatory environment, driven by cytokines like tumour necrosis factor-alpha (TNF-α) and interleukins released by dysfunctional adipose tissue, exacerbates liver damage by activating signalling cascades (e.g., JNK, IKK) that impair insulin signalling and harm hepatocytes. While PPARα possesses anti-inflammatory properties by inhibiting NF-κB and other inflammatory pathways, its effectiveness is compromised when its expression or activity is decreased by the metabolic dysfunctions inherent in T2D, leaving the liver vulnerable to ongoing lipotoxic stress [60].
PPARγ is a master regulator of adipocyte biology, orchestrating the formation of new adipocytes and the expression of enzymes essential for lipid storage and insulin sensitivity [61–63]. In physiological conditions, adipose tissue efficiently stores lipids to prevent their ectopic accumulation in the liver and muscles. However, in T2D, adipose tissue often becomes dysfunctional, leading to increased circulating free fatty acids, which cause steatosis and exacerbate hepatic insulin resistance. Moreover, dysfunctional adipose tissue releases higher levels of pro-inflammatory cytokines, which can hinder PPARγ activity, further worsening insulin resistance and affecting adipocyte differentiation [64]. Clinically, PPARγ agonists like thiazolidinediones (TZDs) have been studied for their ability to improve insulin sensitivity and reduce hepatic gluconeogenesis by enhancing the deposition of lipids in their physiological site, i.e., the adipose tissue, which contributes to reducing the ectopic lipid burden in the liver [65]. Nevertheless, these drugs can also have unwanted side effects such as weight gain, edema, and an increased risk of heart failure in certain patients, highlighting the delicate balance between the benefits and drawbacks of pharmacological PPAR modulation.
Although less extensively studied, PPARβ/δ is now recognized as a potentially influential modulator of energy balance and inflammation in T2D-related MASLD. PPARβ/δ is ubiquitously expressed in mammalian tissues and can promote fatty acid uptake and oxidation in both muscle and liver, thereby supporting systemic insulin sensitivity and metabolic flexibility [66]. Additionally, PPARβ/δ has demonstrated anti-inflammatory actions by inhibiting key inflammatory mediators, which may protect against obesity-related MASLD. However, disturbances in PPARβ/δ signalling, driven by chronic nutrient overload, insulin resistance, or interaction with other concurrent cofactors, can attenuate these favourable effects [66]. For mitochondria to effectively handle the increased load of lipid substrates in T2D, crosstalk with transcriptional co-activators such as PGC-1α is essential [67]. PPARβ/δ partners with PGC-1α to maintain mitochondrial biogenesis and lipid oxidation, but T2D-related epigenetic changes or co-activator dysfunction can undermine this adaptive mechanism, causing incomplete fatty acid oxidation and fuelling further lipotoxicity in the liver [67].
A key dimension of T2D-related MASLD is the interaction between lipid metabolism and inflammation, which Ertunc and Hotamisligil [68] have aptly termed “metaflammation”. In this process, PPARs act in conjunction with innate immune signalling pathways. The accumulation of lipotoxic lipid intermediates triggers endoplasmic reticulum stress responses and oxidative stress, activating JNK and IKK. These cellular responses to metabolic stress interfere with insulin receptor substrate phosphorylation, inhibiting downstream insulin signalling and promoting AP-1-mediated inflammatory gene expression [68]. This persistent sterile inflammation, along with other pro-fibrotic factors, contributes to the progression from simple steatosis to steatohepatitis, characterized by hepatocyte ballooning, mild lobular inflammatory changes, and potential development of chicken wire fibrosis. When PPAR isoforms are sufficiently active, their anti-inflammatory properties can help slow down this histological progression [69]. However, in T2D, epigenetic and post-translational modifications often reduce PPAR availability or ligand affinity, allowing chronic inflammation to progress in an uncontrolled manner.
Adipokines, hepatokines, and myokines secreted by adipose tissue, liver, or muscles, respectively, also directly impact hepatic PPAR function [70]. For example, adiponectin activates AMP-activated protein kinase (AMPK) and PPARα, serving as insulin sensitizers and promoting fatty acid oxidation while suppressing hepatic gluconeogenesis [70]. In T2D, reduced levels of adiponectin weaken PPARα signalling and exacerbate lipid accumulation in the liver. Meanwhile, leptin levels are elevated in obesity and T2D. Although leptin plays a crucial role in regulating appetite and energy expenditure, supranormal levels, indicating leptin resistance, can have negative effects. Hyper-leptinaemia can trigger hepatic fibrogenesis sustained by activated hepatic stellate cells through JAK/STAT and TGF-β pathways [71]. Therefore, the overall impact of adipose-derived signals on hepatic metabolism and inflammation often depends on a delicate balance of biochemical mediators and their interactions with PPARs.
