 Original Article
Original Article

Affiliation:
1Department of Bioengineering, Izmir Institute of Technology, 35430 Izmir, Turkey
2Department of Orthopedics, University Medical Center Utrecht, 3584 CX Utrecht, the Netherlands
3Department of Orthopedics and Traumatology, Dokuz Eylul University, 35340 Izmir, Turkey
Email: a.karaozenler@umcutrecht.nl
ORCID: https://orcid.org/0000-0001-8302-913X
Affiliation:
3Department of Orthopedics and Traumatology, Dokuz Eylul University, 35340 Izmir, Turkey
ORCID: https://orcid.org/0000-0001-8169-3539
Affiliation:
4Department of Chemical Engineering, Izmir Institute of Technology, 35340 Izmir, Turkey
Email: fundatihminlioglu@iyte.edu.tr
ORCID: https://orcid.org/0000-0002-3715-8253
Explor BioMat-X. 2025;2:101345 DOI: https://doi.org/10.37349/ebmx.2025.101345
Received: March 31, 2025 Accepted: August 11, 2025 Published: September 11, 2025
Academic Editor: Omid Akhavan, Sharif University of Technology, Iran
The article belongs to the special issue Nature-Based Biomaterials for Biomedical Applications
Aim: The decellularization process aims to remove cellular components from the tissues while preserving the ultrastructural composition of the extracellular matrix (ECM). Decellularization of bone is gaining attention as a biological scaffold due to its unique histoarchitecture, which consists of both organic and inorganic compounds. This study aims to develop a biological bone ECM using a novel decellularization method for bone regeneration.
Methods: Rabbit and rat bone tissues were decellularized using a novel process that combines physical, chemical, and enzymatic methods with 0.1% SDS. Bone tissues were evaluated in terms of histology, biochemistry, and biomechanical tests, both before and after decellularization. Additionally, decellularized bone substitutes were recellularized with preosteoblast cells to assess the cytotoxic effect of the decellularization process.
Results: Our method effectively removes cellular components while preserving both organic and inorganic compounds. We achieved a 95% in DNA content for rabbit bone and 92% for rat bone. The biochemical and biomechanical properties remained unchanged, and mineralization features were preserved after decellularization. The cell culture results revealed that decellularized bone extracellular matrix (dbECM) is biocompatible, bioactive, and provides a suitable environment for cell growth.
Conclusions: This study demonstrates that our novel decellularization method effectively develops biological bone ECM containing both organic and inorganic compounds while utilizing minimal chemical concentration and incubation time. It is foreseen that the resulting decellularized bone could serve as a biological substitute, providing a favorable microenvironment for bone regeneration.
Bone tissue engineering aims to repair defective bone tissues with scaffolds that can mimic the host tissue. Three-dimensional scaffolds should provide a similar morphological structure and porosity, suitable mechanical strength, biodegradable and bioactive, capable of mimicking bone tissue. The scaffold is expected to be non-toxic and biocompatible to ensure safe integration when implanted in the defective tissue [1]. Also, scaffold replaced the defect site is expected to be osteoconductive and osteoinductive [2–5]. In recent years, several synthetic and natural polymers and methods have been evaluated for the development of biomaterials for bone tissue engineering and regenerative medicine approaches. Synthetic polymers [poly(lactic acid), polycaprolactone, poly(glycolic acid), polyurethanes)] show restricted bioactive moieties to improve biocompatibility and regenerative capacity [6, 7]. To overcome this deficiency, functionalization methods have been used and mixed with the natural polymers or bioactive agents to produce composite biomaterials with physically and biologically better than single material-based ones [8–12]. As far as the bone is concerned, mineralization is a major important factor as well as biocompatibility and biodegradability for bone regeneration [13]. Therefore, biomaterials including hydroxyapatite (HAp), tricalcium phosphate, or derivatives are preferred due to improving the mineralization and mechanical properties to mimic the bone tissue. Moreover emerging approaches in tissue engineering, such as the use of extracellular matrices through decellularization, hold great potential for overcoming these challenges. [14–16].
Biological scaffolds developed by the decellularization method are widely studied due to the lack of immunological reactions, their mechanical strength is similar to native tissue, and the microstructure of the tissues is maintained [17]. The main purpose of the decellularization method is to remove the cellular components from the tissue without damaging the extracellular matrix (ECM) and biomechanical stress, as well as decreasing the antigenicity of the tissues. The natural ECM structure obtained by the decellularization method is a promising candidate as a more ideal carrier in terms of containing collagen, glycosaminoglycan (GAG), matrix proteins, and growth factors [16, 18]. Decellularization methods and their successful results were reported for the small intestine [14], the urinary bladder [19], the liver [20], the dermis [21], and esophagus transplants [22]. Tissues treated with different solutions to remove vital immunogenic cells can provide an optimum decellularized biological scaffold material for tissue engineering [23, 24]. The success of the decellularization process is largely related to tissue structures, methods used, chemicals, and their concentrations.
