 Review
Review

Affiliation:
1Department of Nutritional Sciences, Molecular Nutritional Science, University of Vienna, A-1090 Vienna, Austria
ORCID: https://orcid.org/0000-0002-3356-4115
Affiliation:
2Department of Liver and Digestive Diseases, Groupe Hospitalier Public du Sud de l’Oise, 60100 Creil, France
ORCID: https://orcid.org/0000-0002-8442-4526
Affiliation:
3Qidong Liver Cancer Institute, Qidong 226200, Jiangsu, China
ORCID: https://orcid.org/0000-0003-4102-8934
Affiliation:
4Department of Pharmacology, Toxicology & Therapeutics, University of Kansas Medical Center, Kansas City, KS 66160, USA
ORCID: https://orcid.org/0000-0002-3167-5073
Affiliation:
5Center for Cancer Research, Medical University of Vienna, A-1090 Vienna, Austria
ORCID: https://orcid.org/0000-0002-6074-7144
Affiliation:
6Department of Molecular and Cellular Medicine, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
7Liver Unit, Hospital Clínic i Provincial de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
8Centro de Investigación Biomédica en Red (CIBEREHD), 28029 Madrid, Spain
ORCID: https://orcid.org/0000-0002-2652-6102
Affiliation:
4Department of Pharmacology, Toxicology & Therapeutics, University of Kansas Medical Center, Kansas City, KS 66160, USA
ORCID: https://orcid.org/0000-0002-8695-6980
Affiliation:
9Department of Medical Imaging, Groupe Hospitalier Public du Sud de l’Oise, 60100 Creil, France
Affiliation:
10Department of Internal Medicine, Azienda Ospedaliero-Universitaria di Modena, 41100 Modena, Italy
ORCID: https://orcid.org/0000-0001-9886-0698
Affiliation:
11Biosciences Institute, University of Newcastle, NE2 4HH Newcastle, United Kingdom
ORCID: https://orcid.org/0000-0003-0950-243X
Affiliation:
12Liver Research Unit, Medica Sur Clinic and Foundation, Mexico City 14050, Mexico
13Faculty of Medicine, National Autonomous University of Mexico, Mexico City 04360, Mexico
ORCID: https://orcid.org/0000-0001-5257-8048
Affiliation:
14Department of Laboratory, Groupe Hospitalier Public du Sud de l’Oise, 60100 Creil, France
Affiliation:
15Department of Gastroenterology and Hepatology, University Medical Center Groningen, University of Groningen, 9713 GZ Groningen, The Netherlands
ORCID: https://orcid.org/0000-0002-4764-0246
Affiliation:
16Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy
ORCID: https://orcid.org/0000-0003-2473-3535
Affiliation:
17Research Laboratory in Translational Gastroenterology, University Hospital Aachen, 52074 Aachen, Germany
ORCID: https://orcid.org/0000-0002-7122-6379
Affiliation:
18Institute of Biochemistry, Food Science and Nutrition, The Hebrew University of Jerusalem, Rehovot 7610001, Israel
ORCID: https://orcid.org/0000-0001-5407-0440
Affiliation:
2Department of Liver and Digestive Diseases, Groupe Hospitalier Public du Sud de l’Oise, 60100 Creil, France
Affiliation:
6Department of Molecular and Cellular Medicine, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
7Liver Unit, Hospital Clínic i Provincial de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
8Centro de Investigación Biomédica en Red (CIBEREHD), 28029 Madrid, Spain
19Department of Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Email: checa229@yahoo.com
ORCID: https://orcid.org/0000-0003-3422-2990
Explor Dig Dis. 2026;5:1005110 DOI: https://doi.org/10.37349/edd.2026.1005110
Received: November 28, 2025 Accepted: January 18, 2026 Published: February 09, 2026
Academic Editor: Jae Youl Cho, Sungkyunkwan University, South Korea
Digestive diseases comprise a diverse range of illnesses, which are prevalent worldwide and represent an important health issue. This is particularly relevant for the impact of metabolic dysfunction-associated steatotic liver disease (MASLD) due to its close association with the obesity pandemic, contributing to the escalation of MASLD as the most common form of chronic liver disease, and the main cause of liver cancer. Not only does MASLD reflect the deterioration of liver health, but it also has far-reaching consequences for the development of extrahepatic digestive diseases. Along with the progression of liver and digestive diseases to liver, colorectal and pancreatic cancer, the onset of inflammation in diseases of the digestive tract, drug-induced liver injury, and cholestasis, drives and contributes to the rise of these diseases in the future, which merit the attention of clinical and translational research to increase our understanding of the pathogenic mechanisms underlying these disorders in order to improve the diagnosis, management, and treatment. With this goal in mind, the current collaborative review gathers experts in a wide range of liver and digestive diseases to provide an up-to-date overview of the mechanisms of disease and identify novel strategies for the improvement of these important health issues.
Digestive diseases are very common worldwide and account for considerable health care use and represent a high social impact and economic burden to Western countries. Since 2000, the increase in diseases of the digestive tract has been exponential and alarming. Approximately 40% of the adult global population is estimated to suffer from gastrointestinal disorders, which implies a serious economic impact and significant social cost. A substantial disparity in the burden of digestive diseases exists among countries with different developmental levels. A recent study has shown that in the US, the digestive disease burden has escalated, with higher rates of mortality in men versus women, especially higher in Blacks compared to Whites, while Hispanics show lower rates [1]. In a recent report from the United European Gastroenterology, more than 350 million people are believed to live with digestive disorders in Europe, with higher burdens in central and eastern Europe than in western and southern European countries. Although limited data is available for low to middle-income countries, the 2019 Global Burden of Diseases, Injuries, and Risk Factors Study [2] provides a comprehensive overview to understand the state of digestive diseases.
Epidemiological studies show that the main risk factors for digestive diseases are the use of alcohol or drugs, smoking, and high body mass index. In fact, obesity is a major driver of digestive diseases, including metabolic dysfunction-associated steatotic liver disease (MASLD), causing an increased burden of associated digestive disorders, such as gastrointestinal, kidney, and cardiovascular events, with poor prognosis and low survival rates of patients with obesity-associated cancers.
The cost of billions of US dollars associated with gastrointestinal health is not the worst scenario of the impact of the disease in society, but rather this spectrum of diseases extends beyond the financial and physical burden on patients, health care professionals, and the overall healthcare system. In order to reverse these alarming trends, there is an urgent need to address these burdens through basic, clinical, and epidemiological research, as well as health education of populations with digestive disorders to avoid poor lifestyles and prolong survival time.
In this collaborative, expert-driven review, we summarize the most common forms of digestive diseases, from chronic liver disease to gastrointestinal disorders, and provide insightful suggestions for future developments for the design of strategies aimed at preventing the onset of these diseases and providing a more efficient treatment for the public health concern related to liver and digestive diseases.
The historical definition of NAFLD, created in the 1980s, was widely used until 2022 [3] when Eslam turned the negative definition (i.e., nonalcoholic) into a positive diagnostic criterion: MAFLD [4]. This development was continued in 2023, when a novel definition was coined: MASLD, which requires concurrent steatosis and at least one cardio-metabolic criterion [5]. This effort also accounts for the patients with MASLD, who consume moderate amounts of alcohol. This condition is termed metabolic dysfunction-associated alcohol-related liver disease (MetALD) [6].
Several scholars have highlighted the limitations of the MASLD [7, 8] and MetALD definitions [9] while supporting head-to-head comparative studies [10]. When comparisons have been made, they often suggested that MAFLD may more selectively identify patient populations at risk of severe liver-related and extra-hepatic outcomes [11]. Conversely, MASLD is more inclusive and captures larger strata of patients [11].
MASLD affects 38% of adults and up to 14% of children and adolescents, and the prevalence rate in adults is projected to exceed 55% by 2040 [12]. Groups at high risk of MASLD comprise those with obesity, type 2 diabetes (T2D), and other features of the metabolic syndrome [13]. The fact that MASLD often develops secondary to specific endocrinopathies is increasingly being recognized [14].
MASLD is modified by gender and hormonal status. It is more common in men than in premenopausal women while postmenopausal women’s MASLD rates are similar to men’s [15]. On the other hand, women have a higher risk of advanced fibrosis than men, especially after the age of 50 years [16].
Currently, in the US, MASLD is the leading indication for liver transplantation in women and subjects with hepatocellular carcinoma (HCC) [12]. However, only a subset of subjects with MASLD will progress to advanced forms of liver disease, and the leading cause of mortality among MASLD patients is cardiovascular disease (CVD) [12]. Like other liver disease etiologies, surveillance for early detection of HCC is warranted among those with MASLD and advanced fibrosis, cirrhosis, or portal hypertension [17].
Further to liver-related outcomes and to CVD, accumulating data suggest that MASLD may be associated with certain extra-hepatic cancers, such as those of the gastrointestinal tract and of the urinary system [18, 19]. The stage of liver fibrosis and gender modulate the risk of hepatic and extra-hepatic outcomes [20, 21]. A growing body of studies supports chronic kidney disease as another extra-hepatic outcome whose risk parallels the severity of MASLD [22]. The presence of both conditions also increases the risk of CVD [23].
Although Mendelian-randomization studies do not invariably support a cause-and-effect relationship between MASLD and these extra-hepatic outcomes [24, 25], clinicians should be aware of the risks of CVD and extra-hepatic cancers among those with MASLD as a part of a holistic approach to this systemic disorder [13, 26].
Hepatocytes are the central hub of lipid metabolism of the human body since they take up and secrete lipids into the bloodstream, carry out de novo lipogenesis as well as degrade lipids via β-oxidation. An imbalance in these pathways results in intrahepatic accumulation of neutral fat that becomes evident as steatosis [27] and can be detected histologically as well as by multiple imaging methods [28]. While ultrasound is most widely used in the clinical routine, magnetic resonance imaging with assessment of proton density fat fraction is the most accurate non-invasive method [28].
Unlike the adipose tissue, the liver is not physiologically devoted to the accumulation of fat and the accumulation of intrahepatic fat has the potential for triggering lipotoxic phenomena such as cell degeneration (i.e., ballooning) and cell death. The ongoing stress may trigger pro-inflammatory and pro-fibrotic cascades and thereby lead to metabolic dysfunction-associated steatohepatitis (MASH), an advanced form of MASLD, and subsequently to MASH-fibrosis or MASH-cirrhosis [29]. MASLD results from an intricate organ crosstalk with adipose tissue and the intestinal tract playing key roles [30]. The former acts as a supplier of free fatty acids (FFAs) and of hormonal substances, such as leptin and adiponectin that affect the hepatocellular lipid handling [31]. The intestinal tract is tightly connected to the liver via portal circulation and both organs work closely in the digestion and handling of nutrients. However, MASLD patients often display an increased gut permeability and intestinal dysbiosis that leads to portal circulation of gut-derived toxins and hepatic inflammation [32].
Steatogenic and fibrotic progression of MASH are intimately and bi-directionally associated with insulin resistance (IR) in the context of systemic metabolic dysfunction in subjects with either features or the full-blown metabolic syndrome [33]. Additionally, age, sex, reproductive status, hormonal influxes, and lifestyle factors (diet, smoking, alcohol, physical exercise, sedentary behavior) are key modifiers of the distribution of body fat, of intrahepatic balance of pro- and anti-lipogenic, inflammatory, and profibrogenic pathways [34]. Notably, MASLD displays a strong heritable trait, and genetic studies shaped our understanding of MASLD pathogenesis [35]. On a population level, a variant in patatin-like phospholipase domain-containing 3 (PNPLA3) constitutes the strongest genetic contributor to MASLD and highlights the importance of lipid droplet handling within hepatocytes, which is supported by other MASLD-related variants such as MBOAT7 [35]. In particular, the PNPLA3 I148M seems to accumulate on lipid droplets and to impair their degradation [36]. However, genetic studies also highlight the complexity of the condition with processes like secretion of very low-density lipoproteins (TM6SF2, ERLIN1), lipogenesis (GCKR, ApoE), diversion of triglycerides (MTARC1), and many others being affected [35].
Bile acids (BAs), produced from cholesterol in hepatocytes, are involved not only in bile formation and fat digestion, but also serve as signaling molecules involved in metabolic processes such as lipid and glucose homeostasis [37, 38]. Disruption of BA homeostasis leads to the progression of MASLD and development of MASLD-HCC [39]. Recent studies have found that interleukin-1 receptor 1 (IL1R1), gut microbiota, and BA changes synergistically contribute to the development of MASLD [40], suggesting pathogenic cross-talks between BA metabolism, gut dysbiosis, and “metaflammation”, i.e., sterile metabolic inflammation, in MASH. BAs are elevated in MASLD, particularly ursodeoxycholic acid (UDCA), taurocholic acid (TCA), chenodeoxycholic acid (CDCA), taurochenodeoxycholic acid (TCDCA), and glycocholic acid (GCA). However, BA profiles vary owing to geographic regions or disease severities [41]. Notably, TCA, TCDCA, taurolithocholic acids, and glycolithocholic acids exhibit a potential ability to identify MASH [41]. Trafficking of cholesterol to mitochondria through steroidogenic acute regulatory protein 1 (STARD1), the rate-limiting step in the alternative pathway of BA generation, not only promotes the progression from steatosis to MASH by sensitizing hepatocytes to inflammatory cytokines-induced cell injury [42] but also plays a key role in the pathogenesis of MASLD-HCC (Figure 1) [37]. This is particularly relevant for MASH and HCC development as the synthesis of BA through the classic pathway controlled by CYP7A1 is repressed by endoplasmic reticulum (ER) stress. Finally, fibroblast growth factor (FGF) 21, a potent regulator of glucose and lipid homeostasis, acts as a negative regulator of BA synthesis [43], paving the way for targeted pharmacological interventions in MASLD/MASH arena, as discussed below. Another pathogenically and therapeutically relevant alteration in MASLD/MASH is dysfunctional farnesoid X receptor (FXR)-FGF19 feedback signaling. This leads to elevated BA production, higher pericentral biliary pressure, and pericentral micro-cholestasis that may account for raised GGT serum concentrations in a proportion of MASLD/MASH individuals [38, 44, 45].
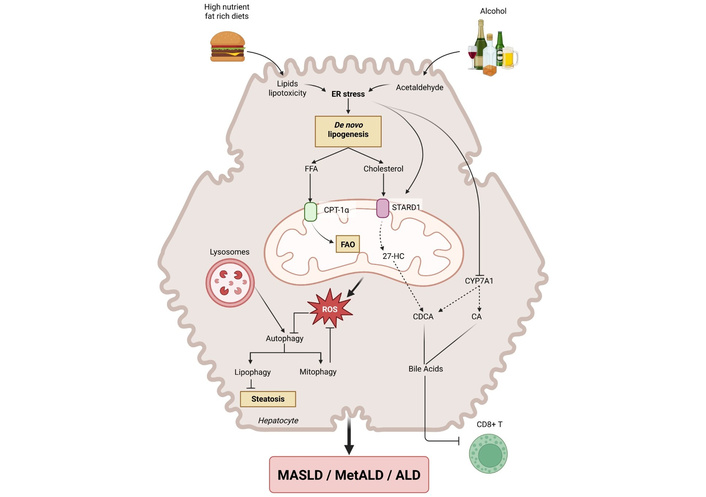
Interplay of players involved in the progression of MASLD and ALD. Consumption of high-nutrient diets and alcohol converges in common mechanisms that synergize to induce the overall phenotypic changes of MASLD and ALD, as well as MetALD. The ingestion of dietary fats and the oxidative metabolism of ethanol alter ER inducing ER stress, which emerges as a critical trigger to cause increased de novo lipogenesis, including free fatty acids (FFAs) and cholesterol via activation of transcription factors SREBP1c and SREBP2, respectively, with the ultimate impact in the onset of hepatic steatosis. ER stress in turn induces STARD1 expression, which translocates cholesterol to the mitochondrial inner membrane for metabolism by CYP27A1 to generate the oxysterol 27-hydroxycholesterol that acts as the precursor of the primary bile acid, chenodeoxycholic acid (CDCA). ER stress blocks the expression of CYP7A1, the rate-limiting step in the synthesis of bile acids through the classic pathway, causing the reprogramming of bile acid metabolism from the classic to the alternative pathway. Besides the stimulation of CDCA, the translocation of cholesterol to mitochondria impairs mitochondrial function and antioxidant defense stimulating the generation of reactive oxygen species (ROS). Defects in autophagy by alterations of the function of lysosomes in part by changes in their physico-chemical properties lead to impairment of lipophagy and mitophagy, perpetuating the induction of steatosis and mitochondrial dysfunction. In addition, the increase in cholesterol content impairs the function of immune cells particularly CD8+ lymphocytes. The figure was generated by BioRender under the agreement number SN298QD1XP. “Interplay of players involved in the progression of MASLD and ALD” created in BioRender. Fernández-Checa, J. C. (https://BioRender.com/8wflzpu) is licensed under CC BY 4.0. ER: endoplasmic reticulum; CPT-1: carnitine palmitoyltransferase-1; STARD1: steroidogenic acute regulatory protein 1; FAO: fatty acid β-oxidation; CA: cholic acid; MASLD: metabolic dysfunction-associated steatotic liver disease; MetALD: metabolic dysfunction-associated alcohol-related liver disease; ALD: alcohol-related liver disease; SREBP: sterol regulatory element binding proteins.