Together, these factors underscore that T2D-related MASLD is not simply a disorder of hepatic lipid overload but rather a systemic disease of energy mismanagement, insulin resistance, and chronic inflammation, in which PPARs play a central coordinating role. Hyperinsulinemia and hyperglycaemia provoke transcriptional changes, such as increased SREBP-1c activity and decreased adiponectin signalling, which accelerate hepatic steatosis and inflammation. At the same time, PPARα’s ability to keep pace with fatty acid influx dwindles, PPARβ/δ struggles to maintain metabolic flexibility in muscle and liver, and the capacity of PPARγ to provide a safe lipid-storage reservoir in adipose tissue is constrained by inflammation and adipocyte dysfunction [72]. These processes culminate in a vicious circle that propagates T2D-related MASLD and elevates the risk of severe liver-related complications in a proportion of cases.
In exploring therapeutic approaches, researchers have evaluated various PPAR agonists to address the hepatic and systemic effects of T2D [73, 74]. Fibrates, which specifically activate PPARα, can slightly reduce hepatic steatosis by enhancing β-oxidation [73]. TZDs that target PPARγ are more effective at improving peripheral insulin sensitivity and may decrease liver fat, but their side effects limit widespread acceptance [75]. Dual PPARα/γ agonists, intended to enhance lipid metabolism and insulin action synergistically, faced safety concerns, leading to investigations on selective PPAR modulators (SPPARMs) that maximize benefits while minimizing adverse effects like weight gain and cardiovascular issues [76]. Recent studies have indicated that the naturally occurring flavonoid alpinetin shows promise as a therapeutic option for T2D treatment [77]. Additionally, PPARβ/δ agonists are being recognized for their insulin-sensitizing properties, but further evaluation is needed on their long-term safety and efficacy in hepatic disease [73]. Current advancements in medicinal chemistry, along with a growing understanding of PPAR interactions with co-activators, co-repressors, and epigenetic regulators, offer hope for more personalized interventions tailored to individual patient phenotypes.
Despite the notable role of pharmacotherapy, lifestyle interventions are paramount in the field of T2D-related MASLD. Weight loss, caloric restriction, reduction of excess carbohydrate consumption, and physical exercise can independently restore, at least partially, normal PPAR activity by correcting hyperinsulinemia, reducing inflammatory stress, and draining ectopic lipid pools [78]. Exercise particularly enhances muscle PPARα- and PPARβ/δ-driven lipid oxidation, reducing plasma free fatty acids that would otherwise exacerbate hepatic steatosis. Sustained caloric restriction can lead to a net improvement in adipose tissue health, supporting PPARγ function and lowering systemic inflammation [79]. These interventions, however, require patient engagement and long-term adherence, and are most advantageous if complemented by pharmacological agents that support or amplify PPAR-driven improvements in metabolic homeostasis.
Overall, the multifaceted roles of PPARs in the molecular pathogenesis of T2D-related MASLD illustrate how defects in nuclear receptor signalling, coupled with disruptions in insulin action, adipokine profiles, and inflammatory pathways, converge to drive liver disease progression. PPARα, PPARβ/δ, and PPARγ each contribute uniquely to hepatic lipid flux, adipose remodelling, and systemic insulin sensitivity, and their impairment under metabolic stress aggravates the cycle of lipotoxic injury and chronic inflammation characteristic of MASLD. As research has increasingly revealed epigenetic and post-translational layers of regulation, it has become clear that restoring PPAR function depends on a wide array of factors, including nutrient sensing, co-regulator availability, and inflammatory status [73, 78, 79]. These insights foster a more nuanced approach to therapy that exceeds simply activating or inhibiting a single PPAR isoform to addressing cofounding elements like SREBP-1c, NF-κB, or AMPK. Novel drug candidates strive to refine the therapeutic index by selectively targeting PPAR subsets in specific tissues or by combining PPAR modulation with other pathways, such as those acted upon by GLP-1 receptor agonists, SGLT2 inhibitors, or bile acid-related nuclear receptors like FXR, to achieve comprehensive metabolic control [80].
