 Review
Review

Affiliation:
1Department of Immunology, Ophthalmology and ENT, Complutense University School of Medicine, 28040 Madrid, Spain
†These authors share the first authorship.
ORCID: https://orcid.org/0000-0001-7606-4682
Affiliation:
1Department of Immunology, Ophthalmology and ENT, Complutense University School of Medicine, 28040 Madrid, Spain
2Department of General Surgery, No. 924 Hospital of Joint Logistics Support Force of PLA, Guilin 541002, Guangxi Zhuang Autonomous Region, China
†These authors share the first authorship.
ORCID: https://orcid.org/0009-0005-1108-155X
Affiliation:
1Department of Immunology, Ophthalmology and ENT, Complutense University School of Medicine, 28040 Madrid, Spain
†These authors share the first authorship.
ORCID: https://orcid.org/0000-0003-3086-1971
Affiliation:
1Department of Immunology, Ophthalmology and ENT, Complutense University School of Medicine, 28040 Madrid, Spain
3Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), 28029 Madrid, Spain
4Instituto de Investigación Sanitaria Gregorio Marañón (IiSGM), 28007 Madrid, Spain
ORCID: https://orcid.org/0000-0003-1390-8002
Affiliation:
1Department of Immunology, Ophthalmology and ENT, Complutense University School of Medicine, 28040 Madrid, Spain
3Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), 28029 Madrid, Spain
4Instituto de Investigación Sanitaria Gregorio Marañón (IiSGM), 28007 Madrid, Spain
Email: fcubero@ucm.es
ORCID: https://orcid.org/0000-0003-1499-650X
Explor Dig Dis. 2023;2:83–99 DOI: https://doi.org/10.37349/edd.2023.00020
Received: February 11, 2023 Accepted: March 22, 2023 Published: June 28, 2023
Academic Editor: Wen-Xing Ding, University of Kansas Medical Center, USA
The article belongs to the special issue Drug-induced Liver Injury: From Bench to Clinical Application
The pathogenesis of drug-induced liver injury (DILI) is still in an early stage of research. However, investigators have shown that both oxidative stress and endoplasmic reticulum (ER) stress play a significant role in the pathological mechanism. However, there is little in-depth literature about these two mechanisms. In order to prevent and improve the clinical symptoms of DILI, it is particularly important to study its pathogenesis. In this review article, the role of ER and oxidative stress in DILI is thoroughly discussed.
Drug-induced liver injury (DILI) plays an essential role both in the clinic and in drug development [1]. The incidence of DILI has continued to increase, and, it has become a major public health problem [2]. There are two types of toxic-related effects, intrinsic and idiosyncratic [3, 4]. Intrinsic DILI is caused by drugs or chemicals directly and its related hepatotoxicity is mostly predictable, reproducible, dose-dependent, and also can be different among individuals [5]. Acetaminophen (APAP) is one of the most widely used painkillers in the world. Although it is a safe drug, APAP is a dose-dependent hepatotoxin which is the most common cause of this type of DILI [6–8]. On the other hand, idiosyncratic DILI causes injury in an unpredictable way, not strictly dose-dependent. It is determined by the interaction of environmental and unique host characteristics, usually occurring in less than 1 of every 10,000 exposed individuals, and it is associated with a long latency period (from a few days to several months) [9, 10]. Clinical observation has blurred the line to distinguish the different forms of hepatotoxicity. Therefore, unless stated otherwise, the term DILI is used for both intrinsic and idiosyncratic lesions in this review.
The pathogenesis of DILI is a complex process, which can develop from asymptomatic liver laboratory abnormalities to acute liver failure (ALF) and death [8]. Hepatotoxicity is a major cause of ALF in Western countries [11], which results in cell death and inflammation in liver. At present, it is speculated that the occurrence of DILI mainly involves immune mechanism and non-immune mechanism [12]. The immune mechanism involves allergic reactions caused by drug properties, dosage, and individual differences. Exposure of hepatocytes to drugs or their reactive metabolites may bring out a series of specific intracellular stress responses and adaptive mechanisms, which will activate the innate and adaptive immune systems. The innate and adaptive immune systems are involved in the process of cell death induced by idiosyncratic DILI, activating death receptor (DR) signaling [13]. The non-immune mechanism mainly involves the metabolic reaction of drugs: direct and indirect damage to hepatocytes. Toxins directly impact hepatocytes likely inducing oxidative organelle stress [such as endoplasmic reticulum (ER)] leading to cell death. Recent research showed that the crosstalk between drug-responsive metabolite-mediated intracellular stress responses and cytokine-mediated pro-apoptotic signaling are important components of DILI pathophysiology [1, 14, 15]. Tumor necrosis factor (TNF) seriously enhances liver injury caused by various exogenous substances [16–18]. Drug-induced ER stress and the oxidative stress response are sensitive to TNF-mediated hepatotoxicity [1].