Obesity, unhealthy diet and sedentary lifestyle are key contributors to both T2D and MASLD. In line with that, lifestyle interventions aiming at dietary changes and increased physical activity constitute an established therapeutic approach [46]. It has been repeatedly demonstrated that weight loss of 5–7% can reverse steatosis while higher weight loss can reverse fibrosis. However, many participants undergoing lifestyle intervention fail to lose weight, and many regain weight after the end of the study [46]. Because of that, many advocate for bariatric surgery as the more reliable method to achieve long-term weight loss [47]. While this invasive method is associated with potential side effects such as dysphagia, abdominal discomfort, or nutritional deficiencies, large studies with long-term follow-up demonstrated that it decreases the overall mortality as well as the rate of major adverse cardiovascular events (MACEs) and major adverse liver outcomes [48, 49]. As an alternative approach, several weight-reducing drugs have been recently developed. Among them, glucagon-like peptide 1 receptor agonists (GLP-1 RAs) and sodium-glucose co-transport 2 inhibitors (SGLT2is) are commonly used in individuals with diabetes and are associated with reduced MACEs and overall mortality [50–52]. Among GLP-1 RAs, semaglutide was tested in a phase 2 randomized, double-blind, placebo-controlled trial (RCT), showing a higher rate of resolution of MASH than placebo [53]. These results were strengthened by a phase 3 RCT where semaglutide improved liver histology in more patients than the placebo arm [54]. Similarly, encouraging results were reported for SGLT2is, in that dapagliflozin outperformed placebo in improvement of histological MASH in a recent RCT [55]. While GLP-1 RAs remain the best studied class of drugs leading to weight loss due to decreased food intake, their effect can be strengthened by combination with glucagon receptor and/or glucose-dependent insulinotropic polypeptide agonists. Among such drugs, the dual agonists tirzepatide and survolutide were able to resolve MASH without worsening fibrosis in the corresponding phase 2 RCTs [56, 57]. These data remarkably demonstrate the usefulness of weight-reducing approaches in MASLD. While these drugs show beneficial extrahepatic effects, none of these agents are currently approved for treatment of MASLD and their prescription is currently reimbursed only in patients with diabetes in most countries.
As the leading liver disease worldwide, MASLD became a coveted pharmaceutical target and yielded unprecedented insights into the relevance of several signaling cascades. Recently, resmetirom, a liver-directed, thyroid hormone receptor beta (THR-β)-selective agonist, became the first FDA-approved MASH agent and highlighted the importance of endocrine signaling in MASLD [58]. Earlier studies demonstrated impaired THR-β function and the corresponding decrease in β-oxidation of fatty acids in MAFLD. The approval of resmetirom is based on a phase 3 RCT where it was superior to placebo in both MASH resolution and improvement of liver fibrosis [59]. FGF21 is another hormone regulating glucose and lipid metabolism as well as insulin sensitivity. While FGF21 agonist pegozafermin led to improvement in liver fibrosis in a phase 2b RCT, the FGF21 agonist efruxifermin did not reduce fibrosis in subjects with MASH-related cirrhosis [60, 61]. Peroxisome proliferator-activated receptors (PPARs) belong to widely studied nuclear receptors that play an important role in liver metabolism, inflammation, and fibrogenesis. Their broad hepatoprotective effects are substantiated by recent clinical trials that led to approval of two PPAR agonists for treatment of primary biliary cholangitis (PBC) [62, 63]. In a phase 2b RCT, the pan-PPAR agonist lanifibranor significantly improved a histological MASH score without worsening liver fibrosis [64].
The advancement of liver-directed RNA silencing approaches (RNAi) [65] opened the possibility of personalized treatment of MAFLD directed at the key genetic risk factors. In that sense, the treatment with the PNPLA3 RNAi JNJ-75220795 resulted in decreased liver steatosis in two independent phase 1 RCTs [66], while the HSD17β13 RNAi associated with improved alanine aminotransferase levels [67]. Finally, a repurposing of FDA-approved drugs might also be beneficial in MASLD as shown in a recent RCT that reported a decreased steatosis in subjects treated with low-dose aspirin for 6 months [68].
Finally, since existing findings indicated that the type rather than the amount of fat emerged as an instigator of lipotoxicity and sensitization to advanced stages of MASLD, with cholesterol playing a key role [42, 69, 70], it has been shown that statins may be another pharmaceutical approach for MASH. In this regard, Yun et al. [71] reported that statins are significantly associated with reduced liver-related event risk in patients with MASLD, especially among those with elevated alanine aminotransferase levels, suggesting a viable preventive strategy for such a population.
Until recently there was a clear distinction between patients suffering from MASLD and patients with ALD, which was mainly based on the average daily or weekly alcohol intake (old definitions: NAFLD: women < 10 g of pure ethanol per day; men < 20 g of pure ethanol per day; ALD: women > 50 g of pure ethanol per day; men > 60 g of pure ethanol per day) [13]. However, this differentiation excluded a large group of (overweight) patients showing clear signs of steatotic liver disease, who were not consuming alcohol in amounts that would be considered ALD patients, but who were ingesting an amount of alcohol that would exceed the criteria for MASLD (women: 20–50 g, men: 30–60 g) [5]. Indeed, it is rather common for high-calorie foods to be consumed alongside moderate and even higher alcohol intake. Moreover, it has been suggested that surveys on self-reported alcohol consumption may be similar to energy reporting in overweight and obese individuals [72–74]. Together, this may result in misleading estimations of intake and subsequently of the prevalence of MetALD. Therefore, the recent estimates of the National Health and Nutrition Examination Survey (NHANES) 2017–2020 consider that steatotic liver disease was prevalent in 37.85% of individuals, of whom 32.45% were diagnosed with MASLD, 1.17% with ALD, and 2.56% with MetALD [75]. Also, data from the NHANES 2017–2018 have revealed marked disparities in the prevalence of MetALD and ALD in the US, which were related to ethnicity and living circumstances, e.g., food safety, suggesting the involvement of other factors [76, 77]. Using data from the UK Biobank, Schneider et al. [78] reported that 10.8% of SLD cases could be attributed to MetALD, indicating that the prevalence of MetALD may be higher in Europe than in the US. Interestingly, the average alcohol intake per person in individuals over 15 years has been reported to be rather similar between UK (9.7–10.7 L in 2024–2025) and the US (8.7–9.97 L in 2024–2025) [79, 80], while the prevalence of overweight or obesity seems to be even higher in the US than in the UK [81]. Stockwell et al. [82, 83] demonstrated in several studies that self-completed questionnaires using quantity-frequency and graduated-frequency methods may be subject to significant underreporting, which can be partially overcome by combining these assessments with an evaluation of beverage-specific yesterday consumption (BSY). However, it needs to be noted that the latter methods do not capture long-term drinking patterns. Taken together, further studies assessing the prevalence of MetALD among patients with MASLD are needed using other, more objective tools to complement 24-h recalls, food frequency questionnaires, and CAGE and AUDIT-C assessments of alcohol intake, in order to achieve a better overview of prevalence and related risk factors. Indeed, in recent years, biomarkers of alcohol intake such as phosphatidylethanol, ethyl glucuronide, ethyl sulfate, and fatty acid ethyl esters have become widely available and have been reported to be useful additions when assessing alcohol ingestion [84–86]; however, they, too, may only reflect alcohol intake in the recent past.
As the amount of alcohol consumed differs markedly between patients with ALD (alcohol intake women: > 350 g/week, men: > 420 g/week) and patients with MetALD (alcohol intake women: > 140 g/week, men: > 210 g/week), there is an ongoing discussion as to whether these patients require a different treatment, especially since patients with ALD may suffer from the condition without additional cardiometabolic risk factors while these are present in patients with MetALD [87]. Indeed, while MetALD and ALD are both chronic, progressive, and potentially terminal liver diseases, they are also markedly different. For instance, acute alcohol-associated steatohepatitis, which is found in heavy and long-term drinkers, is characterized by significant inflammation, deranged liver function, and high mortality rates (20–50% at 28 days in severe cases) [88, 89]. Data on the prevalence of acute steatohepatitis in MetALD patients and its outcome are scarce. Nevertheless, it has been demonstrated that in women, MetALD is associated with a higher hazard for all-cause mortality (+83%), which is thought to stem from their greater susceptibility to developing more severe ALD at lower levels of alcohol intake [90]. This has been linked to differences in gastric alcohol metabolism by alcohol dehydrogenase (ADH), lower body water, and estrogen-related differences in the susceptibility to intestinal barrier dysfunction and macrophage activation by lipopolysaccharide (LPS) [91–94]. Moreover, excessive alcohol intake in MASLD patients has been shown to be associated with a higher mortality risk than in non-drinkers [95]. Furthermore, especially extended binge drinking (at least 13 days/year) has been found to be related to a higher risk of mortality in a survey in the US [96]. However, as the number of studies on MetALD patients and models is still limited, no specific treatment besides abstaining from alcohol and changing lifestyle is currently available for the treatment of MetALD.
As acknowledged in many publications, MASLD and ALD are two clearly different pathological conditions (for an overview, see [97, 98]). It has also been suggested by the results of many studies that MASLD and ALD share several clinical and mechanistic features [97, 98]. For instance, disturbances in lipid metabolism resulting in intracellular lipid accumulation in hepatocytes have been shown to be integral in both MASLD and ALD [99]. Furthermore, in both diseases, this accumulation of lipids has been associated with ER stress and mitochondrial dysfunction, ultimately resulting in cell damage and death (Figure 1) [100]. The activation of hepatic stellate cells (HSCs) triggers not only further inflammatory alterations but also enhances collagen synthesis and deposition, which has been linked to hepatocellular damage in both diseases [101]. Additionally, alongside the observed alterations in the liver lipid synthesis and subsequent stellate cell activation, both MASLD and ALD have also been associated with changes in intestinal microbiota composition and intestinal barrier dysfunction, subsequently leading to increased translocation of bacterial toxins as well as viral compounds [102, 103]. The latter have been linked to an activation of Toll-like receptor (TLR)-dependent signaling cascades, which lead to inflammation but also promote lipid accumulation and stellate cell activation in liver tissue. Indeed, in model organisms, the treatment with antibiotics has been shown to diminish both the development of MASLD and ALD [104, 105]. Whether a combination of a lifestyle/genetic factor promoting the development of MASLD and ALD exacerbated the different alterations, e.g., the disturbance of lipid metabolism, ER stress, and intestinal barrier dysfunction through additive or synergistic effects remains to be determined. The results of recent studies suggest that in the setting of IR, which is frequently found in MASLD patients, alcohol metabolism via ADH-dependent metabolism is impaired [106, 107]. Indeed, in these studies, it was shown that through TNFα- and c-Jun N-terminal kinase (JNK)-dependent signaling cascades, the phosphorylation status of ADH in liver tissue is altered, subsequently leading to a loss of enzyme activity. It was further shown that in mice with MASLD, alcohol clearance was significantly lower than in MASLD-free control mice, further suggesting that alcohol is maintained longer in the system, which could evoke further damage [106]. This needs to be determined in larger human studies, as it may also have implications not only with respect to organ damage but also cognition and subsequently the use of machinery and behavior in traffic. Although gastric ADH may be an important player in alcohol metabolism and hence a determinant of the availability of alcohol in the circulation, once reaching the liver, the oxidative metabolism of alcohol is accounted for by cytochrome P450 2E1, whose expression is induced by alcohol consumption. Nevertheless, the implication of the impaired alcohol metabolism via ADH in MetALD in humans deserves further investigation.
In summary, the interaction of MASLD and alcohol consumption is still poorly understood, despite affecting a large yet unknown number of patients with steatotic liver diseases in many countries. Further studies are needed to assess the number of individuals affected using better, more reliable tools. Studies in patients and animals with MASLD have reported impaired ADH activity; however, the impact of this impairment on alcohol elimination in humans remains to be determined. Furthermore, there is limited data assessing the interaction between MASLD and elevated alcohol consumption at the organ and cellular levels. As our understanding of the molecular mechanisms is still limited, there are few therapies beyond lifestyle interventions such as abstinence and weight reduction.
Universally recognized as the powerhouse of cells, mitochondria generate ATP in the respiratory chain to accomplish myriad cell functions. Despite this ancestral function, they are highly dynamic organelles that act as metabolic hubs, regulating calcium homeostasis, heme synthesis, and the genesis of crucial intermediary metabolites in the tricarboxylic acid (TCA) cycle, some of which act as the building blocks for the synthesis of lipids required for membrane assembly [108–111]. Therefore, it is not surprising that alterations in mitochondrial function play a determinant role in the pathogenesis of metabolic liver diseases, such as ALD and MASLD. Although the etiology of ALD and MASLD is different, they share common biochemical characteristics, including not only liver injury, oxidative stress, steatosis, inflammation, and fibrosis, as discussed in the preceding section Metabolic liver diseases, but also mitochondrial dysfunction. Indeed, alterations in mitochondrial function are a common and determinant event for ALD and MASLD, opening the perspective that both could be considered as mitochondrial diseases [112, 113]. An emerging aspect to keep in mind is that besides the prevalence of ALD, alcohol consumption also occurs in the context of obesity and MASLD, leading to a new paradigm where alcohol intake coexists with metabolic alterations and cardiovascular risks, coined MetALD and covered in the section Metabolic liver diseases, which accounts for most cases of chronic metabolic liver disease and alcohol consumption. An additional open question sparked by the contrast of findings between human and mouse is whether the loss of mitochondrial function is a cause or a consequence of disease progression. In this narrative, we will briefly summarize the link between mitochondria and lipid metabolism and its involvement in the onset of steatosis and progression towards advanced stages, including alcohol-mediated steatohepatitis and MASH.
The imbalance between the availability of lipids in the liver and their metabolism (e.g., catabolism and transport) contributes to hepatic steatosis, a reversible condition that occurs in both ALD and MASLD that can progress to advanced stages, such as alcoholic steatohepatitis and MASH that ultimately can culminate in HCC [114–116]. Adipose tissue lipolysis and hepatic de novo lipogenesis triggered by IR can overwhelm compensatory mechanisms to cope with the excess of fat present in hepatocytes, mainly the packaging of triglycerides into lipoproteins for excretion, and impaired fatty acid β-oxidation (FAO) in mitochondria. Fatty acids can be oxidized at different carbons (α, β, and γ), with the β-oxidation being the process that takes place in mitochondria through the respiratory chain, which requires the assistance of carnitine palmitoyltransferase-1 (CPT-1) for the import of long-chain fatty acids through the mitochondrial inner membrane (MIM) for catabolism in the respiratory chain [117]. In this regard, mitochondrial dysfunction has been described in patients with IR, and impaired mitochondrial respiratory chain components have been described in human MASH [118, 119]. In support of the contribution of impaired mitochondrial β-oxidation to hepatic steatosis, inhibition of CPT-1 by malonil-CoA contributes to fatty acid accumulation as triglycerides in hepatocytes [120, 121]. Although IR is a hallmark and a driver of MASLD pathogenesis and the onset of hepatic steatosis as the initial stage of the disease, whether impaired β-oxidation contributes to IR is controversial. For instance, increased hepatic FAO by adenovirus-mediated CPT-1 overexpression in diabetic db/db mice has been recently reported as a molecular therapy for obesity and IR [122]. However, CPT-1 inhibition in skeletal muscle has been shown to improve insulin signaling in diet-induced obese mice, and unchanged CPT-1 activity has been reported in MASH patients [123, 124]. In analogy to MASLD, alcohol oxidative metabolism disrupts mitochondrial function and hence impairs oxidative phosphorylation both in experimental models and patients with ALD, contributing to the onset of alcohol-mediated hepatic steatosis [113, 114, 125]. Thus, in addition to mechanisms leading to increased de novo lipogenesis triggered by ER stress, impaired mitochondrial FAO and decreased CPT-1 expression can further contribute to fatty acid accumulation and steatosis both in ALD and MASLD (Figure 1) [126, 127]. A remaining question is whether the decreased FAO is a specific process or reflects a general impairment in mitochondrial function, which needs to be clearly established vis-à-vis the species-dependent alterations in mitochondrial function in ALD [113], and the opposing outcome of mitochondrial dysfunction between experimental models and patients with MASLD (see below).
