 Review
Review

Affiliation:
1Service d’Hépato-Gastroentérologie, CHU Dupuytren, 87042 Limoges, France
2Unité INSERM U-1248, Pharmacologie & Transplantation, Faculté de Médecine et de Pharmacie, Limoges University, 87042 Limoges, France
Email: paul.carrier@chu-limoges.fr
ORCID: https://orcid.org/0000-0001-9750-2506
Affiliation:
1Service d’Hépato-Gastroentérologie, CHU Dupuytren, 87042 Limoges, France
2Unité INSERM U-1248, Pharmacologie & Transplantation, Faculté de Médecine et de Pharmacie, Limoges University, 87042 Limoges, France
ORCID: https://orcid.org/0000-0001-6039-1355
Affiliation:
3Service d’Hématologie clinique et de thérapie cellulaire, Centre national de référence Amylose AL et autres maladies de dépôts d’immunoglobulines monoclonales, CHU Limoges, 87042 Limoges, France
ORCID: https://orcid.org/0000-0001-7514-9414
Affiliation:
4Service de Maladies infectieuses, CHU Limoges, 87042 Limoges, France
ORCID: https://orcid.org/0000-0002-5341-2872
Affiliation:
5Service d’Hépato-Gastroentérologie, d’Addictologie et de Nutrition, Groupe hospitalier public du Sud de l’Oise, Centre hospitalier Creil, 60100 Creil, France
ORCID: https://orcid.org/0000-0002-8442-4526
Affiliation:
3Service d’Hématologie clinique et de thérapie cellulaire, Centre national de référence Amylose AL et autres maladies de dépôts d’immunoglobulines monoclonales, CHU Limoges, 87042 Limoges, France
Email: arnaud.jaccard@unilim.fr
ORCID: https://orcid.org/0000-0001-8065-3199
Affiliation:
1Service d’Hépato-Gastroentérologie, CHU Dupuytren, 87042 Limoges, France
2Unité INSERM U-1248, Pharmacologie & Transplantation, Faculté de Médecine et de Pharmacie, Limoges University, 87042 Limoges, France
ORCID: https://orcid.org/0000-0002-6951-0784
Explor Dig Dis. 2025;4:100589 DOI: https://doi.org/10.37349/edd.2025.100589
Received: June 22, 2025 Accepted: August 13, 2025 Published: August 28, 2025
Academic Editor: Nahum Méndez-Sánchez, National Autonomous University of Mexico, Mexico
Amyloidosis is a rare disease, corresponding to a deposition of proteins in various tissues. Amyloid light-chain (AL) amyloidosis can involve the liver in 17% to 45% of patients. Diagnosis of liver disease is based on specific criteria, coupling alkaline phosphatases and hepatomegaly. Liver stiffness is altered in cases of heart involvement, and overall, in cases of liver involvement. Liver biopsy is generally avoided due to an important bleeding risk. Treatment is essentially based on stem cell transplantation and chemotherapy, with large progress during the last decade. Liver involvement recovery is generally diagnosed with a reduction in alkaline phosphatases and in liver size.
Amyloidosis is a rare and heterogeneous disease characterized by the deposition of beta-fibrillar proteins in various tissues. It was first described by Rudolf Virchow in 1853 [1]. Sixty forms of amyloidosis have been identified, 27 of which are potentially associated with human diseases. The clinical presentation can be systemic or, in some cases, restricted to a single organ.
The liver is primarily affected in amyloid light-chain (AL) amyloidosis, the most common form, which results from the deposition of monoclonal immunoglobulin light chains [2]. Amyloid A (AA) amyloidosis, typically associated with chronic inflammatory conditions, is caused by the deposition of serum AA (SAA) protein. While it can lead to gastrointestinal manifestations, the liver is usually spared. Finally, in hereditary transthyretin amyloidosis (ATTR), the liver is not directly affected by the disease but is the site of transthyretin production.
Amyloidosis is a rare disease with a prevalence ranging between 1/17,000 and 1/50,000 in Europe and the USA [3]. AL amyloidosis accounts for approximately 80% of cases [4]. The disease is most diagnosed between the ages of 50 and 70, with fewer than 10% of cases occurring before the age of 50. In the study of Kyle et al. [5], men were involved in 54% of cases. Among AL amyloidosis cases, 75% to 80% involve lambda light chains, while the remaining 20% to 25% involve kappa light chains [6]. Overall, the incidence of amyloidosis appears to be increasing [7].