The liver is a highly dynamic metabolic organ that plays a key role in drug and xenobiotic metabolism and detoxification, protecting potential toxic chemicals against organisms [19, 20]. Under pathological conditions, the following three main mechanisms (oxidative stress, ER stress, and mitochondrial dysfunction) can affect drug metabolism of the liver, and drug metabolites further aggravate liver injury. Bioactivation mechanisms of drugs metabolites and the detoxification and excretion processes of xenobiotics are crucial for understanding the mechanism of DILI [8, 21].
The occurrence of oxidative stress is a sign of DILI, which can activate cell death related pathways [22, 23]. The harmful compounds produced by the oxidation reaction of the body possess strong oxidative properties, damage the tissues and cells of the body, and cause chronic diseases and aging. Oxygen free radicals involved in oxidation reaction mainly exist in organisms and are collectively called reactive oxygen species (ROSs) which are short-lived molecules containing unpaired electrons, highly reactive oxygen-containing molecules that induce DNA damage and activate the DNA damage response (DDR) [24, 25]. ROS is a by-product of normal metabolism and plays an important role in cell signal transduction and homeostasis. Some DILI-causing drugs increase ROS accumulation through a variety of mechanisms, ultimately inducing oxidative stress [26]. It is noteworthy that the increase in ROS generation directly induces ER stress, which activates ER-related apoptosis that is characterized by the release of ER calcium and the activation of caspase-12 [27]. In addition, other small molecules, reactive nitrogen species (RNSs), can also lead to oxidative stress. With the help of the enzymatic activity of the nitric oxide synthase (NOS), RNS mainly come from arginine. NOS produces nitric oxide free radical (NO•) and then reacts with superoxide anion radical (O2•–) to produce peroxynitrite (ONOO−), which is a powerful oxidant and nitrifier [28, 29]. Furthermore, the mechanism of DILI cell death may involve lipid peroxidation (LPO) accumulation and ferroptosis.
The excessive accumulation of ROS is the main cause of oxidative stress, which can be caused by the DILI-causing drugs through different mechanisms [26]. Mitochondria are the main sites for ROS, which includes peroxides, hydrogen peroxide (H2O2), and hydroxyl groups, produced in the cell (Figure 1) [30, 31]. Under physiological conditions, most of the electrons donated to mitochondrial outer membrane (MOM) migrate downward to cytochrome c oxidase, where they react with O2 and H+ to yield H2O2. However, some electrons may react directly with O2 to form O2•–, and spontaneously disproportionate to O2 and H2O2 with manganese superoxide dismutase (MnSOD) [32]. H2O2 is detoxified into H2O2 via mitochondrial glutathione (GSH) [33] peroxidase and peroxiredoxins, or active myeloperoxidase reacts with iron (Fe2+) to become the highly reactive hydroxyl radical (OH•) [31, 34–36]. ROS is not only a signal transduction molecule under pathological conditions, but also important for maintaining physiological balance, depending on the intensity and duration of oxidative stress. Low-intensity ROS participates in immune regulation and signal transduction, maintains energy level, kills bacteria and parasites, and eliminates toxins, and high-level ROS promotes cell apoptosis or autophagy which cause damage to normal cells and tissues [37, 38].