Besides different morphology (macro vs. microvesicular), steatosis reflects the accumulation of a variety of lipids, including triglycerides, fatty acids, and cholesterol (free and esterified). Pioneering studies using nutritional and genetic models of liver steatosis established that the accumulation of free cholesterol, particularly in mitochondria sensitized hepatocytes to inflammatory cytokines, promoting the transition from simple steatosis to steatohepatitis [42]. The trafficking of cholesterol to mitochondria is governed by STARD1, the founding member of a family of proteins with cholesterol binding domain, whose inactivation results in fatal adrenal lipoid hyperplasia and inability to synthesize steroid hormones, illustrating the critical role of this protein in steroidogenic tissues to metabolize cholesterol in the MIM to pregnenolone, the precursor of steroid hormones and neurosteroids in hypothalamic neurons [128, 129]. The basal levels of STARD1 in the liver are low, but under alcohol intake leading to ALD and ER stress underlying the nutritional overload of MASLD, STARD1 expression is markedly overexpressed leading to mitochondrial cholesterol trafficking to the MIM, where it is metabolized by CYP27A1 to 27-hydroxycholesterol [37, 69, 130]. Besides the increase of this oxysterol, which acts as a precursor of CDCA, the primary BA synthesized in the mitochondrial alternative pathway, STARD1-mediated cholesterol trafficking results in accumulation of cholesterol in the MIM, which results in far-reaching consequences, such as disruption of mitochondrial physical properties, leading to decreased membrane fluidity that impairs mitochondrial antioxidant defense, illustrated by the depletion of mitochondrial glutathione (GSH) [131–135]. In addition, cholesterol accumulation in MIM interferes with the assembly of respiratory supercomplexes, underlying the impairment in the respiratory capacity, stimulates the generation of superoxide anion resulting in oxidative stress, and stabilizes hypoxia inducible factor-1 (HIF-1), leading to metabolic reprogramming with impaired oxidative phosphorylation and stimulated glycolysis [136]. In support of the role of STARD1 in metabolic liver disease, increased expression of STARD1 at the mRNA and protein levels has been described in models and patients with ALD and MASH [37, 69, 130]. The pool of cholesterol in mitochondria plays a multifactorial role in fostering the transition from simple steatosis to steatohepatitis, namely by promoting hepatocellular injury and oxidative stress, inflammation, and liver fibrogenesis secondary to the activation of HSCs, underlying the onset of MASH. Mitochondrial cholesterol also contributes to the MASH-driven HCC not only by inducing the reprogramming of BA metabolism, but also by switching the synthesis of BAs from the classic pathway to the alternative mitochondrial pathway, which unlike the former is insensitive to BA-mediated repression of CYP7A1 [37]. While STARD1-dependent BA synthesis stimulates carcinogenesis through the induction of transcription factors involved in the stemness, self-renewal, and inflammation, the enrichment of mitochondria in cholesterol and subsequent impact on mitochondrial membrane fluidity impairs chemotherapy that targets the mitochondrial apoptotic pathway by restraining the mitochondrial outer membrane permeabilization and subsequent engagement of the apoptosome [135]. Interestingly, the expression of CYP7A1 decreases in ALD, but patients with alcoholic hepatitis exhibit increased levels of BAs, which correlate with the clinical score [137]. These results imply that alcohol-induced STARD1 expression mediates the cholestasis accompanying the progression of alcohol-induced steatosis to alcoholic hepatitis. Furthermore, acute-on-chronic model of ALD disclosed not only an early induction of STARD1 preceding the onset of hepatic steatosis and liver injury, but the overexpression of STARD1 exhibited a zonal pattern, with a predominant expression in perivenous (PV) hepatocytes, coinciding with the area where CYP2E1 is mostly expressed that correlates with the depletion of mitochondrial GSH and the onset of oxidative stress [130]. These findings indicate that chronic alcohol feeding caused a zone-dependent morphological change in the shape and number of mitochondria, inducing an increase in the number of mitochondria in the PV area with respect to the periporal (PP) zone. Thus, while steatosis is characterized by the accumulation of different types of lipids, the increase in cholesterol and its trafficking to mitochondria has emerged as a key player in both ALD and MASLD.
As succinctly described above, the impairment of mitochondrial function described in patients with ALD and MASLD has been associated with disease progression, suggesting that loss of mitochondrial function is a cause of the disease. However, this concept has been challenged in both diseases by the availability of experimental studies using genetic models of mitochondrial dysfunction. For instance, mice with liver or muscle specific deletion of apoptosis inducing factor, a mitochondrial protein originally described as a crucial player in apoptosis, and later shown to play a pivotal role in the maintenance of a functional respiratory chain, exhibited impaired oxidative phosphorylation activity, as expected, with defects in Complex I and IV of the respiratory chain, that upon high fat diet consumption, exhibited increased insulin sensitivity, a leaner phenotype, and resistance to steatosis and MASH progression compared to control littermates [138]. Similar findings were reported in mice with deletion of mitochondrial transcription factor A (TFAM) in skeletal muscle and adipose tissue, exhibiting generalized defects in mitochondrial function, expected from the crucial role of TFAM in mitochondrial physiology, that was accompanied by increased glucose uptake in skeletal muscle due to increased glucose transporter 1, while deletion in adipose tissue resulted in resistance to diet-induced obesity, IR, and hepatosteatosis [139, 140]. Taken together, these findings suggest that mitochondrial dysfunction and defects in the respiratory chain are protective against diet-induced obesity, IR, and MASLD. In the context of ALD, intriguing studies have shown that chronic alcohol intake, either orally or intragastrically in mice, caused increased state III respiration associated with enhanced levels of Complex I, IV, and V incorporated into the respiratory chain that reflected the effects of alcohol in the expression of PGC1α, the master regulator of mitochondrial biogenesis [141]. These findings imply that enhanced mitochondrial respiration in mice fed an alcohol-containing diet stimulated the replenishment of NAD+ from NADH oxidation to accelerate the oxidative metabolism of ethanol. These findings contrast with the pioneering studies in rats, which show depression of mitochondrial respiratory capacity with defects in the synthesis of subunits of the main respiratory complexes such as NADH dehydrogenase, cytochrome b-c1 (Complex III), and the ATP synthase complex (Complex V). Interestingly, the increased susceptibility of mice vs. rats to alcohol-induced liver damage implies that the stimulation of the mitochondrial respiratory capacity contributes to enhanced alcohol metabolism and subsequent induction of oxidative stress and recruitment of downstream players involved in ALD. Thus, further research will be required to establish whether the impairment of mitochondrial function described in patients with ALD and MASLD contributes to the onset of disease or is a consequence of the disease progression.
BAs were historically regarded as detergents required for the emulsification and absorption of dietary lipids. This simplistic view has dramatically evolved over the past two decades, with BAs now recognized as multifunctional signaling molecules that participate in systemic metabolic regulation, immune homeostasis, and host-microbiome interactions [142, 143]. BAs act not only as digestive surfactants but also as hormones that regulate glucose and lipid metabolism, energy expenditure, inflammation, and fibrosis through nuclear and membrane-bound receptors such as FXR, Takeda G-protein receptor 5 (TGR5), vitamin D receptor (VDR), pregnane X receptor (PXR), and sphingosine-1-phosphate receptor 2 (S1PR2).
The relevance of BA biology to clinical hepatology and gastroenterology has grown substantially. Dysregulated BA metabolism contributes to bile acid diarrhea (BAD), cholestatic liver disorders, inflammatory bowel disease (IBD), Clostridioides difficile infection (CDI), colorectal neoplasia, and MASLD, as mentioned above (see section Metabolic liver diseases). Evidence indicates that changes in BA quantity and quality are closely linked to MASLD severity and fibrosis progression.
Beyond their detergent role, BAs are potent signaling molecules. FXR senses BA levels in hepatocytes and enterocytes. In the ileum, FXR induces FGF19, which acts in the liver to suppress CYP7A1, limiting BA synthesis. FXR also regulates lipid metabolism and inflammation. TGR5 is expressed in enteroendocrine L-cells, where it induces GLP-1 release, linking BAs to glucose homeostasis. It also reduces inflammation in macrophages. VDR and PXR regulate detoxification and immune responses, with PXR being particularly important for xenobiotic metabolism. S1PR2 mediates direct BA signaling in HSCs, linking BAs to fibrosis [144].
The gut microbiota diversifies the BA pool through deconjugation, dehydroxylation, and epimerization reactions. Bile salt hydrolases (BSHs), expressed by Lactobacillus and Bifidobacterium spp., deconjugate taurine- and glycine-conjugated BAs. The 7α-dehydroxylating bacteria such as Clostridium scindens convert CA to DCA and CDCA to LCA. Other taxa epimerize CDCA to UDCA, a therapeutically important hydrophilic BA [145]. These microbial conversions alter BA hydrophobicity, cytotoxicity, and receptor affinity.
The gut microbiota and BAs exist in a dynamic, bidirectional relationship that influences host physiology. On the one hand, intestinal microbes modify BAs through enzymatic transformations; on the other hand, BAs act as antimicrobial agents that shape the composition of the microbiota. This mutual regulation is increasingly recognized as a critical factor in digestive and metabolic diseases.
As mentioned, BSHs modify primary BAs driven by Clostridium scindens and related taxa [145–147]. Secondary BAs influence immune cell differentiation. For instance, isoallolithocholic acid promotes regulatory T cell development, while taurine-conjugated BAs can activate the NLRP3 inflammasome, driving inflammation. BA signaling through FXR in intestinal epithelial cells maintains barrier integrity by regulating tight junction proteins and antimicrobial peptide production. TGR5 activation in macrophages reduces pro-inflammatory cytokine release, linking BA signaling to mucosal immunity [147, 148].
In health, balanced BA-microbiome interactions support homeostasis. In disease, dysbiosis leads to altered BA pools. Patients with IBD exhibit reduced microbial diversity and decreased secondary BA levels, weakening FXR/TGR5 signaling and perpetuating inflammation [149]. In MASLD, dysbiosis favors taurine-conjugated BAs and reduces FXR agonists, promoting metabolic dysfunction and fibrosis [150, 151]. Importantly, fecal BA signatures are being investigated as noninvasive biomarkers to stratify disease activity in IBD, MASLD, and colorectal cancer (CRC) [152, 153].
BAD occurs when BA reabsorption in the ileum is defective or feedback regulation via the FXR-FGF19 axis is impaired. Primary BAD, often underdiagnosed, is linked to inadequate FGF19 signaling, leading to unchecked CYP7A1 activity and hepatic BA overproduction [154]. Secondary BAD results from ileal resection, Crohn’s disease, or cholecystectomy.
Diagnostics include serum 7α-hydroxy-4-cholesten-3-one (C4), with values > 48 ng/mL suggestive of BAD, and fasting FGF19 < 145 pg/mL. The SeHCAT retention test has excellent sensitivity and specificity. Fecal BA quantification remains a gold standard but is cumbersome [155].
First-line therapy is BAs sequestrants, though tolerability is limited. FXR agonists and FGF19 analogs represent emerging therapies that target the underlying pathophysiology [156].
Cholestasis results in the intrahepatic retention of hepatotoxic BAs. In PBC, UDCA is standard, but many patients are incomplete responders [157]. Obeticholic acid is approved as a second-line therapy but is limited by pruritus. In primary sclerosing cholangitis (PSC), norUDCA showed significant benefit in a 2025 phase 3 trial [158]. Recent findings indicated an increased expression of STARD1 in patients with PBC, suggesting that the mitochondrial alternative pathway of BA synthesis could play a role in PBC pathogenesis [159].
In pediatric cholestasis, ileal bile acid transporter (IBAT) inhibitors such as maralixibat and odevixibat are approved, reducing pruritus and serum BA levels [160].
IBD is characterized by altered BA pools with reduced secondary BAs, weakening FXR/TGR5 signaling. This contributes to barrier dysfunction and inflammation. Experimental studies show that BA supplementation or intestine-restricted FXR agonists restore epithelial barrier integrity and modulate immunity [149, 161]. Microbiome-targeted therapies are also under investigation [162].
Primary conjugated BAs such as taurocholate stimulate spore germination, while secondary BAs such as DCA and LCA inhibit growth. Antibiotic-induced dysbiosis reduces secondary BAs, predisposing to CDI. Fecal microbiota transplantation restores BA profiles and reduces recurrence [146, 162].
Secondary BAs, particularly DCA, promote tumorigenesis by inducing oxidative stress, DNA damage, and Wnt/β-catenin signaling [163]. Epidemiological studies show higher fecal BA load in Western diets and CRC patients [152]. Loss of FXR in colonic tissue amplifies tumorigenesis.
As mentioned above, in MASLD, BA metabolism is profoundly altered, with elevated conjugated and 12α-hydroxylated BAs. Dysregulated FXR-FGF19 signaling leads to excessive BA synthesis, while taurocholate activates stellate cells via S1PR2, promoting fibrosis [150, 164, 165]. Dysbiosis shifts microbial BA metabolism toward pro-fibrogenic profiles, worsening inflammation [151].
Serum conjugated BAs (GUDCA, GCDCA, TCDCA) correlate with histologic severity and fibrosis. Therapeutics including FXR agonists, FGF19 analogs, and resmetirom (THR-β agonist approved in 2024), illustrate how BA modulation is central to MASLD management [166–168].
Multiple serum, fecal, and imaging-based biomarkers have emerged to quantify BA metabolism. Serum C4 and FGF19 are useful for diagnosing BAD, while serum BA panels predict fibrosis in MASLD [150, 151, 155]. Fecal BA profiling characterizes IBD, CRC, and CDI, while SeHCAT is highly sensitive for BAD diagnosis [155, 165].
BA-targeted therapies range from UDCA to novel small molecules, biologics, and microbiome interventions. NorUDCA showed benefit in PSC, while obeticholic acid, cilofexor, and tropifexor reduce fibrosis but are limited by side effects [157, 158, 167]. FGF19 analogs and engineered mRNA restore BA signaling but raise safety concerns. IBAT inhibitors are approved in pediatric cholestasis [160]. Microbiome-directed therapies, including FMT, restore secondary BA production in CDI. Resmetirom, a THR-β agonist, was FDA-approved in 2024 for MASH [168], but whether this agent affects BA homeostasis or not remains to be established.
Challenges include assay standardization, balancing therapeutic efficacy with safety, and addressing patient heterogeneity [167–169]. Microbiome therapy outcomes remain variable, and long-term safety of FXR agonists and FGF19 analogs requires further study. Future directions include plasma BA panels in clinical practice, approval of norUDCA, development of microbiome-based BA modulators, and AI-driven BA modeling for personalized therapy.
BAs have emerged as central modulators of digestive and metabolic diseases. Advances in mechanistic understanding, biomarker discovery, and targeted therapies highlight their dual role as pathogenic drivers and therapeutic opportunities. In MASLD, specific BA signatures contribute to fibrogenesis, making them attractive diagnostic and therapeutic targets. The next decade promises integration of BA-focused strategies into precision hepatology and gastroenterology.