The hallmark of amyloidosis is the abnormal folding of soluble precursor proteins [8].
AL amyloidosis is considered a multi-organ disease, with the heart, spleen, peripheral nervous system, kidneys, and gastrointestinal tract being the most frequently affected. It results from the abnormal production of light chains by clonal plasma cells, typically in elderly patients. These light chains are thermodynamically and kinetically unstable, leading to self-aggregation into misfolded oligomers. These oligomers subsequently form cross-beta amyloid fibrils, which are 8–10 nm in diameter as observed by electron microscopy [9]. These fibrils exert toxic effects on organs by interacting with serum amyloid P component (SAP) and glycosaminoglycans [10]. Their accumulation disrupts tissue architecture, leading to organ dysfunction through both a mass effect and a proteotoxic mechanism involving precursor protein oligomers [11]. In the liver, amyloid proteins accumulate in the space around hepatocytes, especially in sinusoids and portal areas, leading to distortion and compression of hepatocytes. Sinusoidal deposition can compress the hepatocytes, leading to hepatocellular atrophy and can efface the overall architecture. These modifications can lead to liver congestion, giving an increase in liver stiffness. In cardiac involvement, activation of the p38 MAP kinase pathway has been associated with increased cell apoptosis and elevated pro-BNP (pro-brain natriuretic peptide) levels.
A genetic influence is observed in approximately 50% of cases, with a chromosomal translocation t(11;14) involving the immunoglobulin heavy-chain locus (IgH) and the oncogene cyclin D1 [12]. In 10% of cases, diploidy, a common feature in multiple myeloma, is reported [13]. Additionally, mutations in the IGLV gene family may contribute to amyloid fibril formation.
There are no well-defined risk factors for AL amyloidosis, except for monoclonal gammopathy of undetermined significance (MGUS), which confers a relative risk of 8.8. AL amyloidosis is diagnosed in 10% to 15% of multiple myeloma patients, and Congo red-positive deposits in subcutaneous fat aspirates or bone marrow are observed in 38% of these patients [14]. Agent Orange, an herbicide used during the Vietnam War, was suspected to increase the risk of amyloidosis, but this association has not been confirmed [15].
Liver involvement is described in 17% to 45% of patients diagnosed with AL amyloidosis [3, 16, 17]. Specific data concerning age and gender are limited concerning liver involvement, just a Chinese study suggests a ratio of male-female near 3:1 [18]. The fact that patients with AL amyloidosis are old can impact liver function and protein homeostasis, which may influence amyloid deposition.
Autopsy studies report a prevalence of 80% [6]. Isolated liver involvement is rare, occurring in only 1% of cases, while in 5% of patients, the liver is the dominant affected organ. Without treatment, the median survival in cases of hepatic amyloidosis is approximately 8.5 months [19]. Two-thirds of patients with liver involvement also present with cardiac or renal amyloidosis, which has a greater impact on survival.
The classical clinical presentation includes hepatomegaly, typically exceeding 15 cm on imaging, and an elevation of alkaline phosphatase levels above 1.5 times the upper normal limit [20]. However, diagnosis can be challenging in cases of isolated hepatomegaly or isolated elevation of alkaline phosphatase [21–23]. Other, less common presentations include cholangitis, portal hypertension, or, in rare cases, spontaneous hemorrhagic rupture [24–26]. There is evidence that amyloidosis can compromise liver vascular integrity, leading to fragility and hemorrhage. Also, it can lead to ascites formation.
Amyloidosis is often not considered initially, with the correct diagnosis suspected in only 26% of cases before liver biopsy. Furthermore, transaminases (AST and ALT) are not considered sensitive markers for liver amyloidosis, in contrast to alkaline phosphatase. There is no strong evidence that particular liver diseases increase the risk of AL amyloidosis.
Liver biopsy carries a high risk of bleeding and is generally contraindicated or avoided due to factor X deficiency, loss of von Willebrand factor, and amyloid deposition within the walls of blood vessels, all of which increase hemorrhagic risk [27, 28].
Imaging techniques such as ultrasound and computed tomography (CT) scan are not specific but are useful for differential diagnosis and follow-up (Figure 1). In 1988, 123I-labeled SAP scintigraphy was introduced as a non-invasive method for detecting amyloid deposits in the liver, kidneys, adrenal glands, and spleen [29]. Lovat et al. [30] demonstrated that SAP scintigraphy is highly specific and sensitive, but its availability is limited, and the cost is high.