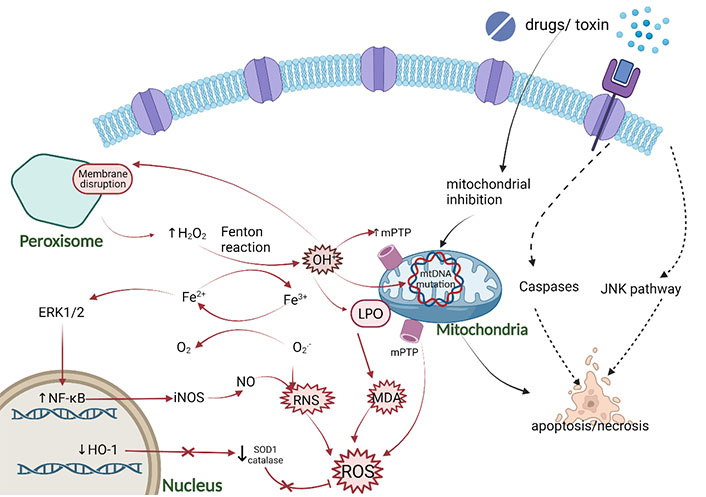
Oxidative stress mechanism caused by DILI. The main pro-oxidation mechanism is characterized by the production of OH• from H2O2 and Fe2+ through the Fenton reaction. The main source of H2O2 is the peroxisome of long-branched fatty acids β-oxidation. OH• can cause LPO of organelle membranes, damage mitochondrial metabolism through the production of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine(mPTP) and mitochondrial DNA (mtDNA) mutation, increase the pro-apoptotic activity, and produce malondialdehyde (MDA). In addition, chronic Fe2+ overload may also activate nuclear factor-kappa B (NF-κB) enhancing the production of inducible nitric oxide synthase (iNOS), resulting in the increase of nitric oxide (NO), which leads to the reaction with the superoxide anion RNS. Figure was created with BioRender.com. ERK1/2: extracellular signal-regulated kinase 1/2; HO-1: heme oxygenase-1; JNK: c-Jun N-terminal kinase; SOD1: superoxide dismutase 1
The liver is a crucial organ for the production of ROSs, which are either converted from endogenous products during liver metabolism or by intake (e.g., APAP). APAP is a non-steroidal anti-inflammatory drug (NSAID) with definite hepatotoxicity in clinical application [12]. After rapid absorption, APAP is mainly metabolized through phase II reaction (sulfation or glucuronidation), converting into non-toxic compounds and excreting into urine. At present, the major reason of hepatotoxicity caused by APAP is its metabolite N-acetyl-p-benzoquinoneimine (NAPQI). A small amount of APAP is metabolized by cytochrome P450 isoenzyme 2E1 (Cyp2E1) to produce intermediates, which produce NAPQI. NAPQI is directly excreted into bile by interaction with GSH to play a detoxification role. After overdose, APAP overwhelms phase II reaction pathway. After NAPQI depletes GSH, excessive NAPQI interferes with the complex I/II of mitochondrial electron transfer chain (ETC), resulting in the leakage of electrons from ETC to O2, thus forming O2•– [39, 40]. The interaction of NAPQI with target DNA and protein in mitochondria and the formation of protein adducts lead to oxidative stress, mitochondrial dysfunction [26, 41]. Furthermore, other drugs produce ROS through different mechanisms. Diethylaminoethoxyhexestrol, perhexiline, amiodarone, and tamoxifen selectively accumulate and reach high concentrations in the mitochondrial matrix, thereby blocking the transfer of electrons along the MOM, increasing production of ROS [42–44].
As mentioned earlier, cytochrome P450 (Cyp450) plays a major role in the detoxification process of APAP. The latest literature also shows that the liver injury caused by polymedication in coronavirus disease 2019 (COVID-19) patients is also considered to be concentrated in the catalytic cycle of Cyp450 with its isoforms and the production of ROS in the oxidative stress environment [45]. In some cases of idiosyncratic DILI caused by drugs, anti-Cyp450 was detected in the blood of patients, representing an association between these drugs and Cyp450 in line with the proposed involvement of the adaptive immune system [46]. Oxidative stress in idiosyncratic DILI includes mitochondrial stress and microsomal stress, which may be related to ROS produced by Cyp450 in these subcellular domains of hepatocytes, but the specific ROS type involved has not been determined [46].
Posttranslational modifications involving various active nitrogen species have a common precursor, NO that is the most well-known free radical acting as a signaling molecule. RNS includes nitrogen free radicals and non-free radicals, which are derived from NO and O2•– by iNOS and nicotinamide adenine dinucleotide phosphate (NADPH), respectively (Figure 1) [47, 48]. Under steady-state, NOS expression is regulated by signal pathways including mitogen-activated protein kinase (MAPK) and JNK/signal transducer of activators of transcription (STAT), which indicates that the inducible production of NO is strictly controlled [49]. In the pathological state, O2•− reacts with NO to increase ONOO– via an up-regulation of NOS and endothelial NOS [50]. Upon APAP overdose, excessive NAPQI can also induce the formation of OH• free radicals and react with NO to form ONOO– [51]. Since O2•− hardly passes through the hepatocyte membrane, the production of ONOO– only occurs in mitochondria. The ONOO– can react with GSH to detoxify [52]. Consequently, GSH is exhausted due to these excessive reactions, resulting in the accumulation of ONOO– [53]. High reactivity and powerful oxidant ONOO– can cause nitration of protein tyrosine residues, resulting in mtDNA damage and membrane pore opening [53]. In addition ONOO– reacts directly with carbon dioxide producing nitrosoperoxycarbonate (ONOOCO2–), which decomposes into nitrogen dioxide (NO2) and carbonate (•CO3–) with a reactivity similar to that of OH• radicals [54]. The production of •CO3– and OH• radicals and the capacity of ONOO– to react with metal centers leads to hepatocyte toxicity [49].