DILI is the most common cause of acute liver failure in the USA and European countries [170]. Most cases of acute liver failure in the US are caused by acetaminophen (APAP) overdose [171]. APAP is considered safe at therapeutic doses. However, an overdose of APAP can lead to the accumulation of APAP protein adducts, resulting in mitochondrial damage, DNA fragmentation, hepatocyte necrosis, sterile inflammation, and potentially liver failure [172]. Many adaptive and survival mechanisms can counteract these adverse effects, but the failure or insufficiency of these mechanisms can ultimately lead to liver failure [172].
One of these adaptive and protective mechanisms is autophagy. Autophagy is an evolutionarily conserved intracellular degradation pathway involving lysosomes that break down proteins, lipids, and organelles such as excess ER, ribosomes, and damaged mitochondria [173, 174]. Autophagy is often activated and serves as an adaptive response to adverse stresses such as starvation or exposure to xenobiotics, functioning as a cell survival mechanism by providing nutrients and building blocks to maintain cellular homeostasis [175–178].
There are three main types of autophagy: macroautophagy (referred to as autophagy), microautophagy, and chaperone-mediated autophagy (CMA). These types differ in how cargoes are delivered to lysosomes [179]. Autophagy involves the formation of a double-membrane autophagosome that encloses the autophagic cargo and transports it to a lysosome, where the membranes fuse to create an autolysosome that degrades the autophagic cargo with lysosomal acidic hydrolytic enzymes [173]. Microautophagy involves lysosomes directly engulfing autophagic cargos, bypassing the formation of autophagosomes [180, 181]. CMA involves the recognition of cellular proteins that contain the pentapeptide motif (KFERQ) by cytosolic chaperones such as the heat shock cognate protein of 70 kDa (HSC70). These chaperone proteins then bind to lysosome-associated membrane protein type 2A (LAMP-2A), which triggers LAMP-2A multimerization. This results in the formation of a translocation complex that transports CMA substrates across the lysosomal membrane for degradation [182–184]. Microautophagy and CMA have been previously reviewed for their mechanisms and role in liver pathophysiology [185, 186]. However, their link to DILI has not been thoroughly studied. Below, we will focus on the current understanding of autophagy mechanisms and their possible roles in DILI, specifically in APAP-induced liver injury (AILI).
Autophagy provides several mechanisms to protect against AILI. First, autophagy can help remove the APAP-AD likely through the autophagy receptor protein SQSTM1/p62 because genetic deletion of p62 slows down the clearance of APAP protein adducts in hepatocytes [187, 188]. Pharmacological inhibition of autophagy/lysosomal functions by chloroquine delays lysosomal clearance of APAP protein adducts and increases liver injury in mice [187, 188]. In contrast, activating autophagy pharmacologically safeguards against AILI in mice. However, it is important to recognize that after a single, severe overdose of APAP, autophagy can rescue cells at the periphery of the necrotic area [188], leading to a reduced area of necrosis [187–189]. In contrast, after multiple low doses of APAP, autophagy can completely prevent liver injury [189], suggesting that the protective effect of autophagy is dose-dependent.
While pharmacological manipulation of autophagy has led to a clear conclusion regarding the protective role of autophagy against AILI, results from studies using genetic deletion of essential autophagy-related genes in mice are complex. Liver-specific Atg5 knockout mice are resistant to AILI due to the compensatory activation of the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway and repletion of hepatic glutathione pools, while unc-51-like autophagy activating kinase 1 and 2 (Ulk1/2) double knockout mice inhibit JNK activation to protect against AILI [190, 191]. The difference between the results from genetic autophagy-deficient mouse models and the pharmacological manipulation of autophagy on AILI may be due to chronic versus acute autophagy deficiency. Increased Nrf2 activation is likely a secondary effect of long-term autophagy inhibition in mouse livers, serving as an adaptive response driven by the activation of non-canonical Nrf2 pathways. Second, mitochondrial damage is an early key event leading to hepatocyte death after APAP overdose [192]. Autophagy can assist in removing damaged mitochondria induced by APAP via selective mitophagy, likely through the PINK1-PARKIN pathway [193]. Third, APAP impairs hepatic transcription factor EB (TFEB), a master regulator of gene expression of autophagy-related genes and lysosomal biogenesis genes. Genetic or pharmacological activation of TFEB promotes the clearance of APAP-AD and protects against AILI in mice [194, 195]. Fourth, APAP-induced hepatocyte necrosis can lead to the release of damage-associated molecular patterns (DAMPs), which further activate Kupffer cells (KCs), resulting in the release of cytokines and chemokines that recruit immune cells, such as neutrophils and circulating monocytes. The role of inflammation in AILI remains debatable, as both protective and harmful effects have been reported, depending on the dose of APAP and the stage of AILI (early phase vs. late phase) [196]. Restricting the expansion of necrotic areas and removing necrotic cells are crucial for liver repair and recovery from AILI [197]. Increasing evidence suggests that activated macrophages and neutrophils play a crucial role in removing necrotic cells and promoting liver repair and regeneration. The expression of ATG5 decreased in bone marrow-derived macrophages (BMDM) in aged mice, and these mice were more susceptible to thioacetamide (TAA)-induced liver injury compared with young mice. Restoration of ATG5 in BMDM protects against TAA-induced liver injury in mice [198]. Moreover, myeloid-specific Atg5 knockout mice also had increased liver injury and inflammation after alcohol exposure [199]. In addition, macrophage ATG16L1 deletion inhibits liver regeneration by promoting and sustaining cGAS-STING pathway activation in mice [200]. While these findings suggest a protective role of immune cells (in particular macrophage autophagy) against AILI, future studies are needed to investigate the role of macrophage autophagy in AILI using these macrophage-specific autophagy-deficient mice. In conclusion, current evidence clearly supports a protective role of autophagy against AILI. Future studies to determine whether and how activating autophagy would promote necrosis resolution and liver regeneration, and to identify new potent autophagy activators, are important for translating these findings to clinical applications.
The pancreas is a unique organ that has both endocrine and exocrine functions. Pancreatitis is characterized by increased necrosis of acinar cells, the exocrine cells in the pancreas, followed by a local and systemic inflammatory response, which can have a varied clinical presentation and course of progression. Acute pancreatitis is a common gastrointestinal condition with an annual incidence of 34 per 100,000 person-years in developed countries without successful treatments. Chronic alcohol consumption accounts for 17% to 25% of acute pancreatitis cases worldwide, although hypertriglyceridemia, hypercalcemia, viral infections, genetics, and autoimmune diseases may also contribute to pancreatitis [201–203].
Pancreatic exocrine acinar cells have high rates of protein synthesis to produce and secrete large numbers of digestive enzymes during the feeding process. To meet the demand for protein synthesis and secretion, acinar cells have a highly enriched ER and vesicle trafficking system. Maintaining the ER, other organelles, and protein homeostasis is thus crucial for regulating acinar cell functions. When the regulation of organelle and protein homeostasis is disrupted, it can cause ER stress, mitochondrial damage, and improper intracellular trypsinogen activation, ultimately leading to acinar cell death and the onset of pancreatitis. As a cellular organelle and protein quality control mechanism, autophagy is therefore vital for maintaining organelle homeostasis and adaptation to protect the acinar cells. Below, we briefly review the current understanding of autophagy and its role in maintaining acinar cell homeostasis and function in pancreatitis.
Accumulation of large vacuoles in acinar cells has been frequently observed in human and experimental pancreatitis. These large vacuoles are LC3 positive and morphologically identified as large autolysosomes, likely resulting from impaired autolysosomal functions. The expression levels of both LAMP-1 and LAMP-2, two lysosomal membrane proteins, decreased in five different rat and mouse models of pancreatitis [204]. More importantly, genetic deletion of LAMP-2 in mouse acinar cells causes impaired autophagy in the exocrine pancreas, leading to the accumulation of large vacuoles in acinar cells and spontaneous pancreatitis [204]. Deletion of Atg7 in pancreatic epithelial cells (likely both acinar cells and endocrine cells) using Pdx-Cre in mice leads to chronic pancreatitis with increased inflammation and fibrosis [205]. In another mouse model, systemic deletion of Atg7 using a doxycycline (DOX) inducible mechanism in mice also led to pancreatic damage resembling mild acute pancreatitis [206]. Deletion of Atg5 specifically in acinar cells using Elastase-Cre in mice only leads to mild pancreatic injury, but these mice are more susceptible to pancreatitis induced by cerulein or Lieber-Decarli liquid diet [207]. Ethanol exposure also impaired autophagy by stabilizing ATG4B to promote alcohol-associated pancreatitis [208]. Vacuole membrane protein 1 (VMP1) is an ER phospholipid scramblase that is crucial for regulating autophagosome closure and the secretion of very low-density lipoprotein (VLDL) proteins [209–213]. An early study shows a correlation between increased VMP1 expression and acinar cell vacuolization in arginine-induced acute pancreatitis in rats [214]. Subsequent studies showed that the loss of acinar cell VMP1 in mice led to spontaneous pancreatitis associated with increased ER stress and Nrf2 activation [215]. It is unclear, however, whether overexpression of VMP1 also impairs autophagy in acinar cells, but deleting VMP1 in these cells indeed severely impairs autophagy, as shown by increased pancreatic LC3-II levels and p62, probably due to impaired autophagosome closure [215]. Further research will be required to dissect the causal role of VMP1 in pancreatitis. TFEB is a master regulator for the gene expression of autophagy and lysosomal biogenesis. TFEB is downregulated in human pancreatitis and experimental pancreatitis induced by either cerulein or alcohol. More importantly, pharmacological or genetic activation of TFEB protects against cerulein or alcohol-induced pancreatitis in mice [207, 216].
Autophagy may help protect against the development of pancreatitis through several mechanisms. First, due to the high translational capacity of acinar cells, increased protein synthesis can lead to the accumulation of misfolded proteins and ER stress. These misfolded proteins can be efficiently removed by autophagy, which helps alleviate ER stress. Second, trypsinogen is stored in zymogen granules within acinar cells, but these granules can become fragile, resulting in their release into the cytosol and subsequent activation of trypsinogen. This activation can cause acinar cell death. Autophagy can selectively remove these zymogen granules via zymophagy, thereby preventing trypsin activation and cell death. While the specific receptor for selective zymophagy has not been identified, increased VMP1 has been associated with zymophagy in AR42J cells [217]. Third, excess ER material can be removed through ER-phagy to reduce ER stress. Though specific ER-phagy receptors have not yet been identified for pancreatitis, VMP1 has several potential LIR domains that may serve as ER-phagy receptors and warrant further investigation. Fourth, damaged mitochondria contribute to acinar cell death and pancreatitis, but these organelles can be removed via selective mitophagy. The increased accumulation of damaged mitochondria, resulting from impaired autophagy, promotes experimental pancreatitis [218]. Mitophagy is regulated by the well-known PINK1-PARKIN pathway and involves a group of specific mitophagy receptor proteins [219, 220], although whether the PINK1-PARKIN pathway and these mitophagy receptor proteins play a role in the pathogenesis of pancreatitis remains unclear.
In conclusion, autophagy protects acinar cells from adverse conditions by maintaining organelle and protein homeostasis through multiple quality control pathways. Future research should aim to identify the specific receptors involved in these forms of autophagy and determine whether pharmacological targeting of these pathways could be beneficial against pancreatitis.
One of the most fundamental questions in the study of metabolic disease progression is how triglyceride accumulation—classically considered a “safe” mechanism for energy storage—can ultimately sensitize hepatocytes to cell death and promote tissue damage. Steatotic liver disease encompasses a wide pathological spectrum, ranging from simple steatosis, defined by excess triglyceride deposition in hepatocytes, to more advanced stages characterized by necroinflammation, fibrosis, and eventual hepatic failure [221–224].
Lipotoxicity is defined as the exposure of cells to increased levels of (toxic) lipid species, often in the context of metabolic liver diseases such as MASLD (often in combination with IR, which can aggravate the lipotoxicity and/or dyslipidemia). The primary target cell of lipotoxicity in the liver is the hepatocyte, although non-parenchymal cell types in the liver also respond to exposure to increased levels of lipids [225, 226].
Lipotoxicity is often discussed in the context of hepatocyte damage. The effects of (excess) lipids on non-parenchymal liver cells, such as HSCs, KCs, and liver sinusoidal endothelial cells (LSECs), have been less studied. FFAs can activate KCs via their TLRs. HSCs lose their lipid content during activation, potentially aggravating the exposure of other liver cell types to lipids. In contrast, direct effects of FFAs on HSCs have not been explored in detail yet. LSECs may very well be the first target of excessive lipid content in the liver [227]. The response of LSECs to FFAs is different from hepatocytes: The unsaturated fatty acid (UFA) oleate is protective in hepatocytes but toxic in LSECs. The response to FFAs also varies with the type of endothelial cells: LSECs respond differently to FFAs than vascular endothelial cells [228]. Finally, lipotoxic cell death of hepatocytes may lead to secondary activation (inflammation, fibrogenesis) of KC and HSCs. Lipids may also induce hepatocyte senescence, leading to the induction of the senescence-associated secretory phenotype, contributing to an inflammatory response of the non-parenchymal cells [229].
Recent studies demonstrate that the deleterious effects induced by lipids are partially regulated by the interaction of saturated fatty acids (SFAs) with LPS. Obesity increases sensitivity to endotoxin-mediated liver injury, which is manifested in fatty liver sensitivity to acute inflammation and injury [230–232]. Therefore, a better understanding of the direct and indirect effects of lipids (and lipotoxicity) on non-parenchymal cells, and the interplay between hepatocytes and non-parenchymal cells, is needed.
In addition to increased levels of lipids in metabolic liver diseases, the role of altered lipid profiles should also be investigated in more detail: It has been shown that UFAs like oleate can mitigate the toxic effects of SFAs like palmitate. In addition, the effects of UFAs/SFAs may be different in non-parenchymal cells like LSECs [228]. This observation suggests that modifying the lipid profile in patients with metabolic diseases may be a target for intervention, although results have been disappointing until now. This may be due to a lack of targeting and/or attaining sufficient levels of ‘beneficial’ lipids in the target cells. In this regard, targeted delivery to liver cells, using nanocarriers, may be an option to explore in the future.
Another interesting finding in in vitro studies is that lipotoxicity in hepatocytes is characterized by a lack of lipid droplet accumulation rather than an excessive accumulation of lipid droplets [225, 226]. This is counterintuitive given the observation of excessive accumulation of lipid droplets in hepatocytes in MASLD. The SFA palmitate is lipotoxic to hepatocytes but does not induce lipid droplet accumulation, whereas the UFA oleate is not toxic and induces lipid droplet accumulation. Moreover, oleate attenuates the lipotoxic effect of palmitate and increases lipid droplet accumulation. A possible explanation is that increased lipid accumulation initially is a protective mechanism to sequester toxic lipid species. However, prolonged and excessive exposure to lipids, as in MASLD patients, may exceed the limit of ‘safe’ lipid droplet storage, leading to changes in lipid droplet dynamics and metabolism resulting in lipotoxicity. More studies are needed to elucidate the role of lipid droplets in the pathogenesis of MASLD, focusing on size, distribution, composition, and dynamics [226, 233]. Another aspect is that not all UFAs behave equally in terms of the mechanisms of lipotoxicity. For instance, myristic acid, another UFA, is not lipotoxic by itself but potentiates palmitic acid-induced lipotoxicity by sustaining the synthesis of ceramide through the myristoylation of ceramide desaturase [234]. In this regard, diets enriched in myristic and palmitic acid markedly induce MASH by channeling the use of palmitic acid in the generation of ceramide de novo. Thus, different lipotoxic lipids may induce cell death through specific pathways.
The exposure of hepatocytes to excessive amounts of lipids induces stress in various organelles, such as mitochondria and the ER, and oxidative stress. The effects of excessive lipid exposure, particularly FFAs, on mitochondria have been described extensively in recent reviews [225, 226, 229]. Impairment of mitochondrial function, in particular β-oxidation and ATP production, increases the generation of ROS and depletes cellular antioxidants. At the early stage of MASLD, exposure to a high influx of lipids increases mitochondrial oxidation; however, this oxidation is incomplete, and increases the production of ROS and oxidized (toxic) lipid intermediates [125, 235], leading to the further impairment of mitochondrial function and, eventually, cell death [236, 237]. Diminished mitochondrial function itself also increases ROS generation, thus creating a vicious cycle of mitochondrial dysfunction and oxidative stress.