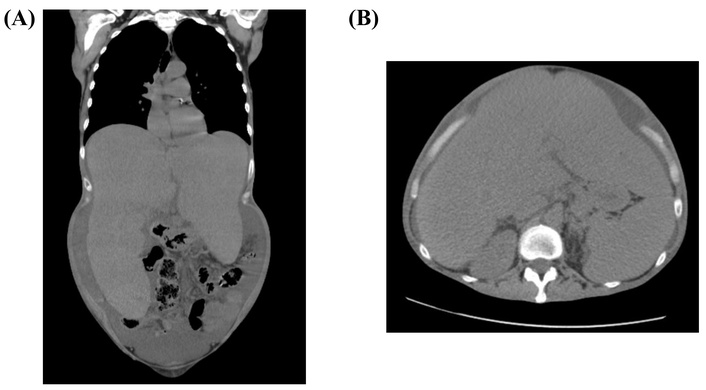
CT-scan showing an important liver size increase. Patient deceased from her liver involvement. (A) Sagittal section; (B) axial section
Given that amyloid fibril deposition increases liver stiffness, non-invasive elastography methods have been explored [24, 31, 32]. The first study, conducted at our hospital in 2011 on 41 patients, showed a significant increase in liver stiffness in amyloidosis: the median stiffness was 27.4 kPa in liver amyloidosis (with or without cardiac involvement), 10.4 kPa in cardiac amyloidosis (with or without liver involvement), compared to 4.8 kPa in controls, 6.8 kPa in HCV patients, 11 kPa in patients with cardiac disease, and 6.1 kPa in AL amyloidosis without cardiac or hepatic involvement [33]. Extreme values (up to 75 kPa, the maximum measurable stiffness) were also observed. We proposed a threshold of 17.3 kPa for liver amyloidosis with good specificity. However, liver stiffness can also be elevated in isolated cardiac involvement (4–38 kPa), making interpretation more complex in these patients. A German study by Brunger et al. [34], conducted on 70 AL amyloidosis patients, identified a threshold of 14.4 kPa, with 50% sensitivity and 74% specificity, using SAP scintigraphy as the gold standard. Notably, their median alkaline phosphatase level was 88 IU/L (range 70–135), differing from standard reference values. These findings were confirmed in the French national registry, which also observed a trend toward increased liver stiffness via FibroScan [35]. The values are near, but the samples were different. Also, further studies are warranted, and we would probably keep a value around 15–17 kPa as a good criterion for liver involvement. Recent studies and case reports have explored MRI and shear wave elastography, but sample sizes remain limited [36, 37].
In suspected liver amyloidosis, the diagnostic approach should include exclusion of other causes of cholestasis or hepatomegaly, confirmation of amyloidosis, and consideration of transient elastography or advanced imaging techniques. All is summarized in Table 1 and Figure 2.
Liver involvement in AL amyloidosis: clinical features, complications, and outcomes
| Clinical aspect | Findings/characteristics |
|---|---|
| Prevalence of liver involvement | Observed in approximately 70% of symptomatic AL amyloidosis patients |
| Diagnosis criteria | Hepatomegaly, elevated alkaline phosphatase above 1.5N |
| Specific complications | Portal hypertension, ascites, hepatic failure, and rare spontaneous hepatic rupture |
| Investigations | Liver stiffness measurement (≥ 14.4–17.3 kPa) |
| Liver biopsy, carefully, showing amyloid deposits | |
| Prognosis | Generally good with recent treatments |
| High mortality with cardiac and hepatic failure | |
| Treatment | Essentially, anti-plasma cell chemotherapy (e.g., daratumumab), emerging CAR-T cells, siRNA, and autologous transplant |
AL: amyloid light-chain; CAR-T: chimeric antigen receptor T; N: normal limit; siRNA: small interfering ribonucleic acid
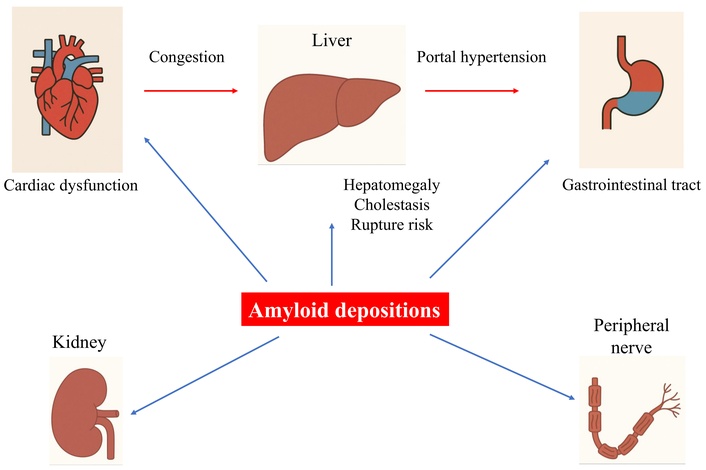
Schematic diagram showing the involvement of different organs in AL, with major highlights in liver-related complications. AL: amyloid light-chain
The diagnosis of amyloidosis is primarily based on clinical examination, as symptoms are often non-specific. Common manifestations include heart failure, periorbital purpura, edema, macroglossia, and hepatomegaly.