The JNK is a subclass of the MAPK family, which was confirmed to play a vital role in APAP hepatotoxicity by mediating APAP-induced mitochondrial targeting and oxidative stress (Figure 1) [55, 56]. JNK has 10 subtypes from three genes, only JNK1 and JNK2 genes are widely expressed in the liver [57]. During JNK activation the first step occurs when the O2•− production from mitochondrial respiratory complex III spills into the cytosol and triggers a signaling cascade [58]. Then, NAPQI and subsequent mitochondrial-derived ROS activates upstream kinases, including protein kinase C (PKC), MAPK, such as mixed lineage kinase 3 (MLK3) and apoptosis signal-regulated kinase 1 (ASK1), eventually leading to JNK activation. MLK3 is activated by oxidative stress and regulates the early stage of JNK activation; however, ASK1 controls the late stage of JNK activation induced by APAP [13, 59, 60]. The activated JNK [phospho-c-Jun N-terminal kinase (p-JNK)] combines with its target SH3 domain-binding protein (Sab) on the mitochondrial membrane, leading to a self-sustaining activation circuit of ROS production and JNK activation, which eventually leads to the opening of mitochondrial membrane permeability transition pore (MPT) [61]. After the toxic stress of APAP, JNK phosphorylates Sab, which causes Sab to release type 6 protein tyrosine phosphatase (PTPN6) [62, 63]. The released PTPN6 is activated and transferred to the intima, where it dephosphorylates and inactivates Src in the membrane gap. With the deactivation of Src, the ETC chain is blocked and the generation of ROS is enhanced [13]. Switching on MPT induces the collapse of mitochondrial membrane potential, thus ceasing ATP synthesis and releasing the membrane proteins which initiate necrosis [64].
A great quantity of experiments demonstrated that hepatocytes can be protected from cell death by knocking down, knocking out, or inhibiting JNK/Sab to interfere with this pathway at any point [59–61, 65]. The key point is that docking site of Sab or inhibiting the binding between JNK and Sab can protect against APAP toxicity, indicating that the interaction of these two proteins is the key step of cell death [13, 66]. In Sab knockout mice, the mitochondrial translocation of p-JNK was totally inhibited regardless of various isoforms of JNK [66, 67]. Therefore, in animal studies, fomepizole can attenuate APAP-induced liver injury by inhibiting JNK during post-metabolic treatment, and its interference with Cyp2E1 metabolism [68, 69]. The latest research found that platanoides may reduce oxidative stress and nitrification stress by preventing continuous JNK activation, which can effectively prevent APAP-related hepatotoxicity [23]. Collectively, this evidence demonstrated that JNK plays an important role in activating oxidative stress and triggering DILI, which also provides targeted targets for clinical treatment.
Lipid peroxide acts as a vital role in the balance of oxidative stress between pro-oxidation and antioxidant activities, LPO may be involved in the mechanism of cell death during DILI [8]. Lipids present hydrophobicity in cells and serve as metabolic fuel, supporting membrane structure and selective cell membrane transport. When the oxidative metabolism in mitochondria is abnormal, the electronic leakage increases the uncoupling of oxidative phosphorylation, resulting in the production of oxygen free radicals which may induce LPO reactions (Figure 1) [70]. LPO is a free radical reaction process, which is triggered by the formation of OH• radicals (Fe2+-dependent Fenton reaction) from H2O2 combined with the production of lipid radicals, leading to the destruction of polyunsaturated fatty acids (PUFAs) [71]. LPO triggered subsequently rapid destruction of membrane potential and ionic gradient, and promoted cell necrosis. This is also one of the mechanisms of DILI caused by anti-epileptic drugs [12]. Mitochondrial oxidative metabolism is blocked by drugs, resulting in abnormal metabolism of free fatty acids (FFAs), increased anaerobic fermentation, lactic acid accumulation, and generation of ROS. ROS oxidizes liposomes and causes LPO, induces mtDNA damage by changing mitochondrial membrane permeability, further promoting liver injury [72]. Furthermore, experimental models of APAP-induced hepatotoxicity showed that vitamin E pre-treatment can prevent massive LPO induce to liver damage [73]. However, this is a matter of debate since there are conflicting results and it is still uncertain whether vitamin E protects against liver injury caused by APAP [74–76]. Research showed that animals fed a regular diet with sufficient fat-soluble antioxidants to prevent a robust increase of LPO, therefore LPO was not the relevant injury mechanism of APAP-induced liver injury in quantity, in addition, LPO was the cause of the injury rather than the outcome [73, 77–80].