Another target of (toxic) lipids in the context of metabolic diseases is the ER. The initial, protective response to lipid exposure of the ER is activation of the unfolded protein response (UPR). However, sustained activation of the UPR leads to ER stress. The UPR is composed of three branches: the transmembrane proteins RNA-dependent protein kinase-like ER eukaryotic initiation factor-2α kinase (PERK), the activating transcription factor 6 (ATF6), and the inositol-requiring ER-to-nucleus signaling protein-1 (IRE1α). The activation of these branches of the UPR counteracts ER stress, but sustained activation of these pathways leads to a pro-inflammatory response and, eventually, cell death. The role of the ER in lipid metabolism and lipotoxicity is complex and bidirectional: Not only is the ER a target for lipotoxicity, but the ER is also involved in lipid homeostasis.
De novo lipogenesis takes place in the ER, where excess free SFAs are incorporated into phospholipids of the ER membrane, leading to Ca2+ release, mitochondrial permeability impairment, and activation of pro-inflammatory and cell death pathways [225, 226, 238]. This causes ER stress and compromises normal ER function, triggering the UPR [225, 226, 238]. Activation of the UPR and ER stress can also increase lipogenesis: ER stress activates the transcription factor sterol regulatory element binding proteins (SREBPs), leading to enhanced lipid synthesis [239–242]. SREBP transcription factors are master regulators of hepatic lipid metabolism [239–242]. In normal conditions, they are attached to ER membranes as inactive precursors. Upon activation, e.g., by low sterol levels, these precursors are cleaved, and a water-soluble fragment then translocates to the nucleus [239–242]. Activation of the PERK-p-eIF2α signaling pathway promotes lipid accumulation via SREBP activation [241–243]. In addition, ATF4 and ATF6 can activate SREBPs and play an important role in lipid homeostasis during ER stress, mainly by increasing hepatic lipogenesis [243–246].
The excessive incorporation of SFAs into ER membranes in conditions of lipid overload reduces the presence of other lipid species in the ER membranes, such as sphingomyelin and cholesterol [247]. Moreover, SFA overload in the ER increases the ratio of phosphatidylcholine (PC)/phosphatidylethanolamine (PE), resulting in the inhibition of sarco/ER calcium ATPase (SERCA) activity and disrupting ER calcium homeostasis. In normal conditions, the ER is characterized by a very high Ca2+ concentration, maintained by the SERCA-ATPase. This regulation is important for ER function since many chaperone proteins and enzymes involved in protein folding and maturation processes are dependent on high Ca2+ levels [238].
Lipotoxicity, caused by the accumulation of FFAs, cholesterol, ceramides, and other toxic lipid intermediates, is a central driver of hepatocyte injury in steatotic liver disease and steatohepatitis. The hepatotoxic effects of lipids converge on regulated cell death (RCD) pathways, including apoptosis, necrosis, necroptosis, autophagy-dependent death, pyroptosis, and ferroptosis. These processes interact, shaping inflammation, fibrosis, and disease progression.
Apoptosis is the most extensively studied RCD pathway in the liver and is strongly driven by lipotoxicity. SFAs such as palmitate induce ER stress, mitochondrial dysfunction, and oxidative injury, activating the intrinsic apoptotic pathway. This involves mitochondrial outer membrane permeabilization, cytochrome c release, apoptosome formation, and caspase-9/-3/-7 activation. The extrinsic pathway, mediated by death receptors (e.g., TNFR, FAS), is also sensitized by lipotoxicity: FFAs upregulate death ligands and decrease anti-apoptotic proteins, enhancing caspase-8 activation. In hepatocytes (type II cells), mitochondrial amplification via BID cleavage is required, again highlighting the lipid-mitochondria connection. Apoptosis contributes directly to inflammation and fibrosis by releasing apoptotic bodies that activate Kupffer and stellate cells [248–250].
Toxic lipids induce mitochondrial overload, ROS production, and calcium imbalance, all of which favor mitochondrial permeability transition (MPT). In lipotoxic hepatocytes, sustained opening of the MPT pore collapses the membrane potential and leads to necrotic rupture, releasing DAMPs. Free cholesterol and SFAs destabilize mitochondrial membranes, lowering the threshold for pore opening. This necrosis amplifies sterile inflammation in steatohepatitis and may synergize with apoptosis to drive mixed injury patterns.
Lipotoxic conditions frequently create an environment that favors necroptosis. Inhibition of caspase-8 by lipotoxic stress (via oxidative modifications or c-FLIP regulation) allows receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and RIPK3 to form the necrosome, which phosphorylates MLKL to disrupt membranes. Experimental models have shown that SFAs increase RIPK3 expression in non-parenchymal cells and sensitize hepatocytes to necroptotic injury. Necroptosis contributes to inflammatory amplification in MASH, since ruptured cells release pro-inflammatory lipids and DAMPs that further activate immune pathways.
Autophagy normally serves a protective role in lipotoxicity, clearing lipid droplets (lipophagy) and damaged mitochondria (mitophagy). However, excessive lipid load can overwhelm autophagy or impair lysosomal function, shifting the balance toward injury. In some models, blocking autophagy exacerbates lipotoxic apoptosis and necrosis, while in others, dysregulated or excessive autophagy contributes to hepatocyte loss. For example, impaired autophagic flux in steatohepatitis worsens lipotoxic stress by preventing the clearance of lipid peroxides and damaged mitochondria. Thus, autophagy in the lipotoxic liver is double-edged: protective under moderate stress but potentially death-promoting when overloaded.
Lipotoxic hepatocytes release mitochondrial DNA and oxidized lipids that act as danger signals, activating inflammasomes (NLRP3, AIM2). SFAs promote potassium efflux and ROS, key triggers of inflammasome assembly. Activated caspase-1 and gasdermin D (GSDMD) pores cause pyroptotic lysis, releasing IL-1β, IL-18, and lipid adducts that fuel hepatic inflammation. Lipotoxicity thus links hepatocyte pyroptosis to stellate cell activation and fibrosis. Importantly, lipid-induced pyroptosis also amplifies immune cell activation in non-parenchymal compartments of the liver.
Ferroptosis is an iron-dependent form of RCD defined by the accumulation of lipid peroxides when GSH-dependent repair systems, particularly glutathione peroxidase 4 (GPX4), are compromised [251–253]. Hepatocyte ferroptosis is blocked by both iron chelators and lipophilic antioxidants, underscoring the central role of cysteine/GSH metabolism [254–257]. In experimental models, methionine- and choline-deficient diets highlight ferroptosis as a major driver of steatohepatitis, with inhibition of ferroptosis attenuating hepatic lipid peroxidation and cell death [258]. Pharmacological induction of ferroptosis (e.g., with RSL3) exacerbates hepatic injury by reducing GPX4 activity and increasing 12/15-lipoxygenase expression [259]. Conversely, GPX4 deficiency in hepatocytes causes progressive liver injury that can be rescued by vitamin E supplementation, further linking lipid peroxidation to cell death in MASLD [260]. Early mechanistic studies implicated ceramide generation from palmitoyl-CoA and serine, as well as inducible nitric oxide synthase (iNOS) activity in lipid-induced hepatocyte death [261]. More recently, however, NO has emerged as a natural inhibitor of ferroptosis, reframing its role from purely cytotoxic to potentially cytoprotective [262, 263]. Prior studies support this view, showing that the NO donor S-nitroso-N-acetylcysteine, but not nitrite, attenuates ferroptosis in hepatocytes [264], thereby linking cellular thiol status, NO signaling, and survival responses.
NO has emerged as an additional modulator of ferroptosis in hepatocytes. In steatotic liver and endotoxemia models, iNOS-derived NO protected against hepatocellular injury, with iNOS-deficient mice exhibiting heightened susceptibility [265]. Mechanistically, NO may suppress ferroptosis by blocking 15-lipoxygenase-mediated lipid peroxidation [263], although selective LOX inhibition has shown limited efficacy in hepatocytes [264, 266–268]. NO also promotes hepatic glucose production under stress, suggesting a dual metabolic and survival function [269]. Importantly, steatosis may impair endogenous NO activity, thereby compromising this protective pathway and rendering hepatocytes more susceptible to ferroptotic death.
Recent data suggest that steatosis may sensitize hepatocytes to ferroptosis under electrophilic stress. For example, dimethyl fumarate (DMF), a potent activator of the Nrf2 antioxidant pathway, paradoxically induces ferroptosis in steatotic hepatocytes, possibly reflecting system exhaustion or NF-κB inhibition [270–272]. Electrophile stress not only promotes ferroptosis but also enhances lipid accumulation, establishing a vicious cycle of steatosis, thiol depletion, and further oxidative damage. Nrf2 is a master regulator of antioxidant defense and metabolic adaptation in hepatocytes, modulating phase II enzymes, GSH synthesis, and iron metabolism [273]. While Nrf2 activity generally protects against ferroptosis by upregulating GPXs, its downstream effector heme oxygenase-1 (HO-1) can paradoxically promote ferroptosis through iron release [274–276]. Thus, depending on the cellular redox state and iron balance, Nrf2 signaling may be either protective or deleterious in MAFLD progression.
In summary, lipotoxicity acts as a unifying trigger across diverse cell death programs in the liver. Saturated and oxidized lipids promote apoptosis through mitochondrial injury, necrosis via MPT pore opening, necroptosis through caspase-8 inhibition and RIPK signaling, pyroptosis via inflammasome activation, and ferroptosis by driving lipid peroxidation. Autophagy plays a dual role, buffering lipotoxic stress under physiological conditions but failing to offer protection under overwhelming lipid overload. The interplay of these pathways links lipid metabolism to inflammation, fibrosis, and the progression of fatty liver disease.
Taken together, emerging evidence indicates that simple steatosis is not merely a benign state of triglyceride storage but can act as a critical sensitizer to ferroptosis. Electrophilic stress, thiol depletion, impaired NO signaling, and dysregulated Nrf2/HO-1 pathways may converge to create a permissive environment for lipid peroxidation and RCD. This interplay likely represents a key mechanism driving the progression from simple steatosis to steatohepatitis and advanced liver pathology.
Based on the topics discussed in this expert overview, several potential areas of research and interventions can be considered:
Altering the lipid profile. Considering that some lipid species (UFAs, e.g., oleate) have a beneficial effect on SFA-induced lipotoxicity, shifting the lipid profile in blood from toxic (SFA) to non-toxic (UFA) may be considered. The excessive levels of lipid species in metabolic disorders are partly derived from dietary intake, and dietary intake could be a target for intervention.
Both mitochondrial and ER stress lead to increased ROS generation and oxidative stress. A logical target for intervention would therefore be antioxidants; however, studies have shown that although antioxidants may be effective in in vitro or animal studies, they are hardly effective in clinical studies.
Since ER stress contributes to ROS generation and since the ER is involved in lipid metabolism, agents that relieve ER stress may be considered for therapy. ER stress relievers such as tauroursodeoxycholic acid, 4-phenylbutyric acid, and the recently described natural compound arctigenin (which also acts at the level of lipid droplet formation) could be considered.
Some natural compounds have been shown to display various beneficial effects in the context of steatosis. E.g., the coumarin-like derivative esculetin has been shown to both decrease lipid accumulation and protect against ROS-induced damage in hepatocytes via different mechanisms.
Liver cancer comprises a heterogeneous group of primary hepatic malignancies, among which HCC and cholangiocarcinoma (CCA) represent the predominant forms. Despite clear biological and clinical differences, recent research has unveiled converging molecular mechanisms and immune landscapes that are reshaping disease classification and therapeutic development. This review aims to bridge bench and bedside perspectives, summarizing the most relevant translational and clinical advances in liver cancer. We highlight how mechanistic discoveries—spanning immunobiology, tumor-stroma interactions, ferroptosis, and AI-assisted precision medicine—are driving a paradigm shift toward biology-guided and patient-tailored interventions in both HCC and CCA.
Global projections estimate that liver cancer incidence may rise from approximately 0.87 million in 2022 to over 1.5 million by 2050 if current prevention efforts stagnate [277]. Although the burden from chronic viral hepatitis is gradually decreasing due to vaccination and antiviral therapy, cases linked to MASLD, alcohol consumption, and aging are sharply increasing [12, 278]. The recent redefinition from NAFLD/NASH to MASLD/MASH more accurately reflects the metabolic and inflammatory basis of disease and has reframed HCC risk models, highlighting that a significant fraction of HCC cases can develop even in the absence of cirrhosis—complicating surveillance paradigms [279].
In contrast, the incidence of intrahepatic CCA (iCCA) has shown a consistent increase in Western countries over the past two decades, while perihilar and distal CCA [extrahepatic CCA (eCCA)] rates have remained stable or declined slightly [280]. Recent population-based analyses from England confirm a continuing rise in iCCA incidence and mortality, largely driven by metabolic comorbidities, aging, and improved diagnostic imaging [281]. Major risk factors for CCA include PSC, hepatolithiasis, recurrent cholangitis, biliary parasitic infections (Opisthorchis viverrini, Clonorchis sinensis), and fibrotic or metabolic liver disease. Molecularly, FGFR2 fusions and IDH1/2 mutations characterize iCCA, whereas HER2 amplification/overexpression and KRAS/BRAF alterations predominate in eCCA [282]. The interplay of environmental and genomic factors underscores CCA’s heterogeneity and its growing overlap with the metabolic and viral risk spectrum of HCC.
Collectively, these shifting patterns underscore a global epidemiologic transition in adult liver cancers, where viral hepatitis-driven carcinogenesis is gradually being replaced by metabolic and aging-related mechanisms. This evolution not only alters the demographic and risk landscape but also calls for revised surveillance strategies and biomarker-driven prevention models tailored to non-viral populations.
Hepatoblastoma (HB) is the most common primary liver malignancy of children, with the majority (90%) of cases diagnosed at under 5 years of age. Although HB is a rare cancer, with an incidence of 1.7 cases per million children, cases have risen in the past few decades, possibly as a consequence of improved survival of premature and low-weight births [283]. HB arises from a small number of genetic modifications of which β-catenin activating mutations are found in the majority of cases, and alterations in the 11p15.5 locus occurring in around 85% of tumours. However, HB is a phenotypically heterogeneous cancer with tumours evolving across a continuum of cellular states arising from a combination of clonal evolution and epigenetic plasticity [284].
Updated EASL (2024) and AASLD (2023–2025) guidelines now emphasize risk-stratified surveillance and treatment allocation [285, 286]. The BCLC 2022 strategy continues to dominate staging, integrating refinements in intermediate-stage management and multidisciplinary decision-making. The LI-RADS v2024 update introduced key improvements in ultrasound-based surveillance, CT/MRI response assessment, and standardized interpretation post-locoregional therapies such as yttrium-90 (Y-90) and stereotactic body radiotherapy (SBRT) [287]. Despite strong recommendations, surveillance underuse remains widespread, particularly in MASLD populations where ultrasound sensitivity is poor. This has driven interest in biomarker-augmented surveillance models, such as the GALAD panel and cell-free DNA methylation assays, alongside risk stratification tools like aMAP and PAGE-B, which are being validated for individualized surveillance frequency and modality [288–290].
The EASL-ILCA Clinical Practice Guidelines (2023) for iCCA have introduced molecularly informed recommendations for diagnosis, staging, and therapeutic allocation, emphasizing systematic next-generation sequencing (NGS) for actionable targets [IDH1, FGFR2, BRAF, HER2, microsatellite instability (MSI)-H/TMB-high, NTRK] and multidisciplinary tumor boards [291]. The 2025 EASL guidelines on perihilar and distal CCA refine imaging, surgical margins, and systemic therapy management, distinguishing iCCA from eCCA both biologically and clinically [292]. The ESMO 2023–2024 consensus similarly advocates for upfront molecular profiling to inform first-line and subsequent treatment decisions [282, 293]. Collectively, these updates reflect a shift from purely anatomic to biology-driven stratification, paralleling the precision-medicine paradigm already transforming HCC.