Regarding organ involvement, based on both biological and clinical criteria (note: items in italics are considered defining criteria) [8, 20]:
Cardiac involvement is defined by an NT (N-terminal)-proBNP level greater than 332 ng/L (in the absence of renal failure or atrial fibrillation) or a diastolic septal thickness over 12 mm without another cause. Clinically, patients may have signs of right heart decompensation, hypotension, dyspnea, or peripheral edema.
Kidney involvement is characterized by a 24-hour proteinuria ≥ 0.5 g/day, predominantly albumin. This may lead to nephrotic syndrome, renal insufficiency, and clinically significant edema.
Neurologic involvement may be peripheral or autonomic. Peripheral neuropathy typically presents as a sensorimotor neuropathy of the lower limbs. Autonomic dysfunction includes impaired gastric emptying, intestinal pseudo-obstruction, voiding difficulties unrelated to direct infiltration, orthostatic hypotension, and erectile dysfunction.
Pulmonary involvement may be histologically proven in patients with respiratory symptoms. Clinical manifestations include interstitial lung disease or pleural effusions.
Initial laboratory tests include serum free light chain (FLC) assay, serum protein electrophoresis, and proteinuria detection. Bone marrow examination is recommended to assess for associated multiple myeloma [8].
The gold standard for diagnosis remains histological examination of affected tissues, demonstrating beta-fibrillar protein deposits, particularly with Congo red staining [38]. Amyloid fibrils appear as amorphous, eosinophilic deposits on hematoxylin-eosin staining, with characteristic apple-green birefringence under polarized light using Congo red. Further characterization of amyloid deposits is performed using direct immunofluorescence or immunohistochemistry with specific antibodies (Figure 3). When deposits are sparse and difficult to detect with Congo red, electron microscopy may be utilized. More advanced techniques such as mass spectrometry, DNA analysis, and protein sequencing are also available [39].
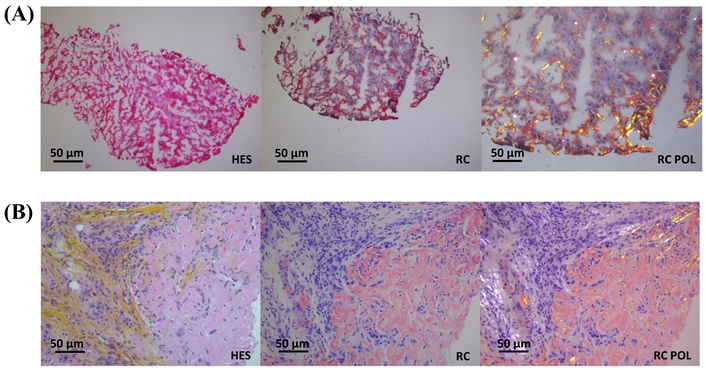
Liver biopsy from amyloid light-chain (AL) amyloidosis. RC (Congo red) staining is characteristic of hepatic amyloidosis. HES (hematein-eosin-saffron), RC, and RC POL (RC in polarized light) staining show significant extracellular deposition, including the RC staining, without and with (for birefringence) POL, showing amyloid deposits. (A) Frozen section staining. Original magnification: ×400. (B) Staining on deparaffinized sections. Original magnification: ×400. Sourced from the CNR Limoges/Poitiers database
Biopsies are usually performed on subcutaneous fat (approximately 70% sensitivity) or minor salivary glands (60% sensitivity) [40, 41]. In some cases, samples may be obtained from the gastrointestinal tract, kidneys, myocardium, or peripheral nerves. Given the high bleeding risk, these sites are preferred over liver biopsy [2]. The combination of abdominal fat and bone marrow biopsy confirms AL amyloidosis in 83% of patients [41].