From a pathophysiological perspective, renaming NAFLD to MASLD reminds us that the development of steatosis and its progression to histologically more advanced stages of liver disease are driven by a constellation of metabolic derangements, of which PPAR dysfunction is a key component. In the pathogenic trajectory of T2D-related MASLD, the effects of PPARs on gene transcription, lipid partitioning, and inflammatory response extend far beyond the liver, heavily involving the adipose tissue, skeletal muscle, and immune cells [54, 81]. Therefore, it is vital to integrate an organ-spanning perspective in research and therapy development. The prospect of personalized medicine approaches, associating genetic and epigenetic markers of PPAR function to guide therapy choices, is appealing. Patients who exhibit robust responses to certain PPAR agonists may benefit significantly from such tailored approaches, whereas others may need combination regimens [82].
The systemic nature and complex pathogenesis of MASLD and MASH help to explain why oversimplified therapeutic approaches have yielded disappointing results [83]. In this context, the idea that different PPARs have varying activities on different cell and tissue targets helps to explain why Pan-PPAR agonists, such as Lanifibranor, can have pleiotropic actions and enhanced therapeutic potential in MASH and MASH-related hepatic and extra-hepatic morbidity [84]. Based on this conceptual background, the most important clinical trials with PPAR-agonists published in the MASLD arena are summarized in Table 1 [85–97].
Therapeutic trials on PPAR agonists in MASLD.
| Author, year [Ref]* | Study design, patients enrolled, duration of treatment | Biochemical response | Liver histology/imaging response, side effects |
|---|---|---|---|
| Laurin et al., 1996 [85] | 16 in the Clofibrate arm, 24 in the UDCA arm, for 1 year. | Decreased ALP in the Clofibrate arm. | No change in the Clofibrate arm. |
| Belfort et al., 2006 [86] | RCT with 6 months of a low-calorie diet with Pioglitazone (n = 26) or diet and placebo (n = 21). | Transaminases decreased in the Pioglitazone arm. | Improved steatosis, inflammation, and ballooning, with no change in fibrosis, weight gain, despite a low-caloric diet in the Pioglitazone arm. |
| Fernández-Miranda et al., 2008 [87] | Fenofibrate (n = 16) for 48 weeks, with no control arm. | Significant decreases were observed in TG, glucose, liver enzymes, and MetS. | Decreased ballooning, no change in grade of steatosis and inflammation/fibrosis. |
| Ratziu et al., 2008 [88] | RCT with Rosiglitazone (n = 32), placebo (n = 31). | Normalized transaminase levels (38% vs. 7%, p = 0.005). | Improved steatosis (47% vs. 16%; p = 0.014), although only half of the patients responded, with no change in other histologic parameters, and weight gain in the Rosiglitazone arm. |
| Sanyal et al., 2010 [89] | RCT with Pioglitazone (n = 80), vitamin E (n = 83), and placebo (n = 84) for 96 weeks. | Transaminases improved in the vitamin E and Pioglitazone arms. | Improvement in NASH compared to placebo (vitamin E, p = 0.001), with Pioglitazone (p = 0.04), both vitamin E and Pioglitazone were associated with significant reductions in steatosis and lobular inflammation. Improvement in fibrosis, with weight gain in Pioglitazone. |
| Torres et al., 2011 [90] | RCT with n = 137, comparing Rosiglitazone and metformin to Rosiglitazone and losartan vs. Rosiglitazone alone for 48 weeks. | Transaminases decreased in all groups. | 108 completed the study, with an overall improvement in all histologic parameters, and no added benefit of metformin (did not prevent weight gain) or losartan. |
| Cusi et al., 2016 [91] | RCT for 18 months followed by an 18-month open-label phase with Pioglitazone (n = 50) or placebo (n = 51). | More normalization in the Pioglitazone arm. | Pioglitazone is associated with a better NAS reduction and resolution of NASH, steatosis, inflammation, and ballooning, with no improvement in fibrosis or weight gain. |
| Ratziu et al., 2016 [92] | Elafibranor 120 mg, Elafibranor 80 mg, and placebo. | Liver enzymes, lipids, glucose profiles, and markers of systemic inflammation were significantly reduced in the Elafibranor 120 mg group. | Elafibranor 120 mg is superior to placebo, with NASH resolution without worsening of fibrosis in 19% vs. 12% in the placebo group (p = 0.045), based on a post hoc analysis for the modified definition. |