Moreover, there is a controversy whether Fe2+ as a promoter of LPO. Studies shown that protein nitration and LPO can lead to APAP hepatotoxicity after Fe2+ overload [81]. PUFAs react with ROS to generate lipid hydroperoxides (LOOHs), which react with Fe2+ via Fenton reaction to produce highly reactive lipid free radicals [80]. Lipid free radicals further react with other PUFAs propagating a chain reaction. If is an uncontrolled episode, it leads to the accumulation of lipid peroxides and induces Fe2+-mediated cell death [82]. In addition, Fe2+ has also been proved to catalyze the nitrification process of ONOO− to proteins such as MnSOD, which is beneficial to oxidative stress and mitochondrial dysfunction by APAP-mediated cell death [83, 84]. Deferoxamine can inhibit ONOO–-mediated protein nitrification, which demonstrates that Fe2+chelators have partial protective effect on APAP-induced liver injury [85]. As mentioned earlier, LPO was not sufficient in quantity to cause APAP-induced cell death. Therefore, under normal conditions, ferroptosis is not a cell death mode related to APAP hepatotoxicity [86], but may affect DILI through classical Fenton reaction and other mechanisms.
The ER is an essential component of endomembrane system of cells which provide a great many of functionality, such as regulating cellular calcium (Ca2+) concentration, involved in lipid synthesis and transportation, and thus accelerating protein folding (Figure 2) [87]. The tight coupling between the subunits of new proteins in the ER lumen and the ER folding ability is necessary for effective protein folding in ER [88]. However, protein folding is prone to failure when various cellular stresses occur, leading to the induction of the unfolded protein response (UPR), which is initiated to restore cellular homeostasis after acute stress exposure [89]. In mammalian cells, UPR transmits survival signals through three primary pathways and those major proteins involved in this stress response; those pathways mentioned above include inositol-requiring enzyme 1 (IRE1), the protein kinase RNA (PKR)-like ER kinase (PERK), and the activating transcription factor 6 (ATF6) which remain inactive by attaching to binding immunoglobulin protein (BiP), also known as 78-kDa glucose-regulated protein (GRP78) conditions [90–93]. The activation modes of the three proteins are not identical. IRE1 and PERK are activated by autophosphorylation, while ATF6 is activated by intra-membrane cleavage, which releases its N-terminal fragment for transcription transactivation [94].