In advanced HCC, atezolizumab-bevacizumab (IMbrave150) and tremelimumab-durvalumab (STRIDE) remain standard first-line regimens [277, 294–296]. The HIMALAYA long-term results confirmed durable survival benefit and manageable safety over more than four years, particularly in patients at risk for bleeding. In contrast, lenvatinib-pembrolizumab (LEAP-002) and cabozantinib-atezolizumab (COSMIC-312) failed to improve overall survival, underscoring the need for biologically informed combinations rather than empirical TKI-IO pairings [297–300]. In 2024, the EMERALD-1 trial established the first positive immunoembolization strategy: Durvalumab plus bevacizumab with TACE significantly improved progression-free survival versus TACE alone, heralding a new era of locoregional-immunotherapy synergy [301]. In the adjuvant space, IMbrave050 demonstrated improved recurrence-free survival with atezolizumab-bevacizumab after curative therapy [302].
For CCA, immunotherapy has become a new standard. The TOPAZ-1 phase III trial showed that durvalumab + gemcitabine/cisplatin improved overall survival compared with chemotherapy alone (median OS 12.8 vs. 11.5 months; HR 0.80), establishing the first IO-chemo combination in biliary tract cancers [303]. Similarly, KEYNOTE-966 confirmed OS benefit with pembrolizumab + gem-cis (HR 0.83; OS 12.7 vs. 10.9 months) [304]. These results cement immune-chemotherapy backbones as the new front-line standard. In MSI-H or TMB-high CCA, anti-PD-(L)1 monotherapy remains recommended per tissue-agnostic approvals [282].
At the locoregional level, selective use of TARE/TACE or HAIC for unresectable iCCA is expanding, with translational studies exploring immune activation and vascular remodeling as combinatorial endpoints, mirroring progress seen in HCC.
Single-cell and spatial multi-omics analyses have redefined the HCC microenvironment, exposing an intricate tumor-immune-stromal ecosystem [305, 306]. Malignant hepatocytes reprogram tumor-associated macrophages (TAMs) into immunosuppressive TREM2-positive phenotypes, which restrict CD8+ T cell infiltration. In preclinical models, TREM2 blockade reactivates antitumor immunity and synergizes with PD-(L)1 inhibition, offering a plausible mechanism behind the clinical heterogeneity in immunotherapy response [307, 308]. Preclinical MASLD-HCC models reveal dysfunctional, metabolically exhausted T cells and myeloid reprogramming that impede immunosurveillance, aligning with clinical observations of attenuated immune checkpoint inhibition (ICI) efficacy in MASLD-related HCC [309, 310]. A deeper understanding of the features of the HCC tumour microenvironment that predict sensitivity to atezolizumab-bevacizumab can enable precision approaches for the selection of ICI. A recent scRNAseq investigation revealed three infiltrating cell types associated with around 40% of responders; these comprised two types of CD8+ T cell (CD8 Temra and CD8 Tex) and pro-inflammatory CXCL10+ macrophages [311]. The other 60% of responders characteristically carried features of genomic instability associated with anti-VEGF response indicative of an “Angiogenesis-driven” subset of patients. Disease progression was mainly associated with low immune infiltrations with enrichment of Notch signalling and progenitor-like states. However, a subset of resistant tumours was identified as immune-enriched but infiltrated by immunosuppressive CD14+ monocytes and TREM-2+ macrophages.
In iCCA, single-cell and spatial transcriptomics have mapped distinct immune niches within a dense desmoplastic stroma populated by cancer-associated fibroblasts (CAFs) driving immunosuppression via TGF-β, CXCL12, and IL-6 signaling [312, 313]. CAF-tumor crosstalk activates PI3K-AKT/Notch pathways, supporting epithelial-mesenchymal plasticity and therapeutic resistance [314]. Murine models highlight the contribution of cancer stem-like cells and myeloid-epithelial interactions to treatment escape and metastasis [315]. Together, these findings underscore the stroma-dominated immune exclusion of CCA and the potential for “stroma-first” strategies that parallel emerging approaches in HCC.
Mechanistic studies since 2022 have identified several tumor-intrinsic vulnerabilities. The ferroptosis axis (GPX4-SLC7A11) represents a key therapeutic target; ferroptosis inducers combined with TKIs or ICIs potentiate tumor death in preclinical models, offering a strategy to overcome sorafenib resistance [316]. Concurrently, Hippo-YAP/TAZ-TEAD signaling and focal adhesion kinase (FAK) pathways are being targeted to modulate tumor stiffness, stemness, and immune exclusion. Inhibition of FAK improves anti-PD-1 efficacy and reshapes the fibrotic stroma in mouse models, laying the groundwork for ‘stroma-first’ clinical approaches [317].
CCA shares similar vulnerabilities. Intrahepatic models demonstrate that ferroptosis resistance—via overexpression of GPX4, SLC7A11, and ACSL3—is associated with poor prognosis and metabolic reprogramming [318]. YAP/TAZ activation sustains proliferation, stemness, and immune evasion in iCCA [319], and its crosstalk with FAK-mechanosignaling contributes to stromal stiffness and tumor invasion [320]. Targeting these pathways may overcome the desmoplastic barrier that limits immune infiltration and drug delivery—suggesting strong parallels between HCC and CCA in mechanobiology-driven immunoresistance.
Together, these data highlight mechanotransduction and ferroptosis resistance as shared therapeutic vulnerabilities across HCC and CCA. Translationally, targeting these pathways could serve as a unifying strategy to overcome immune exclusion and stromal stiffness, paving the way for “stroma-focused” precision therapies in liver cancer.
The only curative treatment for primary liver cancers is surgery, specifically either tumour resection or a liver transplant, the latter arguably being optimal since, in addition to removing detectable tumours, it also removes any unseen micrometastatic lesions and the diseased liver in which the cancer developed. However, up to 70% of cancers treated by surgery recur in both HCC and CAA, including extra-hepatic as well as intra-hepatic growths [321, 322]. This high rate of recurrence is due to a combination of contributory factors including circulating tumour cells, residual intrahepatic tumour cells, or micrometastases, the pro-oncogenic environment of the diseased liver and an immunosuppressed tissue microenvironment, which can persist due to intra-hepatic but also extra-hepatic influences such as the dysregulated gut microbiome [323]. The role of immunity in recurrence is supported by studies that report high circulating platelet-to-lymphocyte ratio (PLR) and/or neutrophil-to-lymphocyte ratio (NLR) with early recurrence in HCC [324]. Elevated PLR is also unfavourable after surgery for CCA [325]. Hence, immunotherapy is being investigated for perioperative approaches with the aim of enhancing anti-tumour immunity for the prevention or delay of tumour recurrence. The IMbrave050 trial examined adjuvant atezolizumab-bevacizumab in the setting of high-risk recurrence HCC patients undergoing tumour resection. While initial results were reported as encouraging, with a 28% reduction of recurrence [302], an updated report of the trial failed to demonstrate a durable benefit [326]. Ongoing trials of adjuvant anti-PD-1 agents (EMERALD-2 and CheckMate-9DX) will help further determine if adjuvant ICI treatment has merit. As CCA is an immunogenic tumour [327], there is good justification for perioperative immune checkpoint therapy in those patients who are eligible for surgery. The ACCORD trial combined post-surgical chemoradiation with camrelizumab (anti-PD-1) and demonstrated OS and RFS efficacy [328].
Neoadjuvant protocols are also being tested in ongoing trials and may have greater efficacy due to the presence of a fully intact tumour antigen-immune axis. The aims of neoadjuvant treatment include pre-operative tumour downsizing, bridging to transplant, and prevention/delay of recurrence. In HCC, MORPHEUS-NEO HCC is evaluating immunotherapy combinations (atezolizumab, bevacizumab, tiragolumab, tobemstomig) as neoadjuvants in resectable HCC [NCT05908786] while PRIME-HCC is investigating the safety and efficacy of nivolumab/ipilimumab combination treatment prior to tumour resection [329]. Similar studies in resectable CCA are determining neoadjuvant treatments with durvalumab plus tremelimumab (anti-CTLA-4) combined with gemcitabine [NCT04989218], tsilezumab (anti-PD-1) plus gemcitabine and cisplatin [NCT06903273], and adebrelimab (anti-PD-L1) plus lenvatanib [NCT06208462].
Genomic and immunologic subclassification is now guiding patient stratification. CTNNB1 (β-catenin)-mutant HCC defines an immune-excluded phenotype associated with poor ICI response; combination strategies (STING agonists, YAP/FAK modulators) are under preclinical and early-phase clinical evaluation [330]. The cGAS-STING pathway, in turn, mediates antigen spreading and myeloid activation when coupled with radiation or oncolytic agents, suggesting translational synergy between RT-STING-IO triads [331].
Molecularly targeted therapy has reshaped iCCA management. Futibatinib (FOENIX-CCA2) achieved durable responses in FGFR2 fusion-positive iCCA, leading to global regulatory approval [332]. Pemigatinib (FIGHT-202) and derazantinib maintain efficacy across updated cohorts [333]. Ivosidenib improved PFS and OS in IDH1-mutant iCCA (ClarIDHy) with sustained long-term benefit [334]. HER2-directed therapies—trastuzumab deruxtecan and zanidatamab—achieve clinically meaningful responses in HER2-amplified CCA [335]. For rare NTRK fusions, larotrectinib and entrectinib remain tissue-agnostic options [336].
Emerging clinical trials are testing IO + targeted therapy combinations (FGFR/IDH inhibitors plus anti-PD-1 or anti-VEGF), while biomarker-guided enrichment strategies are integrating transcriptomic and immune profiling to tailor patient selection. Together, these efforts signify the translational shift toward precision oncology in CCA, comparable to HCC’s biology-informed trial design.
Genomic studies confirm hepatitis B virus (HBV) DNA integration and TERT-promoter mutations as early carcinogenic events and potential biomarkers for minimal residual disease surveillance [337, 338]. Circulating HBV-host junctional DNA and ctDNA methylation assays are now entering prospective trials for post-resection monitoring. Parallel research on the gut microbiome-liver axis shows distinct microbial signatures correlating with ICI efficacy, prompting ongoing efforts to integrate microbiome modulation into immunotherapy regimens [339].
Liquid biopsy approaches in CCA are advancing across multiple analytes: plasma ctDNA, bile cfDNA, and extracellular vesicles enhance diagnostic sensitivity in indeterminate biliary strictures [340, 341]. Panels of exosomal circRNAs show promise for early diagnosis and recurrence monitoring in pilot cohorts [342]. Standardization of preanalytical workflows and prospective multicenter validation remain urgent priorities for clinical translation.
Advanced patient-derived organoids (PDOs), 3D spheroids, and microfluidic liver-on-chip systems now recapitulate tumor heterogeneity, fibrotic microenvironments, and immune cell interactions, enabling more predictive preclinical screening [343, 344]. In vivo, MASLD-faithful murine models are clarifying immune-metabolic crosstalk and testing ferroptosis, stroma-targeted, and myeloid reprogramming interventions. Translationally, GPC3-directed CAR-T and bispecific T-cell engagers, as well as personalized neoantigen DNA vaccines, have shown promising preclinical and early clinical signals, supporting the expansion of immunotherapy beyond checkpoint blockade [345, 346].
iCCA and eCCA organoids, co-culture epithelial-stromal models, and biliary organ-on-chip platforms replicate desmoplasia and plasticity, enabling testing of FGFR/IDH + IO and stroma-targeted combinations with immune and mechanical readouts [312]. Integration of single-cell and transcriptomic data into preclinical pipelines is paving the way for transcriptomic subtype-guided clinical trials.
Artificial intelligence (AI) has rapidly emerged as a transformative tool in HCC research, integrating imaging, genomics, and clinical data to enhance diagnostic and prognostic precision. A recent scientific metric analysis encompassing nearly 4,000 publications reported a 12-fold increase in AI-related liver cancer studies between 2013 and 2022, with predominant applications in imaging-based diagnosis (37%), predictive modeling (19%), and molecular bioinformatics (18%) [347]. Deep learning-based radiomics now enable automated detection and characterization of focal liver lesions across CT, MRI, and ultrasound modalities, achieving diagnostic accuracies exceeding 90% in differentiating malignant from benign lesions [348]. In a large-scale study including over 26,000 ultrasonographic images, convolutional neural networks reached a specificity of 97% for malignancy detection [349]. Beyond diagnostic applications, AI-driven models for survival and recurrence prediction have shown promising performance, with ensemble machine learning algorithms attaining accuracies up to 92% for outcome prediction across different HCC stages [350]. Nonetheless, systematic reviews consistently highlight the limited prospective and multicentric validation of these models, which currently constrains their clinical implementation [351]. As large annotated datasets and federated learning frameworks continue to expand, AI is expected to complement conventional and molecular risk stratification approaches, fostering precision surveillance and personalized therapeutic decision-making in liver cancer.
AI applications are also expanding rapidly in CCA, a heterogeneous and often late-diagnosed malignancy of the biliary tract. Over the past decade, research on AI in CCA has grown significantly, particularly in the fields of diagnostic imaging, preoperative risk assessment, and recurrence prediction [352, 353]. Radiomics and deep learning-based algorithms have demonstrated strong capabilities in distinguishing iCCA from HCC and benign hepatic lesions, with area-under-the-curve values frequently exceeding 0.90 in retrospective studies [354]. AI models trained on ultrasonographic and cross-sectional imaging datasets have also shown high diagnostic accuracy, achieving sensitivities of up to 92% in multicenter analyses [349]. Beyond imaging, integrative AI classifiers that combine transcriptomic signatures with clinical parameters are being developed to predict lymph node metastasis and postoperative recurrence risk; however, most remain at early stages of validation and lack large-scale, multicentric testing. Importantly, clinical translation is beginning to take shape, as illustrated by ongoing trials evaluating AI-guided biopsy approaches in suspected CCA (NCT05374122) [https://clinicaltrials.gov/study/NCT05374122]. Despite these encouraging advances, significant challenges persist—including limited access to standardized, annotated datasets, inter-institutional variability in imaging protocols, and the “black-box” nature of deep learning models. Future research should prioritize multi-omics data integration, development of explainable AI frameworks, and prospective validation studies to advance precision diagnosis and individualized therapy in CCA.
Beyond its diagnostic and prognostic roles, AI holds promise as a translational integrator, capable of merging imaging-derived features with genomic and immune signatures. This convergence could enable real-time, biology-informed clinical decision-making, further bridging the gap between bench discoveries and patient care in liver cancer.
The field of liver cancer is undergoing a convergence of clinical innovation and mechanistic insight. Modern therapy is shifting from empiricism to biology-guided precision—anchored in detailed molecular and immune profiling. The integration of spatial and single-cell data, ferroptosis and stromal mechanobiology, and immune-etiologic diversity is informing both rational combination design and risk-based clinical trial enrichment. Future success will depend on embedding translational endpoints into trials, standardizing MASLD-oriented preclinical models, and bridging molecular discoveries with pragmatic clinical benefit.
CRC is the third most common malignancy and the second leading cause of cancer-related death worldwide. In particular, patients with metastatic stage IV CRC have a very poor prognosis [355]. CRC has served as a paradigm for multistep carcinogenesis with accumulating mutations in APC, KRAS, BRAF, p53, as well as components of the TGF-β and PI3K signaling pathways such as SMAD4 and PIK3CA. Germline mutations in APC are the cause of the familial adenomatous polyposis (FAP) cancer syndrome, an autosomal dominant inherited condition with the formation of numerous colon polyps early in life. In patients with Lynch syndrome (also called hereditary nonpolyposis CRC or HNPCC), which is the most common inherited cancer syndrome in humans, mutations in genes of the DNA mismatch repair system (in particular Mlh1 and Msh2) have been identified [355]. Furthermore, CRC served as a paradigm for the definition of the Immunoscore, a tumor classification system that is based on the composition of immune cells in the stroma and their mode of infiltration [356, 357]. Here, we summarize important current advances and challenges in CRC research and therapy.