| Francque et al., 2021 [93] | RCT for 24 weeks with Lanifibranor 1,200 mg (n = 83), Lanifibranor 800 mg (n = 83), and placebo (n = 81). | Levels of liver enzymes decreased, and most biomarkers of lipid metabolism, inflammatory activation, and liver fibrosis improved with Lanifibranor. | Both the 1,200 mg and 800 mg doses of Lanifibranor were superior to placebo for NASH resolution without worsening of fibrosis (49% and 39%, respectively, vs. 22%), improvement in fibrosis stage of at least 1 without worsening of NASH (48% and 34%, respectively, vs. 29%), and NASH resolution NASH + improvement in fibrosis stage of at least 1 (35% and 25%, respectively, vs. 9%). The dropout rate for adverse events was less than 5% and similar across the trial groups. Diarrhea, nausea, peripheral edema, anemia, and weight gain occurred more frequently with Lanifibranor than with placebo. |
| Gawrieh et al., 2021 [94] | RCT for 16 weeks with Saroglitazar 1 mg (n = 26), 2 mg (n = 25), or 4 mg (n = 27) vs. placebo (n = 28). | The least-squares mean percent change from baseline in ALT at week 16 was –25.5% (5.8), –27.7% (5.9), and –45.8% (5.7), with Saroglitazar 1 mg, 2 mg, and 4 mg, respectively, vs. 3.4% (5.6) in placebo (p < 0.001 for all). Saroglitazar 4 mg improved adiponectin [–0.3 μg/mL (0.3) vs. 1.3 μg/mL (0.3)], HOMA-IR [–1.3 (1.8) vs. –6.3 (1.7)], and TG [–5.3 mg/dL (10.7) vs. –68.7 mg/dL (10.3)] (p < 0.05 for all), lipoprotein particle composition and size, and reduced lipotoxic lipid species. | Compared to placebo, Saroglitazar 4 mg improved LFC [4.1% (5.9) vs. –19.7% (5.6)], and Saroglitazar was well-tolerated. A mean weight gain of 1.5 kg was observed with Saroglitazar 4 mg vs. 0.3 kg with placebo (p = 0.27). |
| Grobbee et al., 2022 [95] | Post-hoc analysis of an RCT enrolling 7,226 T2D individuals with recent CAD assigned to receive aleglitazar or placebo for two years. | LFS, LAP, and FIB-4 decreased with treatment, while scores in the placebo group remained unaffected or increased (p < 0.001). NFS responded differently but remained consistently lower than placebo. In the treatment group, more participants shifted to a lower FIB-4 and NFS category or improved with respect to the LAP cut-off values compared to placebo (p < 0.001 for FIB-4 and LAP, p < 0.004 for NFS). | NA |
| Cooreman et al., 2024 [96] | Secondary and exploratory outcomes of the NATIVE trial (ClinicalTrials.gov NCT03008070) were analyzed. | With Lanifibranor, TG, HDL-C, apolipoproteins, insulin, HOMA-IR, HbA1c, FG, hs-CRP, ferritin, and diastolic BP improved significantly, regardless of diabetes status. Most patients with pre-diabetes returned to normal FG levels. Significant increases in adiponectin levels correlated with improvement in hepatic and CMH markers. | Steatosis improved significantly, independent of diabetes status. Patients had an average weight gain of 2.5 kg, with 49% gaining at least 2.5% weight. Therapeutic benefits were similar regardless of weight change. |
| Tan et al., 2025 [97] | This prospective cohort study enrolled 235 T2D patients with MASLD who received either Chiglitazar or other glucose-lowering medications over a 24-week period. Forty Chiglitazar users, 195 non-Chiglitazar users, and 31 matched pairs were derived after 1:1 PSM. | NA | The adjusted mean reduction in CAP from baseline to 24 weeks was significantly greater in the Chiglitazar group [–28.38 dB/m (95% CI: 36.11 to –20.65)] than in the non-Chiglitazar group [–16.74 dB/m (95% CI: –24.47 to –9.01)], with a between-group difference of –11.64 dB/m (95% CI: –22.38 to –0.90, p = 0.038). LSM changes were similar between groups [difference in LS mean: 0.11 (95% CI: –1.04 to 0.82), p = 0.813]. |
*: These original studies have been identified using the keywords “PPAR” AND “NAFLD” OR “MAFLD” OR “MASLD” OR “NASH” OR “MASH” AND “trial”. Studies regarding primary biliary cholangitis, cardiovascular outcomes unlinked from liver health were excluded. ALP: alkaline phosphatase; ALT: alanine aminotransferase; BP: blood pressure; CMH: cardiometabolic health; CAD: coronary artery disease; CAP: controlled attenuation parameter; FG: fasting glucose; FIB-4: Fibrosis-4; HbA1c: glycosylated hemoglobin; HDL-C: high-density lipoprotein cholesterol; hs-CRP: high-sensitivity C-reactive protein; HOMA-IR: homeostatic model assessment-insulin resistance; LFC: liver fat content; LAP: liver accumulation product; LFS: liver fat score; LSM: liver stiffness measurement; MASLD: metabolic dysfunction-associated steatotic liver disease; MetS: metabolic syndrome; NA: not addressed; NAFLD: nonalcoholic fatty liver disease; NAS: NAFLD activity score; NFS: NAFLD fibrosis score; PPAR: peroxisome proliferator-activated receptor; PSM: propensity score matching; RCT: randomized controlled trial; T2D: type 2 diabetes; TG: triglycerides.