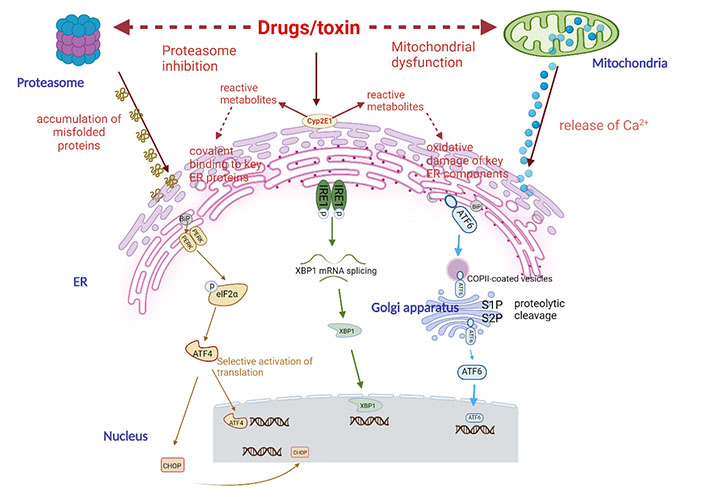
The potential mechanism of drug-induced ER stress. Drugs can induce ER stress through different mechanisms, including proteasome inhibition, mitochondrial dysfunction, and changes in key ER components. The latter mechanism is suspected to be related to drugs, which can be converted into one or several reactive metabolites and can covalently bind with ER protein and/or induce oxidative damage of ER components secondary to oxidative stress. Cyp2E1 is usually involved in the production of reactive metabolites. When ER stress occurs, cells will initiate an adaptive response called UPR. It first activates three effectors, namely PERK, IRE1, and ATF6, and then removes the companion BiP (GRP78) that keeps them in an inactive state. PERK is a phosphorylation and inactivation elongation initiation factor α-subunit of eukaryotic initiation factor 2 (eIF2α) kinases, resulting in a general reduction in protein translation. However, eIF2α, it selectively stimulates the translation of ATF4, a transcription factor with a specific structure (uORF) on its messenger RNA (mRNA). Then ATF4 activates chaperone proteins and protein synthesis involved in autophagy, protein secretion, and amino acid metabolism. IRE1 has kinase activity that leads to its own phosphorylation and activation of ribonuclease (RNase) activity. This leads to the splicing of X-box binding protein 1 (XBP1) mRNA, which is then translated into an active transcription factor. The transcription factor ATF6, which is a non-active precursor and binds to ER membrane, is transferred to Golgi apparatus through the coat protein complex II (COPII) vesicles, where it is cut into active forms by site-1 protease (S1P) and S2P proteases. Then XBP1 and ATF6 will activate the transcription of a group of factors in the nucleus to restore ER homeostasis, including chaperones, folding enzymes, and proteins involved in the degradation of unfolded peptides [ER-associated degradation (ERAD)]. If these mechanisms cannot effectively restore ER and cell homeostasis, UPR will eventually activate the mechanism leading to apoptosis, especially through transcription factor CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP)
As an essential ER stress sensor for UPR in animals and plants, IRE1 detects ER homeostasis and triggers UPR through cytoplasmic kinase domain and RNase domain under the stimulation [95–98]. Under ER stress, IRE1 RNase is activated by conformational changes, self-phosphorylation, and higher oligomerization (Figure 2) [96, 99, 100]. The primary function of IRE1-dependent spliced X-box binding protein 1 (sXBP1) signal transduction is to protect the liver from the stress caused by stimulation [101]. Upon activation of IRE1, multiple downstream signaling of UPR is initiated through unconventional splicing of the transcription factor XBP1 or post-transcriptional modification through controlled inositol-requiring enzyme 1 dependent decay (RIDD) of multiple substrates [97–99]. Splicing occurs through XBP1 mRNA to the translation of sXBP1 protein, which is a transcription factor that induces genes involved in chaperoning proteins and degrades misfolded proteins in ER, thus triggering inflammatory reaction and promoting cell apoptosis mediated by apoptosis ASK1 and JNK [99, 102].
With the main cause of DILI [7, 99, 103]—APAP-mediated hepatoxicity—a great deal of data indicated a role for ER stress. The mechanism of APAP hepatotoxicity is similar in humans and mice [104]. The sXBP1 or unspliced X-box binding protein 1 (uXBP1) in human HepaRG cells was consistent with the findings in mice [105]. Under ER stress, the IRE1α-XBP1 arm of the UPR response plays a critical role in APAP-induced liver injury via upregulating the activity of Cyp450 [106]. In XBP1-deleted mice, overexpression of IRE1 in liver led to reduced JNK activation, and protected against APAP via the inhibition of Cyp450 activity [105–107]. This was linked to the recovery of GSH, which prevented severe oxidative stress [105]. In recent years, increasing studies demonstrated that autophagy is activated to respond to APAP-induced liver injury [108].
Furthermore, IRE1α can also function via the RIDD pathway. IRE1α accelerates the degradation of mRNAs encoding mostly ER-targeted proteins, resulting in a decrease of protein entering the ER membrane during ER stress [109, 110]. Meanwhile, XBP1-deficient mice showed decreased damage caused by APAP due to enhancement of autophagy and activating RIDD [105, 106].