Due to its prognostic significance, cardiac involvement must be assessed in all patients, as it occurs in approximately 80% of cases. Evaluation includes NT-proBNP measurement, electrocardiography, transthoracic echocardiography with an experienced cardiologist, and, if needed, cardiac MRI. Key findings include low-voltage ECG (electrocardiogram), elevated NT-proBNP, myocardial thickening (especially of the interventricular septum), and characteristic features on MRI. 99mTc-phosphate scintigraphy helps distinguish ATTR amyloidosis from AL amyloidosis. NT-proBNP monitoring is also crucial for assessing treatment response. From a hepatogastroenterology perspective, identifying cardiac involvement is important to assess hepatic congestion. Distinguishing between primary liver amyloidosis and secondary hepatic congestion can be challenging. A French study of 200 prospectively enrolled patients with cardiac amyloidosis found that 49% had elevated liver enzymes (> 1.5 times the upper limit of normal). Liver abnormalities included hepatomegaly (40% of cases), hepatic vein dilation (38%), increased hepatic uptake on scintigraphy (liver/mediastinum ratio = 0.7 vs. 0.6, p < 0.001), and sinusoidal dilation on histology (56%). Notably, bilirubin levels had prognostic value, with a threshold of 5 μmol/L associated with reduced 3-year survival (28% vs. 65%) [42].
Gastrointestinal symptoms frequently accompany liver involvement. While symptoms occur in 20% of patients, histological evidence of amyloid deposits is found in 80% of gastrointestinal biopsy samples. Presentations range from diarrhea, weight loss, and abdominal pain to constipation, gastroparesis, perforation, or intestinal obstruction. Treatment relies on conventional antidiarrheal agents such as opium tincture, loperamide, and octreotide [43], as well as evaluation for small intestinal bacterial overgrowth. AL amyloidosis patients are often malnourished, requiring nutritional support.
Other organs may also be involved, including the kidneys and peripheral nerves, while rare cases of amyloid deposition have been reported in the bones, skin, larynx, and adrenal glands [39]. The brain is uniquely spared. Laboratory findings such as plasma cell dyscrasia (elevated immunoglobulin-secreting plasma cells) and isolated factor X deficiency may provide additional diagnostic clues.
Prognosis often depends on the extent of organ involvement and the delay in diagnosis [44]. In liver amyloidosis, a high bilirubin concentration is generally associated with an increased mortality risk [2, 45].
Over the last decades, significant improvements in treatment and prognosis have been observed. Overall survival has increased from 15% in the 1980s to 58% in the 2010s, even before the latest advancements in specific treatments, including protocols based on cyclophosphamide-bortezomib-dexamethasone combined with daratumumab [46].
For specific liver involvement, response criteria are determined by a reduction in alkaline phosphatase levels of more than 50% and a decrease in liver size of at least 2 cm [20, 47]. These criteria, proposed by the Amyloidosis Forum Multi-Organ System Working Group, require further validation in clinical settings [3, 48]. In some cases, the treatment response can be rapid and clinically remarkable. Additionally, a decrease in liver stiffness may be considered a criterion for liver response (personal data).
Improvement depends on response criteria, with response typically occurring within 6 to 12 months but sometimes taking over 24 months [47, 49]. The estimated time for a full liver clinical response is 39 months [47].
Treatment is classically based on three principles: rapid and significant reduction in amyloidogenic protein production, individualized management based on organ involvement and treatment risks, targeting specific organ involvement [8]. Management is coordinated by hematologists, potentially within clinical trials, and follows risk stratification according to the Mayo Clinic standard classification system. This system categorizes patients into low, medium, or high risk based on the following parameters: NT-proBNP less than 5,000 ng/L, troponin less than 0.06 ng/mL, glomerular filtration rate greater than 50 mL/min per 1.73 m2, age less than 65 years, performance status 0 to 2, NYHA class less than III, ejection fraction greater than 45%, systolic blood pressure greater than 90 mmHg at rest and carbon monoxide diffusing capacity (CMDC) greater than 90% [50]. Autologous stem cell transplantation with melphalan is generally preferred for low-risk patients. Depending on disease severity, a regimen of cyclophosphamide, bortezomib (a proteasome inhibitor), and dexamethasone may be used as a first-line treatment, eventually with daratumumab [40, 51]. Other treatments can be suggested in case of failure, such as melphalan, proteasome inhibitors, anti-CD-38 monoclonal antibodies, and immunomodulatory agents [8]. Also, antifibril monoclonal antibodies are an emergent class.