Lin et al. [98] conducted a systematic review of relevant randomized double-blind controlled trials published until July 11, 2024. They found that for histologically proven fibrosis improvement, GLP-1-based poly-agonists were the most potentially effective, followed by FGF-21 analogues, THR-β agonists, Pan-PPAR agonists, and GLP-1R agonists. For MASH resolution in histology, GLP-1-based polyagonists were the most potentially effective, followed by THR-β agonists, GLP-1R agonists, FGF-21 analogues, and Pan-PPAR agonists.
The Pan-PPAR agonist Lanifibranor, at the highest dose, offers the chance to ameliorate hard liver histology outcomes among subjects with noncirrhotic NASH with a high grade of activity [93]. This implies that the activation of all three PPAR isotypes synergizes their therapeutic activity on multiple cell and target organs, thereby improving glucose and lipid metabolism and, simultaneously, also pro-inflammatory and pro-fibrogenic pathways [73, 99, 100].
Conversely, single PPAR agonists, such as various fibrates and glitazones, demonstrate strong anti-MASH activity in preclinical models. However, they only exhibit modest clinical efficacy on severe liver histology endpoints like MASH resolution and fibrosis regression [84]. Moreover, a clinically relevant and specific feature of PPAR agonists in the context of T2D-related MASLD is their positive impact on parameters of glucose and lipid metabolism in individuals with T2D [101]. Chatterjee et al. [101] observed this metabolic enhancement in their study of T2D patients treated with Saroglitazar, a novel dual PPARα and PPARγ agonist. After 14 weeks of treatment, participants experienced significant reductions in fasting and post-prandial plasma glucose, glycosylated haemoglobin, total cholesterol, low-density lipoprotein cholesterol, triglycerides, non-high-density lipoprotein cholesterol, and the ratio of triglyceride to high-density lipoprotein cholesterol. These improvements were observed without notable changes in body weight, blood pressure, high-density lipoprotein cholesterol, and serum creatinine. A concise overview of the isoform-specific dysfunctions, their clinical correlates, and the effects of corresponding agonists is provided in Table 2.
Linking isoform-specific PPAR biology to clinical manifestations and therapeutic outcomes in T2D-associated MASLD.
| Isoform | Predominant expression | Key biological role(s) | Typical clinical consequence(s) when signalling is impaired | Representative therapeutic outcome(s) when the isoform is pharmacologically activated |
|---|---|---|---|---|
| PPARα | Predominant in liver, heart, and muscle | Induces mitochondrial and peroxisomal β-oxidation and ketogenesis, enhancing hepatic FFA disposal | Accumulation of intra-hepatic triglycerides and higher ALT/AST levels increase the risk of simple steatosis progressing to MASH | Fibrate monotherapy modestly lowers liver-fat content and transaminases but shows limited histological reversal of fibrosis in humans (cf. Table 1, e.g., fenofibrate, clofibrate) |
| PPARβ/δ | Ubiquitous; muscle-centric | Promotes fatty-acid uptake and oxidation in skeletal muscle and liver; supports mitochondrial biogenesis via PGC-1α | Reduced metabolic flexibility, peripheral insulin resistance and higher circulating FFAs that spill over to the liver contribute to this progression | Early-phase β/δ agonists improve insulin sensitivity in pre-clinical MASLD, but long-term human data remain scarce; safety and durability are still under evaluation |
| PPARγ | Adipose-dominant; inducible in liver | Governs adipocyte differentiation and safe lipid storage; enhances systemic insulin sensitivity | Adipocyte dysfunction triggers ectopic fat deposition, worsens insulin resistance and leads to an inflammatory adipokine profile | Thiazolidinediones (e.g., Pioglitazone) improve NAS and achieve MASH resolution in ~35–40% of biopsy-proven cases but cause weight gain and fluid retention (see Table 1) |
| Pan-PPAR | Simultaneous α + β/δ + γ | Integrates oxidation (α, β/δ) with adipose buffering and insulin sensitisation (γ), while counter-modulating NF-κB-driven inflammation | The combined dysfunction of these factors fuels lipotoxicity, metaflammation and accelerates fibrosis | Lanifibranor 1.2 g/day resolves MASH without worsening fibrosis in ~49% and improves ≥ 1 stage of fibrosis in ~48% of patients, outperforming placebo in a 24-week RCT |
ALT: Alanine aminotransferase; AST: aspartate aminotransferase; FFA: free fatty acid; MASH: metabolic dysfunction-associated steatohepatitis; MASLD: metabolic dysfunction-associated steatotic liver disease; NAS: nonalcoholic fatty liver disease activity score; PPAR: peroxisome proliferator-activated receptor; RCT: randomized controlled trial; T2D: type 2 diabetes.