Once cell stress reaches its critical threshold, over-activation of the JNK signaling pathway and mitochondrial dysfunction are considered as major cellular events in APAP-induced liver injury [111, 112]. p-JNK translocates to the MOM and finally induces mitochondrial dysfunction which contributes to hepatocyte necrosis [80]. Mitochondrial injury is likely to positively regulate the sustained activation of JNK cell death pathways and the onset of ER stress, which may be an intertwined mechanism [113]. After APAP treatment, the intense JNK phosphorylation and upregulated JNK1 expression in JNK-deleted liver correlated with a sharp increase in serum transaminases [22]. Gunawan’s study [114] showed that disruption of JNK2 instead of JNK1 can suppress liver injury, indicating the importance of JNK2 in hepatotoxicity caused by APAP. Another paper indicated that extensive necrosis occurred in APAP-treated JNK hepatocyte-specific knockout mice, and further demonstrating that combined activity of JNK1 and JNK2 can prevent APAP-induced necrosis by controlling the generation of oxidative stress [114].
PERK is a member of the eIF2α kinase family, which protects cells involved in various stimuli by biasing general protein synthesis, but initiating stress-related protein production [115]. In ER stress, after BiP/GRP78 dissociation, increasing expression of phosphorylated eIF2α/PERK signaling is activated by APAP hepatotoxicity (Figure 2) [94]. PERK temporarily inhibits the translation of general protein via elF2α phosphorylation. Notably, phosphorylated elF2α selectively enhanced mRNA translation including ATF4 transcription factor to mediate UPR [116–119]. During the ER stress regulation, ATF4 activates UPR target gene, which encodes proteins necessary for antioxidant reaction and amino acid synthesis and transport [120]. ATF4 is also a transcriptional activation CHOP, which acts as a transcriptional inhibitor downstream of PERK and IRE1. The main function of CHOP is to induce apoptosis by various kinds of mechanisms in ER stress [117], thereby mediating cell apoptosis via mitochondrial pathway or DR pathway that further exacerbate hepatotoxicity [1, 121]. Conversely, decreased expression of CHOP, BiP/GRP78, and JNK are related to protect against APAP-induced hepatotoxicity which are possibly mediated by increased proliferation [94, 105].
Similar to IRE1 and PERK, ATF6 contains an ER luminal stress-sensing domain and a cytoplasmic enzymatic domain, which is one of ER transmembrane proteins. In response to a sublethal dose of APAP-mediated ER stress, the transcription factor ATF6 is activated (Figure 2) [122]. ATF6 dissociates from BiP/GRP78 to Golgi apparatus, where it is hydrolyzed into the nuclear form of ATF6 [123]. The cleavage transcription factor domain of ATF6 regulates UPR in the nucleus, including CHOP, XBP1, and protein chaperones such as BiP/GRP78 [120, 124, 125]. Additionally, ATF6 forms a heterodimer with IRE1-XBP1 pathway to activate the genes involved in ERAD [126].
Mitochondria can provide energy for cells and are also the place where many important metabolic processes take place [127, 128]. In addition, mitochondria are the main source of intracellular ROS and the regulatory center of cell apoptosis (Figure 3) [129]. When mitochondria are seriously stimulated, it can induce mitochondrial structure and function abnormalities, which display in the following aspects: morphological and structural changes, abnormal energy metabolism, ROS elevates, mtDNA damage, and abnormal mitophagy [32, 43, 44, 130–134]. Recent studies demonstrated the mechanism of DILI is related to the mitochondria damage of hepatocyte, including antipyretics, NSAIDs, antibiotics, immunosuppressive drugs, and antivirals, and others over 300 drugs [26, 135–138]. The metabolic process of APAP in vivo includes the conversion of APAP into the active metabolite NAPQI under the action of Cyp450 [80]. Normally, NAPQI complex can be cleared by GSH, but excessive APAP can lead to excessive production of NAPQI complex, thus depleting GSH and damaging mitochondria [139]. In addition, overdose APAP induces mitochondrial oxidative stress, which results in mitochondrial dysfunction and cell death (Figure 3) [140]. Due to mitochondrial dysfunction induced by APAP hepatotoxicity, the removal of damaged mitochondria through mitochondrial autophagy is a vital mechanism of APAP-induced ALF [141].