Several advances have been made in the molecular characterization of CRC, which will gradually be incorporated into treatment strategies. Starting with the Cancer Genome Atlas (TCGA) initiative, which primarily provided data on bulk RNA expression from tumor samples, methods such as CITE-seq and VisiumHD are now available to analyze protein and RNA expression at the single-cell level and in a spatial context [358]. These techniques are particularly informative for characterization of the tumor microenvironment (TME) because the spatial resolution allows for the assessment of cell-cell interactions in stromal niches [358]. Large-scale data sharing and analytics resulted initially in the identification of the consensus molecular subtype classification of CRC, which assigns a detailed molecular characterization to the originally classified subtypes of cancers with microsatellite instability, the CpG island methylator phenotype, and chromosomal instability [359]. Single-cell sequencing and spatial transcriptomics data are currently being collected, and large datasets from many patients are expected to be available soon. This goes hand in hand with the development of new algorithms for the biological interpretation of the datasets and for the integration of multi-omics data (i.e., the overlaying of multiple omics data onto individual tumor sections). The new methods will enable refinement of CRC classification, identify stromal cell-cell interactions that are indicative of patient prognosis and treatment response as well as uncover treatment resistance mechanisms.
The identification of therapy resistance mechanisms is a main topic in CRC research. The discovery of Yap1/AP-1-driven oncofetal reprogramming in CRC, a process that puts cancer cells back into a fetal-like state, has challenged the view that a specific, canonical Lgr5+ cancer stem cell state is predominant in CRC [360]. Instead, multiple cancer stem cell states might coexist that cooperate in CRC progression and therapy resistance. Although the concept of phenotypic plasticity is well established, it is important to define the specific interplay of plastic stem cell states in tumors to block oncofetal reprogramming for better therapy responses. The acquisition of cancer cell plasticity without additional mutations in cancer genes is also a driver of CRC metastasis. Two studies have identified epigenetic control mechanisms of CRC stem cell plasticity that are associated with metastasis. In one study, the chromatin remodeling factor ATRX was identified as a metastasis suppressor and its loss enhanced lineage plasticity through epigenetic regulation of HNF4 [361]. Another study attributed a similar function to the transcriptional activator/repressor Prox1. Although Prox1 inhibits differentiation and reinforces intestinal stem cell fates in early CRC, metastatic colonization seems to depend on the acquisition of a Prox1-insensitive state [362]. Phenotypic plasticity poses a central challenge in cancer therapy. Therefore, it is important to identify cell states and trajectories that are conserved across multiple patients. Underlying factors such as ATRX or Prox1 could serve as molecular targets to inhibit plasticity [362].
CRC is in close contact with the gut microbiome, which modulates CRC pathogenesis and therapy response. Host genetics is a known factor that influences the composition of the gut microbiome, but the responsible genes are largely unknown. One study investigated the interplay between host genetics, the microbiome, and its influence on CRC progression. It was shown that a single-nucleotide polymorphism in an intron of the Kcnj11 gene, which encodes a potassium channel, is associated with Fusobacterium enrichment and enhances Fusobacterium nucleatum adhesion to CRC cells, thus promoting tumor growth [363]. Fusobacterium nucleatum has long been associated with CRC, but it was not known until recently that only one specific subtype, Fusobacterium nucleatum subspecies animalis clade C2, is the main promoter of CRC [364]. Furthermore, it was demonstrated that the circadian clock impinges on the microbiome to accelerate CRC pathogenesis. Disruption of the clock resulted in dysbiosis with changes in Bacteroides, Helicobacter, and Fusobacterium as well as reduced intestinal barrier function which was associated with CRC progression in mice [365]. The more we learn about the microbiome in CRC, the better we can adapt our fecal microbiota transplantation protocols to address dysbiosis and its negative impact on CRC progression and therapies.
Pioneered by the Clevers laboratory about 15 years ago [366], the cultivation of normal intestinal organoids and PDOs from CRC is now a common cell culture technology. PDOs are grown three-dimensionally and largely retain the genetic and epigenetic characteristics of their primary progenitors [367]. Other cell types such as vascular endothelial cells, fibroblasts, and macrophages can be included in the cultures, thereby reconstructing the TME [368]. Importantly, the PDOs from CRC accurately reflect the responses of cancer cells to drugs or drug combinations from the primary progenitor, enabling precision medicine-based prediction of therapeutic responses even before clinical intervention [369]. Prolonged cultivation of CRC PDOs in the presence of drugs can lead to drug resistance in vitro. Subsequent genomic, transcriptomic, proteomic, and single-cell RNA sequencing analyses of resistant clones have unraveled drug resistance mechanisms such as methylation of CIMP genes that may also operate in patients after long-term therapy [370]. The versatile application of PDOs in research and therapy has enabled the establishment of large organoid biobanks. What used to be freshly frozen cancer tissue is now organoids, which offer the significant advantage that the cancer cells are alive and can be further cultured [371].
AI holds great promise in the field of CRC because of its ability to perform image analysis and bioinformatics of multi-omics datasets to discover new biomarkers and therapeutic targets [372]. AI-driven analysis following machine learning of hematoxylin-eosin (H&E)-stained CRC sections can predict MSI and consensus molecular subtypes (CMS) status and detect histopathological features predictive of treatment responses that may be overlooked by pathologists [373–376]. AI can significantly facilitate the characterization of the immune microenvironment of CRC for patient prognosis and identify stromal immune cell types to predict responses to ICI [377, 378]. AI is now being used to predict patient response to various therapies, including chemotherapy, radiotherapy, targeted therapy, and immunotherapy. By integrating data from image analysis, molecular analysis, and clinical history, AI is able to predict treatment response and side effects and even select the most effective drugs and combination therapies in a personalized manner [372].
Advances in CRC research and therapy also entail new challenges. PDOs, for example, are a valuable tool for pre-screening of treatment responses, but standardized culture protocols are still lacking. They also incompletely model the complex immune microenvironment of CRC. Patient-derived xenografts, preferably in host mice with humanized immune systems, would be a better option, but due to cost and infrastructure constraints, it is unrealistic for them to become a routine clinical tool. Another unsolved challenge is gender-specific medicine for CRC. Gender differences in CRC development, treatment, and treatment-related toxicity have long been recognized. Possible explanations in metabolism, immune responses, hormonal status, and the importance of certain cancer genes on sex chromosomes have been elucidated, but have not yet led to gender-specific, tailored therapies. Furthermore, major challenges remain in the treatment of CRC and there is a worrying increase in CRC incidence rates in people under 50 years of age.
Existing treatment options do not yet fully address our clinical needs. Response rates remain suboptimal and recurrence is common [379]. Since the KEYNOTE-016 trial with pembrolizumab, immunotherapy has become the first-line therapy for patients with microsatellite instability-high/mismatch repair-deficient MSI-H/dMMR CRC who were previously treated with 5-FU, oxaliplatin, and irinotecan. A combination of nivolumab with ipilimumab may be even more effective [380]. Unfortunately, these therapies are not working in patients with MSS CRC. However, data from the phase II NICHE study, which used nivolumab plus ipilimumab as neoadjuvant immunotherapy in patients with early metastatic mismatch repair-proficient CRC, demonstrated a response rate of 26%. Tumors of responders exhibited a high CIN state, a high proliferation index and a higher proportion of Ki67+ CD8+ T cells [381]. BRAF mutations are common in the inflammatory CMS1 subtype of CRC, but targeted monotherapies with BRAF inhibitors have shown limited success. Therefore, BRAF inhibitors such as encorafenib were tested in combination with the EGFR inhibitor cetuximab and the MEK inhibitor binimetinib [382, 383]. The combination of encorafenib with cetuximab has now become the standard of care for patients with BRAFV600E-mutated metastatic CRC (mCRC) after prior systemic therapy. However, no benefit of triple-therapy with binimetinib over doublet-therapy has yet been demonstrated [382]. The combination of BRAF inhibitors with EGFR inhibitors has improved clinical efficacy, but response rates remain low. Therefore, BRAF and MEK inhibitors are being tested in combination therapies with chemotherapeutic agents and with anti-PD-1-based ICI [384]. It remains to be seen whether these approaches will lead to better treatment options for the BRAFV600E CRC subtype. Several experimental inhibitors for targeting the Wnt pathway, the TGF-β pathway or mutant p53 are in the pipeline, but few are expected to reach the clinic. For example, a glutaminase inhibitor was investigated for the treatment of PIK3CA-mutated CRC which is known to be glutamine-dependent, and phase I data showed efficacy [385]. The new generation of RAS inhibitors such as the KRASG12C inhibitor sotorasib or MRTX1133, which targets KRASG12D are promising, but they are quite toxic and resistance is often observed due to secondary KRAS mutations or the activation of bypass signaling pathways. Furthermore, a recent study has shown that inhibition of KRASG12D can induce a transition to a Wnt-Lgr5+ cancer stem cell state that no longer relies on KRAS signaling [386]. Interestingly, considerable progress has been made in the development of Myc inhibitors, a target long considered undruggable. In a recent phase I study in patients suffering from advanced solid tumors, the peptide Myc inhibitor OMO-103 showed promising therapeutic efficacy, which recommends a phase II clinical trial [387].
It is estimated that over 90% of cancer-related deaths are due to multidrug resistance of tumors. A variety of different mechanisms for resistance to chemotherapy, targeted therapy, immunotherapy and radiotherapy have been described [379]. Some of these mechanisms are treatment-specific, while others, such as the induction of autophagy, cancer stem cell plasticity and sustained activation of bypass signaling pathways, are more common [379]. Recent findings emphasize the role of extrachromosomal DNA (ecDNA) [388, 389], the sharp increase in the mutation rate in drug-tolerant persistent cells in CRC due to targeted therapy [390], and the influence of microbiota products such as Fusobacterium nucleatum-derived succinate [391] on CRC resistance. However, questions remain as to how clinicians should address these resistance mechanisms. In preclinical models, ecDNA-containing cancer cells could be targeted, and drug-tolerant persistent cells could be killed by inducing ferroptosis. In Fusobacterium nucleatum, treatment with metronidazole was shown to reduce bacterial counts and improve the CRC response to anti-PD-1 treatment, which is attenuated by succinate. Extrapolating these strategies to human patients remains a major challenge.
There is worrying evidence of a globally increasing incidence rate of CRC in adults under 50 years of age [392]. The pathophysiology of this so-called early-onset CRC (EOCRC) includes MSI features, synchronous metastasis, and a mutational profile that differs from that of CRC in older patients [393]. EOCRC is thought to be mainly caused by lifestyle factors such as the intake of high-fat and highly processed foods, coupled with reduced physical activity and a susceptible genetic background [394]. There is also evidence that the microbiome plays an important role, as a mutational signature of the bacterial toxin colibactin, which is produced by some E. coli strains, has been found in EOCRC. It appears that this signature, which affects the APC gene, was imprinted early in life [395]. The emergence of EOCRC requires further consideration of genetic testing in high-risk patients, adaptation of existing therapies for optimal benefit of patients already suffering from EOCRC, and re-evaluation of the recommended starting age for CRC screening using non-invasive and invasive methods. The measures must be taken in the context of the increased burden on the entire healthcare system, which could be addressed with tailored approaches (e.g., by preferential screening of individuals with a family history of cancer).
In summary, considerable progress has been made recently in CRC research and therapy, but there is still a long way to go before satisfactory treatment responses are achieved. Growing datasets with single-cell sequencing and spatial multi-omics data, as well as the parallel development of AI-based bioinformatics analysis tools will be highly informative. They will identify new therapeutic targets and uncover complex biomarkers for patient prognosis, treatment response, and mechanisms of therapy resistance.
Chronic liver disease affects millions globally, with cirrhosis accounting for 1.5 million deaths annually [396]. Persistent hepatic injury, regardless of etiology, progresses to fibrosis and ultimately cirrhosis, which can lead to portal hypertension, hepatic decompensation, and HCC. Accurate staging of fibrosis is essential for prognosis, monitoring, and treatment [397].
Although liver biopsy is the reference standard, its limitations—procedural risk, cost, sampling error, and interobserver variability—have motivated the development of non-invasive alternatives [398–400]. These include serum biomarkers and imaging-based techniques, both capable of providing reliable fibrosis assessment in routine clinical practice.
Indirect markers: Transaminases, platelet count, and prothrombin time are widely used. Transaminases alone have limited predictive value [401, 402]. An AST/ALT ratio > 1, particularly when combined with thrombocytopenia, increases cirrhosis detection [403–405]. Thrombocytopenia (< 200 g/L) with elevated AST and ferritin > 1,000 µg/L predicts cirrhosis with 82% accuracy in hereditary hemochromatosis [406]. Prothrombin time < 80% has 86% accuracy for cirrhosis, highlighting its utility as a simple, reproducible marker [401]. The advantages and limitations of some of the serum biomarkers used for diagnosis are summarized in Table 1.
Advantages and limitations of serum biomarkers.
| Advantages | Limitations |
|---|---|
| High reproducibility | Limited liver specificity |
| Widely available, inexpensive | Poor discrimination of intermediate fibrosis |
| Outpatient use | Lower accuracy than elastography for cirrhosis |
| Well validated | Proprietary assays are costly; influenced by hemolysis/inflammation |
Direct markers: Ideal biomarkers are liver-specific, minimally influenced by extrahepatic conditions, and sensitive to fibrosis stage [407]. Direct markers include hyaluronic acid (HA) and procollagen III N-terminal peptide (PIIINP). HA thresholds of 100–110 µg/L yield 79–86% sensitivity and 86–89% specificity [401, 408]. PIIINP correlates with fibrogenesis and distinguishes MASH from simple steatosis in NAFLD [409]. Other markers include type VI collagen, undulin, and matrix metalloproteinases. Composite scores:
FIB-4: incorporates age, ALT, AST, and platelet count; AUROC 0.87 for cirrhosis [410, 411].
MAFLD fibrosis score: specific for NAFLD; AUROC 0.84 for advanced fibrosis [412].
Point shear wave elastography (pSWE): pSWE is accurate in chronic HBV and hepatitis C virus (HCV), with an AUROC of 0.88–0.91 for F2–F4 and cirrhosis [417–419]. In NAFLD, AUROC > 0.97 for advanced fibrosis has been reported [420].
Two-dimensional SWE (2D-SWE): AUROC reaches 0.91 for significant fibrosis and 0.95 for cirrhosis in viral hepatitis; thresholds 7.1 kPa (≥ F2) and 10.1 kPa (F4) yield high sensitivity and specificity [421].
Transient elastography (TE): TE is widely validated: AUROC 0.93–0.96 for cirrhosis; cutoffs 11.3–15.6 kPa for F4 [422–424]. XL probes improve applicability in obesity. TE is recommended for fibrosis assessment in NAFLD, with biopsy reserved for uncertain cases [425]. The advantages and limitations of these imaging approaches are summarized in Table 2.
Advantages and limitations of imaging techniques.
| Technique | Advantages | Limitations |
|---|---|---|
| TE | Widely validated; bedside; AUROC > 0.9 for cirrhosis | Dedicated device; less accurate in obesity/ascites; false positives |
| pSWE | Standard US; operator selects ROI; high applicability | Narrow range; quality criteria not fully defined |
| 2D-SWE | Real-time; adjustable ROI; wide stiffness range | Learning curve; inflammation may influence |
| MRE | Whole-liver assessment; high accuracy; obesity/ascites feasible | Expensive; MRI required; iron overload limits |
TE: transient elastography; pSWE: point shear wave elastography; 2D-SWE: two-dimensional shear wave elastography; ROI: region of interest; MRE: magnetic resonance elastography; MRI: magnetic resonance imaging.
MRE provides excellent diagnostic performance, AUROC 0.92–1.0 for cirrhosis [426, 427]. In NAFLD, MRE may differentiate NASH from simple steatosis (AUROC 0.93) [428]. Its success rate (94%) exceeds TE (84%).
In summary, non-invasive assessment of cirrhosis has become standard. Serum biomarkers, composite scores, and elastography each provide valuable information. TE is widely accessible and accurate for advanced fibrosis, whereas MRE offers superior precision but is limited by cost and availability. Combining biochemical and imaging methods optimizes diagnostic performance and minimizes biopsy use. This integrated approach is essential for clinical evaluation and long-term monitoring of chronic liver disease.