In summary, PPAR agonists like Pioglitazone and Lanifibranor offer potential benefits for MASLD and MASH, including improved liver health and insulin resistance. Pioglitazone can improve SLD and inflammation in MASH, especially when associated with T2D. It can reduce the NAFLD activity score (NAS) by 2 points and lead to MASH resolution without worsening fibrosis [102]. Saroglitazar usage decreases alanine aminotransferase (ALT) levels, liver fat content, insulin resistance, and dyslipidemia in MASLD patients [94]. Lanifibranor induces significant reductions in liver fat content in T2D patients with MASLD, while also improving hepatic and muscle insulin resistance and adipose tissue biology [103]. However, side effects and limitations exist, and careful patient selection and monitoring are crucial. While PPAR agonists show promise, more research is needed to optimize their use, identify the best candidates for treatment, and develop strategies to mitigate side effects [104]. In conclusion, the improved liver health associated with the usage of PPAR agonists may be envisaged as follows:
PPAR agonists can help reduce liver fat content, hepatic inflammation, and liver fibrosis in MASLD/MASH, potentially decreasing the risk of progression to more severe liver disease.
By enhancing insulin sensitivity, PPAR agonists have the potential to target the main driver of systemic metabolic dysfunction in MASLD.
Some PPAR agonists have shown promise in resolving MASH without worsening fibrosis.
Moreover, as regards limitations and risks, it must be remembered that:
Pioglitazone must be used with caution in patients with pre-existing heart failure or other cardiovascular conditions, as it is linked to an increased risk of bone fractures and bladder cancer.
While some PPAR agonists show promise, others may not yield significant improvements in all patients, especially those with advanced fibrosis or cirrhosis.
PPAR agonists need to be used long-term to maintain their benefits.
MicroRNAs (miRNAs) have gained prominence as approximately 22-nucleotide, non-coding regulators that fine-tune glucose handling, lipid turnover, inflammatory tone, and angiogenesis. Dysregulated miRNA profiles not only contribute to obesity, T2D, and CVD but also circulate stably in serum, making them attractive non-invasive biomarkers and therapeutic targets for the metabolic-syndrome continuum [105]. In addition to these post-transcriptional insights, a recent study showed that a methanolic leaf extract of Alpinia calcarata (A. calcarata) administered to high-fat-diet mice (200 mg/kg) significantly lowered body-weight gain, visceral-fat mass, total cholesterol, and triglycerides. The extract also down-regulated IL-6, COX-2, MCP-1, TNF-α, GLUT-4, and notably, PPARγ transcripts in adipose tissue, thereby attenuating adiposity and adipocyte inflammation [106]. Together, miRNA-based diagnostics and PPAR-targeting phytochemicals such as A. calcarata represent complementary, rapidly evolving strategies for intercepting the molecular drivers of metabolic syndrome and T2D-related MASLD.