The goal of treatment is to reduce or stop monoclonal protein production, allowing progressive elimination of amyloid deposits from affected organs.
Monitoring is based on the reduction of FLC production. Predictors of survival include: iFLC (involved FLC) value of less than 20 mg per liter and a dFLC (uninvolved FLC) value less than 10 mg per liter, predicting survival. This treatment achieves a high hematologic response rate, with 78% of patients showing at least a good partial response and 50–55% exhibiting organ response 18 months after initiation. If unsuccessful, alternative treatments are available [52, 53]. New therapeutic approaches, such as chimeric antigen receptor T (CAR-T) cells and specific antibodies, are under investigation for refractory cases. Chemotherapy can pose specific liver toxicity risks, and stem cell transplantation may lead to conditions like sinusoidal obstruction syndrome. Even in cases of good hematologic response, measurable residual disease may persist, potentially contributing to residual organ dysfunction [54].
There is no specific treatment aimed at liver amyloidosis involvement. Nevertheless, in case of fluid retention, most often due to heart failure, salt restriction and diuretics are prescribed. In case of portal hypertension rather rare in this situation, specific management is necessary, including diuretics, beta-blockers, and ligation in case of esophagus varices, such as in cirrhosis [55]. Also, data in the literature report a 72% improvement rate of liver involvement criteria in case of standard treatment [56].
ATTR amyloidosis results from transthyretin, a serum transporter primarily for holo-retinol-binding protein and, to a lesser extent, thyroxine [57]. It predominantly affects men over 70 years, leading primarily to cardiomyopathy. In hereditary forms, symptoms appear earlier and manifest as systemic disease, including polyneuropathy and autonomic dysfunction [58].
Although ATTR amyloidosis does not directly involve the liver, it can influence liver stiffness as a secondary effect of cardiac disease. A recent French study reported that 36% of patients with ATTR cardiac amyloidosis exhibit liver stiffness > 10 kPa, which correlates with advanced cardiomyopathy and increased hospitalization risk for heart failure in wild-type ATTR patients [59].
Because the liver is the primary site of transthyretin production, liver transplantation was historically considered to slow disease progression. More than 2,000 patients with ATTR amyloidosis have undergone liver transplantation in a domino program, where they receive a liver from a deceased donor and their explanted liver is transplanted into patients with end-stage liver disease (cirrhosis, hepatocellular carcinoma) [60].
Recent advancements in pharmacotherapy, including tafamidis, inotersen, and patisiran (an interfering RNA), have transformed disease prognosis, reducing the need for liver transplantation [61, 62]. Patients who receive a liver graft from ATTR amyloidosis donors remain at risk of transthyretin-related symptoms, but these usually occur late and can be managed with early screening and intervention [63].
Amyloidosis is a rare systemic disease that often involves multiple organs. While diagnosis is primarily managed by hematologists, hepatologists should recognize liver-related presentations, such as elevated cholestatic enzymes and hepatomegaly. Liver stiffness is frequently increased in cases of liver and/or cardiac involvement, but its precise role in diagnosis and prognosis warrants further study.
AA: amyloid A
AL: amyloid light-chain
ATTR: transthyretin amyloidosis
FLC: free light chain
NT: N-terminal
pro-BNP: pro-brain natriuretic peptide
SAP: serum amyloid P component
Al-Assisted Work Statement: During the preparation of this work, authors used the ChatGPT for organs illustration. After using the tool, authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
PC: Conceptualization, Investigation, Writing—original draft. MDG, JA, AC, JFC, AJ, and VLR: Writing—review & editing. All authors have read and agreed to the published version of the manuscript.
Jean-François Cadranel, who is the Guest Editor and Editorial Board Member of Exploration of Digestive Diseases, had no involvement in the decision-making or the review process of this manuscript. Other authors declare that they have no conflicts of interest.
No ethical approval is required, as data were obtained from routine diagnostic procedures.
No informed consent for participation is required, as data were obtained from routine diagnostic procedures.
No informed consent for publication is required, as data were obtained from routine diagnostic procedures and do not contain patient-identifying information.
The pathological images presented in this study are derived from the internal archives of Centre Hospitalier Universitaire de Limoges and are not publicly available. Requests for accessing the datasets should be directed to the corresponding author.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