Rapid advances in AI, multi-omics, non-invasive diagnostics, and precision therapeutics are reshaping care for liver and digestive diseases. Randomized and real-world evidence show AI-assisted colonoscopy can raise adenoma detection and reduce miss rates, while validated non-invasive tests (NITs)—including elastography and blood-based panels—now underpin risk stratification for steatosis and fibrosis in routine pathways [429, 430]. In parallel, consensus guidance has unified terminology and case-finding strategies for MASLD, aligning hepatology with cardiometabolic care and enabling earlier, guideline-driven intervention [13]. Yet the population burden remains substantial and shifting: Global syntheses document rising morbidity and mortality from metabolic liver disease even as viral hepatitis control improves, underscoring epidemiology’s role in tracking trends, targeting resources, and evaluating public-health impact [431]. Against this backdrop, the ethical agenda is equally urgent: safeguarding privacy and data security in data-intensive research and care, mitigating algorithmic bias, ensuring explainability and accountability for AI systems, and promoting equitable access to high-cost innovations within and across countries. A responsible path forward integrates robust ethical frameworks (Declaration of Helsinki; CIOMS guidelines), contemporary regulatory safeguards (GDPR; HIPAA), and population-health metrics so innovation measurably reduces burden and disparities rather than amplifying them [432–435].
AI systems in endoscopy, imaging, pathology, and risk stratification are increasingly embedded in clinical workflows. Their benefits depend on large, often cross-border datasets; compliance with GDPR/HIPAA therefore requires privacy-by-design governance, clear purpose limitation, and auditable access control. Ethical review should appraise dataset provenance, re-identification risks, model drift monitoring, and subgroup performance reporting to prevent inequities in underrepresented populations [432–435].
Multiple meta-analyses confirm AI-assisted colonoscopy improves adenoma detection; however, a 2025 multicenter observational study suggested a potential “deskilling” effect—endoscopists exposed to routine AI performed worse when subsequently operating without AI. Editorials have urged competency preservation (periodic non-AI sessions, simulation, behavioral training) and post-deployment surveillance as adoption scales [429, 436, 437].
Omics-guided care in HCC, CRC, and MASLD/MASH enhances stratification and targeted therapy, but complicates consent (data reuse/storage, cross-border sharing, familial implications). CIOMS and Helsinki provide guardrails for risk-benefit balance, vulnerable groups, data/samples governance, biobanks, and post-trial access; protocols should use layered, literacy-appropriate consent and clarify policies for incidental/secondary findings [432, 433].
The ethical imperative is to avoid a two-tier system in which advanced diagnostics and therapies cluster in affluent centers. Equity-oriented reimbursement and procurement (tiered pricing, pooled purchasing), tele-expertise, and technology transfer can extend benefits to safety-net settings and low- and middle-income countries (LMICs); equity metrics should be tracked alongside clinical outcomes. A salient, recent example is Egypt’s HCV elimination program [438], which paired nationwide screening with locally manufactured or discounted direct-acting antivirals (DAA) and scaled treatment capacity. Independent evaluations document millions treated at sustainable cost and rapid reductions in HCV prevalence and sequelae—demonstrating that price negotiations, domestic production, and delivery at scale can operationalize affordability and equity.
Developers and institutions should document model intent, training data domains, and failure modes; clinicians should disclose AI involvement and maintain human oversight with clear override pathways. These measures support trust and align with responsible-innovation principles. A recent multicentre observational study in colonoscopy [436] reported a “deskilling” signal among endoscopists routinely using AI, with performance decrement when AI was withdrawn. The finding underscores the need for pre-specified monitoring plans, routine competency maintenance, incident reporting, and subgroup performance audits—practical accountability mechanisms that complement efficacy trials and help prevent unintended harms during post-deployment use.
Contemporary syntheses quantify the shifting aetiologic mix—declining viral hepatitis where vaccination/antivirals scale, and rising metabolic liver disease in parallel with obesity, diabetes, and alcohol—informing prevention, surveillance, and workforce planning [431]. MASLD guidance now recommends pragmatic, stepwise NIT-based case-finding in individuals with T2D/obesity, abnormal liver enzymes, or steatosis on imaging [13].
Translational epidemiology moves beyond accuracy to ask whether innovations improve hard outcomes at acceptable cost, across diverse care levels. For NITs, population-based and health-system studies evaluate referral yield, fibrosis progression prediction, and equity of access; for AI in endoscopy, program metrics (reach, adoption, fidelity, subgroup effects) complement sensitivity/specificity and adenoma detection rate (ADR) [429, 430].
Large cohorts and meta-analyses show that achieving sustained virological response (SVR) after DAA therapy reduces—but does not abolish—HCC risk, especially in advanced fibrosis/cirrhosis, justifying continued surveillance in high-risk subgroups [439–441]. For HBV, accumulating evidence suggests lower HCC risk with tenofovir vs entecavir in some populations, informing first-line antiviral choices and cost-effectiveness models [442–444].
Recurrent findings across digestive diseases—worse outcomes with lower income, education, rural residence, or minoritized status—argue for embedding equity targets into innovation programs (subsidized diagnostics, mobile elastography, community CRC screening with quality assurance). Epidemiologic monitoring should flag “innovation gaps” early (e.g., attenuated benefit of AI in bandwidth-constrained settings) to trigger corrective action [445–447].
Disability-adjusted life years (DALYs), mortality, and preventable fractions support prioritizing high-yield interventions—viral hepatitis elimination, fibrosis case-finding, and quality-assured CRC screening—before niche applications, with periodic reprioritization as epidemiology evolves [13, 431].
Policies should ensure NITs, essential antivirals, and proven screening AI reach safety-net and rural clinics (pooled procurement, tiered pricing, tele-mentoring). Implementation should include equity dashboards (utilization by socioeconomic stratum, geography, race/ethnicity) and remedial actions when gaps emerge [445–447].
Balance early detection against overdiagnosis anxiety and downstream harms; publish transparent thresholds (e.g., fibrosis/steatosis cut-offs), ensure pathways for confirmatory testing, and use shared decision-making aids adapted for health literacy.
Maintain human skills via credentialing, simulation, and deliberate “non-AI” exposure; deploy drift detection, human overrides, and incident reporting; disclose AI use to patients and to institutional review boards during evaluations [436, 437].
Respect local norms and legal frameworks (GDPR/HIPAA); use privacy-preserving technologies where feasible (de-identification with expert determination, data minimization, federated learning). CIOMS/Helsinki emphasizes public accountability, community engagement, and protections for vulnerable groups, which are vital for multi-center registries and biobanks [432–434].
In summary, innovation is accelerating across hepatology and gastroenterology—from AI-assisted detection and image-based elastography to omics-guided therapeutics. Epidemiology quantifies where need is greatest, validates real-world performance, and reveals inequities; ethics provides the guardrails—privacy, consent, fairness, transparency, and just access—so advances reduce rather than entrench disparities. A durable roadmap couples (1) global burden monitoring and equity-aware priority-setting; (2) stepwise, guideline-based deployment of validated NITs and AI with post-deployment safety/competency surveillance; (3) privacy-preserving, GDPR/HIPAA-compliant data governance; and (4) adherence to Helsinki/CIOMS principles throughout research and implementation. Done together, these pillars convert technological promise into measurable gains in survival, quality of life, and health equity for patients with liver and digestive diseases worldwide.
The spectrum of digestive diseases constitutes a complex, heterogeneous mosaic of alterations affecting various organs, such as the liver, pancreas, and digestive tract, which challenge the public health system in Western societies. The prevalence of these diseases is expected to rise in the future due to their association with obesity, the diabetes pandemic, and IR. This is of particular relevance for MASLD and the constellations of alterations affecting extrahepatic organs, including the kidney and heart. Indeed, CVD is the major cause of morbidity associated with MASLD, and the identification of the molecular players linking MASLD to CVD may represent a significant step toward treating cardiovascular events. In addition, the onset of CVD increases the risk to develop chronic liver disease, particularly MASLD, indicating the bidirectional relationship between these organs. This raises the prospect that treating one end of this binomial may result in beneficial effects in the other. Whether this bidirectional linkage is held with other organs remains to be fully established. In the era of high-resolution imaging, omics, and AI-guided discovery, it is anticipated to witness an unprecedented blooming of knowledge that may impact the diagnosis, management, and treatment of highly prevalent liver and digestive diseases. In this regard, the development of microbiome-based BA modulators and AI-driven BA modeling will be expected to evolve into precision hepatology for a more precise and effective treatment of cholestatic liver diseases. Similar expectations concern the identification of specific receptors and pathways involved in ALD/MetALD, lipotoxicity as well as in the regulation of autophagy and DILI management, which will require further and specific genetic models to establish a cause-and-effect relationship. In this regard, although existing evidence suggests a protective role of immune cells, particularly macrophage autophagy, in alcoholic acute-on-chronic liver injury, future studies would require the development of novel murine models with macrophage-specific autophagy deficiencies to more precisely define this mechanistic contribution to alcoholic acute-on-chronic liver injury. The development of next-generation models of liver and gastric cancers such as the PDOs and xenografts emerge as invaluable tools to develop a more effective therapeutic armamentarium and dissect mechanisms involved in resistance. AI-driven analysis following machine learning holds great promise in liver cancer, such as HCC and CCA, as well as in CRC due to its ability to improve image analysis and bioinformatics of multi-omics datasets to discover new biomarkers and therapeutic targets, in addition to its capacity to identify new players involved in the immune microenvironment of HCC, CCA, and CRC. AI-assisted strategies will also impact favorably on the identification of non-invasive assessment of cirrhosis, such as serum biomarkers, composite scores to optimize diagnostic performance, management, and treatment.
ADH: alcohol dehydrogenase
AI: artificial intelligence
AILI: acetaminophen-induced liver injury
ALD: alcohol-related liver disease
APAP: acetaminophen
ATF6: activating transcription factor 6
BAD: bile acid diarrhea
BAs: bile acids
BMDM: bone marrow-derived macrophages
BSHs: bile salt hydrolases
CAFs: cancer-associated fibroblasts
CCA: cholangiocarcinoma
CDCA: chenodeoxycholic acid
CDI: Clostridioides difficile infection
CMA: chaperone-mediated autophagy
CMS: consensus molecular subtypes
CPT-1: carnitine palmitoyltransferase-1
CRC: colorectal cancer
CVD: cardiovascular disease
DAA: direct-acting antivirals
DAMPs: damage-associated molecular patterns
DILI: drug-induced liver injury
eCCA: extrahepatic cholangiocarcinoma
ecDNA: extrachromosomal DNA
EOCRC: early-onset colorectal cancer
ER: endoplasmic reticulum
FAK: focal adhesion kinase
FAO: fatty acid β-oxidation
FFAs: free fatty acids
FGF: fibroblast growth factor
FXR: farnesoid X receptor
GLP-1 RAs: glucagon-like peptide 1 receptor agonists
GPX4: glutathione peroxidase 4
GSH: glutathione
HA: hyaluronic acid
HB: hepatoblastoma
HCC: hepatocellular carcinoma
HO-1: heme oxygenase-1
HSC: hepatic stellate cells
IBAT: ileal bile acid transporter
IBD: inflammatory bowel disease
iCCA: intrahepatic cholangiocarcinoma
ICI: immune checkpoint inhibition
iNOS: inducible nitric oxide synthase
IR: insulin resistance
JNK: c-Jun N-terminal kinase
KCs: Kupffer cells
LAMP-2A: lysosome-associated membrane protein type 2A
LSECs: liver sinusoidal endothelial cells
MACEs: major adverse cardiovascular events
MAFLD: metabolic dysfunction-associated fatty liver disease
MASH: metabolic dysfunction-associated steatohepatitis
MASLD: metabolic dysfunction-associated steatotic liver disease
MetALD: metabolic dysfunction-associated alcohol-related liver disease
MIM: mitochondrial inner membrane
MPT: mitochondrial permeability transition
MRE: magnetic resonance elastography
MSI: microsatellite instability
NAFLD: nonalcoholic fatty liver disease
NHANES: National Health and Nutrition Examination Survey
NITs: non-invasive tests
NO: nitric oxide
Nrf2: nuclear factor erythroid 2-related factor 2
PBC: primary biliary cholangitis
PDOs: patient-derived organoids
PERK: protein kinase-like endoplasmic reticulum eukaryotic initiation factor-2α kinase
PIIINP: procollagen III N-terminal peptide
PLR: platelet-to-lymphocyte ratio
PNPLA3: patatin-like phospholipase domain-containing 3
PPARs: peroxisome proliferator-activated receptors
PSC: primary sclerosing cholangitis
pSWE: point shear wave elastography
PV: perivenous
PXR: pregnane X receptor
RCD: regulated cell death
RCT: randomized placebo-controlled trial
RIPK: receptor-interacting serine/threonine-protein kinase
ROS: reactive oxygen species
S1PR2: sphingosine-1-phosphate receptor 2
SERCA: sarco/endoplasmic reticulum calcium ATPase
SFAs: saturated fatty acids
SGLT2is: sodium-glucose cotransport 2 inhibitors
SREBPs: sterol regulatory element binding proteins
STARD1: steroidogenic acute regulatory protein 1
T2D: type 2 diabetes
TAA: thioacetamide
TCA: taurocholic acid
TCDCA: taurochenodeoxycholic acid
TE: transient elastography
TFAM: mitochondrial transcription factor A
TFEB: transcription factor EB
TGR5: Takeda G-protein receptor 5
THR-β: thyroid hormone receptor beta
TLR: Toll-like receptor
TME: tumor microenvironment
UDCA: ursodeoxycholic acid
UFA: unsaturated fatty acid
UPR: unfolded protein response
VDR: vitamin D receptor
VMP1: vacuole membrane protein 1
We would like to thank Dr. Laura Fábrega for assistance with the preparation of Figure 1 with Biorender.
IB, JFC, JGC, WXD, RE, CGR, HJ, FK, AL, DAM, NMS, CM, HM, CR, PS, OT, and HTZ: Investigation, Writing—original draft, Writing—review & editing. JCFC: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. All authors read and approved the submitted version.
Ina Bergheim, Associate Editor of Exploration of Digestive Diseases; Jean Francois Cadranel, Editorial Board Member and Guest Editor of Exploration of Digestive Diseases; Jian-Guo Chen, Editorial Board member and Guest Editor of Exploration of Digestive Diseases; Wen-Xing Ding, Associate Editor of Exploration of Digestive Diseases; Robert Eferl, Editorial Board member of Exploration of Digestive Diseases; Carmen Garcia-Ruiz, Editorial Board member of Exploration of Digestive Diseases; Hartmut Jaeschke, Editorial Board member of Exploration of Digestive Diseases; Amedeo Lonardo, Associate Editor and Guest Editor of Exploration of Digestive Diseases; Derek A. Mann, Associate Editor of Exploration of Digestive Diseases; Nahum Méndez-Sánchez, Associate Editor and Guest Editor of Exploration of Digestive Diseases; Han Moshage, Associate Editor of Exploration of Digestive Diseases; Chiara Raggi, Editorial Board member and Guest Editor of Exploration of Digestive Diseases; Pavel Strnad, Editorial Board member of Exploration of Digestive Diseases; Oren Tirosh, Associate Editor and Guest Editor of Exploration of Digestive Diseases; Jose C Fernandez-Checa, the Editor-in-Chief of Exploration of Digestive Diseases, had no involvement in the decision-making or the review process of this manuscript. The other authors declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
We acknowledge the support from the [PID2022-142956OB-100, PID2023-148803OB-100, and PID2025-173094OB-I00] from Plan Nacional de I+D funded by the Agencia Estatal de Investigación (AEI) and the Fondo Europeo de Desarrollo Regional (FEDER) and from the CIBEREHD; Plan Nacional sobre Drogas del Ministerio Sanidad, Gobierno de España; 2021 SGR 00491 de la Generalidad de Cataluña; as well as support from the National Cancer Institute [PO1 CA281819-01A1] from the NIH; and funding from the European Horizon’s research and innovation program HORIZON-HLTH-2022-STAYHLTH-02 under agreement [No 101095679] to JCFC.
Funding for this work was provided by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) [IG23117] to Prof. Chiara Raggi. Prof. Raggi is a member of the European Network for the Study of Cholangiocarcinoma (ENSCCA) and participates in the initiative EURO‐CHOLANGIO‐NET and Precision‐BTC‐Network granted by COST ACTION [CA18122, CA22125]. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 8450
Download: 106
Times Cited: 0