There are several areas of heterogeneity that must be acknowledged when interpreting PPAR-centered therapeutics. First, circulating adiponectin and resistin levels, often used as indicators of PPARα/γ activity, show conflicting results across studies [107, 108]. While levels consistently rise with weight loss and TZD therapy, some studies have shown neutral or decreased concentrations after selective PPARα agonism. This suggests that there are influences specific to isoforms, tissues, and assays that complicate their use as universal biomarkers. Second, differences in histological fibrosis outcomes between key trials, such as the lack of effect of Pioglitazone in the PIVENS study compared to the significant improvement seen with Pan-agonist Lanifibranor, highlight how factors like study design (duration, dose, biopsy timing) and patient characteristics (diabetes status, baseline NAS, PNPLA3 genotype) can impact treatment outcomes [109, 110]. These considerations emphasize the need for an integrative pathophysiological framework that explains how systemic insulin resistance, communications between adipose tissue and muscles, and activation of hepatic stellate cells contribute to MASLD progression through isoform-specific PPAR dysfunction. This framework can also guide future strategies for combination therapies or precision medicine approaches (Figure 2).
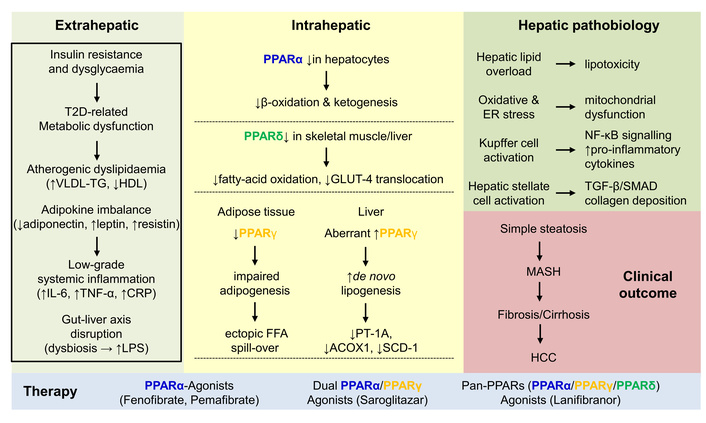
Systemic T2D perturbations and their impact on PPARs. Insulin resistance induces metabolic dysfunction, dyslipidaemia, adipokine imbalance, inflammation, and alterations in the gut-liver axis. These extrahepatic manifestations provoke changes in PPAR isoform expression, which in the liver, lead to steatosis by decreasing β-oxidation and fatty-acid oxidation, while increasing de novo lipogenesis. These changes result in lipotoxicity, mitochondrial dysfunction, and activation of Kupffer cells and hepatic stellate cells. The clinical outcomes can range from simple steatosis, MASH, fibrosis/cirrhosis, or even HCC. Various PPARα agonists, dual-specific agonists (PPARα/γ), or pan-specific agonists (PPARα/γ/δ) are available on the market to reduce high cholesterol and triglyceride levels in patients with diabetes. CRP: C-reactive protein; FFA: free fatty acid; HCC: hepatocellular carcinoma; HDL: high-density lipoprotein; MASH: metabolic dysfunction-associated steatohepatitis; PPARs: peroxisome proliferator-activated receptors; T2D: type 2 diabetes; TG: triglycerides.
T2D amplifies metabolic stress in the liver, adipose tissue, and muscle, thereby blunting the physiological actions of PPARα, PPARβ/δ, and PPARγ on fatty-acid oxidation, mitochondrial biogenesis, lipid storage, and anti-inflammatory signalling. The resulting overflow of free fatty acids, hyperinsulinaemia, and chronic “metaflammation” drive the transition from simple steatosis to MASLD and its progressive form, MASH, with downstream risks of fibrosis, cirrhosis, and HCC. Although single-isoform agonists (fibrates, glitazones) modestly improve steatosis and insulin sensitivity, only Pan-PPAR agonism (e.g., Lanifibranor) has so far achieved both MASH resolution and fibrosis improvement in randomised trials, highlighting the need for coordinated activation of all three receptor subtypes. Going forward, integration of lifestyle intervention, precise selection of PPAR-targeted drugs, and combination therapy with incretin or FXR pathways are expected to maximise clinical benefit while minimising adverse effects in T2D-related MASLD.
Key messages for readers comprise the following:
Insulin resistance in adipose tissue, muscle, and liver disrupts PPAR signalling and initiates the metabolic cascade that drives MASLD progression.
Under normal conditions, PPARα, PPARβ/δ, and PPARγ regulate fatty acid oxidation, metabolic flexibility, and safe lipid storage.
When their function is impaired, steatosis, inflammation, and fibrosis can occur.
In individuals with T2D, increased free fatty acid flux, hyperinsulinaemia, and chronic “metaflammation” overwhelm the protective PPAR pathways.