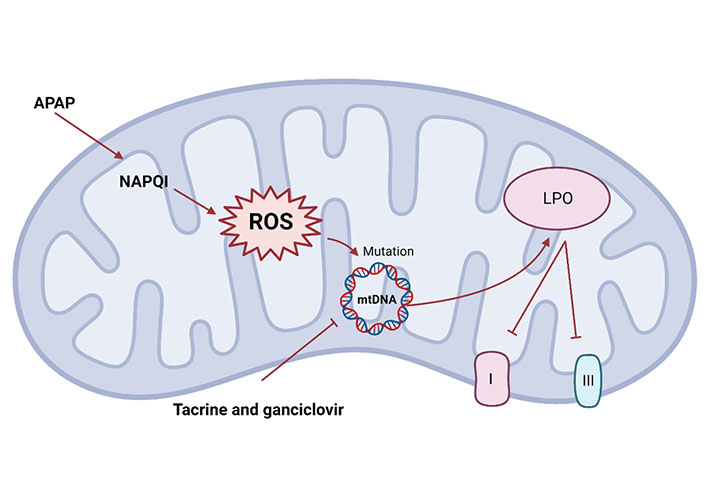
Mitochondrial dysfunction mechanism. Mitochondria stimulation can trigger changes in mitochondrial structure and function abnormalities. Drugs such as APAP or tacrine and ganciclovir can cause mitochondrial cell death, via the activation of ROS which may cause a damage to mtDNA, resulting in LPO and inhibition of ETC complexes I and III
In addition, other drugs have different mechanisms for mitochondrial dysfunction. Tacrine and ganciclovir alter mtDNA homeostasis from different mechanisms, including inhibition of mtDNA replication and translation, to induce mitochondrial damage [132, 142]. Mitochondrial dysfunction caused by the significant increase of ROS, resulting in LPO and inhibition of respiratory chain (complexes I and III) triggering apoptosis, which may explain the hepatotoxicity caused by statins (Figure 3) [143]. The mechanism of liver injury caused by most drugs has not been clearly studied, and the degree of drugs-induced damage to human varies greatly according to individual differences and drug dosage.
DILI is a sustained area of immense clinical and research interest, especially the pathogenesis of DILI is still not completely determined, and the occurrence of DILI is the result of multiple factors. Oxidative stress, ER stress, and mitochondrial injury are the main etiological factors for DILI initiation and the pathophysiological mechanism is complementary and participates in the initiation of immune response. The drug properties, host factors, and environmental conditions determine the susceptibility, phenotypic expression, and results of DILI. Especially with the rapid development of new drug research and the previously unidentified new mechanism of DILI, it is urgent to study the mechanism of DILI and improve the diagnostic efficiency. Continuous research on specific biological diagnostic markers and DILI development mechanisms will contribute to the detection of drug hepatotoxicity and side effects, but there is still a long way to reduce the incidence rate of DILI in clinical practice.
ALF: acute liver failure
APAP: acetaminophen
ASK1: apoptosis signal-regulated kinase 1
ATF6: activating transcription factor 6
BiP: binding immunoglobulin protein
CHOP: CCAAT/enhancer-binding protein homologous protein
Cyp2E1: cytochrome P450 isoenzyme 2E1
Cyp450: cytochrome P450
DILI: drug-induced liver injury
eIF2α: α-subunit of eukaryotic initiation factor 2
ER: endoplasmic reticulum
ETC: electron transfer chain
GRP78: 78-kDa glucose-regulated protein
GSH: glutathione
H2O2: hydrogen peroxide
iNOS: inducible nitric oxide synthase
IRE1: inositol-requiring enzyme 1
JNK: c-Jun N-terminal kinase
LPO: lipid peroxidation
MAPK: mitogen-activated protein kinase
MOM: mitochondrial outer membrane
mRNA: messenger RNA
mtDNA: mitochondrial DNA
NAPQI: N-acetyl-p-benzoquinoneimine
NO: nitric oxide
NOS: nitric oxide synthase
O2•–: superoxide anion radical
OH•: hydroxyl radical
ONOO−: peroxynitrite
PERK: protein kinase RNA-like endoplasmic reticulum kinase
p-JNK: phospho-c-Jun N-terminal kinase
PUFAs: polyunsaturated fatty acids
RIDD: inositol-requiring enzyme 1 dependent decay
RNase: ribonuclease
RNSs: reactive nitrogen species
ROSs: reactive oxygen species
Sab: SH3 domain-binding protein
sXBP1: spliced X-box binding protein 1
UPR: unfolded protein response
XBP1: X-box binding protein 1
HW and XB equally contributed to Conceptualization, Investigation, and Writing—original draft. AHG: Validation, Resources, Investigation, and Formal analysis. YAN: Writing—original draft and Writing—review & editing. FJC: Supervision, Writing—original draft, and Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by the
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Gulcin Cakan-Akdogan ... Ozlen Konu
Alejandro Cueto-Sánchez ... Marina Villanueva-Paz
Qian Wei ... Jinsheng Guo
Miren García-Cortés ... Alberto García-García