While single-isoform agonists like fibrates and glitazones provide limited histological improvement, Pan-PPAR activation with Lanifibranor has shown success in resolving MASH and improving fibrosis in clinical trials.
Lifestyle changes such as weight loss, exercise, and caloric restriction can help lower insulin and free fatty acid levels, partially restoring endogenous PPAR activity and enhancing pharmacotherapy.
Combining PPAR agonists with incretin, SGLT2, or FXR pathways in rational combination therapy may optimize metabolic and hepatic benefits while minimising side effects.
Early non-invasive screening and the use of emerging PPAR-driven biomarkers can help identify patients who are most likely to respond to treatment and objectively monitor treatment outcomes.
Personalised modulation of all three PPAR isoforms provides a comprehensive solution for T2D-related MASLD, addressing liver, cardiometabolic, and inflammatory risks simultaneously.
As crucial regulators strategically positioned at the intersection of energy metabolism, insulin action, and inflammatory responses, PPARs are essential in maintaining hepatic and systemic metabolic homeostasis. When T2D disrupts hormonal and adipokine signals, increases free fatty acid flux, and chronically promotes metaflammation, the protective functions of PPARs, particularly their ability to enhance fatty acid oxidation, regulate lipid storage in adipose tissue, and reduce pro-inflammatory processes, are compromised. This leads to the development of MASLD, where the liver experiences lipid toxicity, oxidative stress, and fibrotic signalling [111]. Characterization of the molecular mechanisms of PPARα, PPARβ/δ, and PPARγ, as well as their interactions with cofactors like PGC-1α, and pathways like SREBPs, NF-κB, AMPK, and adipokines, may pave the way for novel preventive and therapeutic avenues for T2D-related MASLD.
With this pathobiological background, PPAR agonists offer a comprehensive approach to the pathophysiology and histogenesis of MASLD and MASH in the context of the metabolic syndrome featuring systemic metabolic dysfunction [2, 101, 112, 113]. Several drug agents, such as Semaglutide, Tirzepatide, Survodutide, Lanifibranor, Pegozafermin, and Resmetirom, have shown significant promise in resolving MASH and improving fibrosis. However, unresolved issues persist regarding treatment duration, response heterogeneity, and long-term patient compliance [114]. Future directions include:
Extensive research to identify the ideal clinical profile of the best candidate to receive PPAR agonists. Non-invasive PPAR-driven biomarkers need to be identified to predict the individual patient’s response to therapy.
The rational combination of PPAR agonists with other classes of effective drugs against MASH, such as Resmetirom, GLP-1Ras, and SGLT2i, must be assessed.
Strategies aimed at decreasing the burden of unwanted side effects need to be developed.
Continued encouragement of lifestyle modifications is crucial in preventing and managing T2D-related MASLD.
Through these concerted efforts, deeper insights into disease mechanisms can be gained, ultimately improving patient outcomes by leveraging the central and versatile roles of PPARs in hepatic and systemic metabolic regulation.
AMPK: AMP-activated protein kinase
CI: confidence interval
CKD: chronic kidney disease
CVD: cardiovascular disease
HCC: hepatocellular carcinoma
LXR-SREBP-1c: liver X receptor-sterol regulatory element-binding protein-1c
MASH: metabolic dysfunction-associated steatohepatitis
MASLD: metabolic dysfunction-associated steatotic liver disease
miRNAs: microRNAs
MRI: magnetic resonance imaging
NAFLD: nonalcoholic fatty liver disease
NAS: nonalcoholic fatty liver disease activity score
PPARs: peroxisome proliferator-activated receptors
SLD: steatotic liver disease
T2D: type 2 diabetes
TNF-α: tumour necrosis factor-alpha
TZDs: thiazolidinediones
The supplementary tables for this article are available at: https://www.explorationpub.com/uploads/Article/file/100590_sup_1.pdf.
The authors are grateful to Sabine Weiskirchen for preparing Figure 1 of this article. AI-Assisted Work Statement: During the preparation of this work, authors used Free AI Editing for language polishing (not content creation); no generative AI writing tools were used. After using the tool, authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
AL and RW: Conceptualization, Methodology, Software, Data curation, Supervision. Validation, Writing—original draft, Writing—review & editing. Both authors have read and agreed to the published version of the manuscript.
Amedeo Lonardo, who is the Associate Editor and Guest Editor of Exploration of Digestive Diseases, and Ralf Weiskirchen, who is the Guest Editor of Exploration of Digestive Diseases, had no involvement in the decision-making or the review process of this manuscript.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