Different methods have been used for decellularization, including physical treatments (repetitive freeze-thaw, high pressure), chemical treatments (ionic, non-ionic solutions, hypotonic, hypertonic buffers), and enzymatic treatments (trypsin, collagenase, protease) [23, 25]. Repetitive freezing/thawing and high-pressure treatments can leave cell debris, and as a result, potential immunogenic reactions may occur. The combination of Triton-X with sodium dodecyl sulfate (SDS) has been reported to be highly effective in removing cellular components from tissues, however, this treatment may also lead to the loss or degradation of extracellular matrix components such as collagen and glycosaminoglycans (GAGs) [26]. SDS and other detergents or solvents like Triton-X or tri-n-butyl phosphate (TnBP) remove all cells and cellular debris [27, 28]. These frequently used chemicals are biocompatible and have been shown to support different levels of cellular growth [29]. When Triton-X, TnBP, and SDS are compared, it is seen that SDS is more effective in removing cell and cellular residues [29, 30]. In addition, it is seen that among the methods used, the best results are obtained by the chemical decellularization method; however, biomechanical properties were observed to be affected due to the deterioration in the ECM structure of the tissue [31]. Changes in biomechanical properties can adversely influence cellular activity and compromise the regeneration capacity of the tissue. The development of a decellularized bone scaffold has been motivated by the need to improve the biocompatibility of allograft bone and the benefit of preserving the natural structure of bone.
Recently, several studies have been reported on bone tissue decellularization. These studies indicate that demineralization and decellularization processes are often applied in combination. The inorganic components that provide mineralization of tissue are damaged while separating the cellular components [32]. On the other hand, high amounts of detergent (0.5%, 1%, 2.5%) and mixing of different detergents were used for decellularization of the bone tissue [32–36]. These processes can cause damage in both organic and inorganic structures of bone. Bracey et al. [5] used chemical decellularization and oxidation methods to obtain osteoinductive bone scaffolds as well as to investigate the osteoinductive potential of decellularized bones in comparison to the demineralized scaffolds. It was reported that decellularized bone extracellular matrix (dbECM) had greater ALP enzyme activity and BMP2 expression, which indicated that decellularized scaffolds possessed osteoinductive potential compared to demineralized scaffolds [5]. High amounts of detergents cause breakdown of the ECM proteins and disrupted tissue morphology. Mattioli-Belmonte et al. [37] used both decellularization and demineralization methods with various chemicals such as sodium hypochlorite (for decellularization), hydrochloric acid, ammonia, and ammonium oxalate (for demineralization and decalcification). It was reported that ALP, Sox-2, osteocalcin, and Octamer-binding transcription factor, which are involved in the mineralization process, were decreased [37]. Besides, demineralization causes the removal of inorganic compounds, which are important for osteoinductivity [3, 5, 38, 39]. After demineralization, insufficient cellular functions can occur during bone regeneration [40]. Mineralization is the distinctive feature of bone tissue; however, this feature has not been considered in many decellularization studies.
The aim of the study is therefore to develop an effective decellularization method that preserves the natural organic and inorganic histoarchitecture of the bone tissue. Our novel decellularization method consists of physical, chemical, and enzymatic steps and enables decellularization of the bone with a minimum detergent concentration with a minimum incubation time. After the decellularization process, dbECM was recellularized to obtain a biological substrate that allows cell adhesion and proliferation. The schematic representation of the presented study is illustrated in Figure 1. For this purpose, rabbit and rat bone tissues were decellularized with 0.1% (w/v) SDS for 12 days and investigated histologically, biochemically, and biomechanically after decellularization. Moreover, cytocompatibility and osteoinductivity of the dbECM were evaluated by using MC3T3-E1 cells.
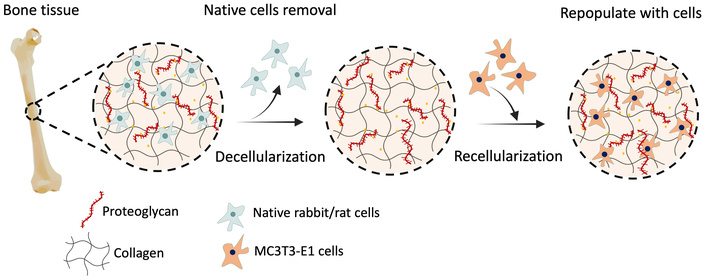
Schematic representation of decellularization and recellularization of the bone ECM.
Rabbit and rat femur bones were used for decellularization. New Zealand White Rabbits (weight, 2.5–3.0 kg; female) and rats (weight, 150–200 g; female) were obtained from Dokuz Eylul University, Department of Laboratory Animal Science (Izmir, Turkey). All animal experiments were performed in accordance with the protocol accepted by the Experimental Animals Ethical Council of Dokuz Eylul University. The rabbits and rats were euthanized using an overdose of pentobarbital (200 mg/kg, IP for rats and 150 mg/kg, IV for rabbits), in accordance with AVMA Guidelines for the Euthanasia of Animals [41]. Rat and rabbit bone tissues were cut into 1 cm3 pieces using an autopsy saw, and one of the adjacent pieces was used as a control, and the other as a sample. Bone tissues were stored in –20°C until further use.
Decellularization was performed with a novel technique involving the combination of physical, chemical, and enzymatic methods with a minimum detergent concentration for 12 days. The novel technique firstly starts with a physical treatment method that was subjected to three freeze-thaw cycles, freezing at –80°C for 3 h and thawing at room temperature for 2 h. Then samples were incubated in a hypotonic buffer at 37°C for 24 h, followed by 0.1% (w/v) SDS in the presence of 0.1% (w/v) EDTA at 45°C for 48 h with agitation as a chemical method. These steps in the chemical method were repeated as three cycles. The samples were washed in 1× PBS, and then incubated twice in a nuclease solution consisting of DNase I in 50 mM tris-HCl and 50 mg/mL BSA buffer for 4 h at 37°C in the enzymatic treatment step. Hypertonic buffer was used as a final incubation step, then samples were washed with 1× PBS for 24 h at room temperature. For the sterilization step, samples were incubated in 0.01% peracetic acid for 3 h. Finally, bone tissues were washed twice in PBS at 37°C and 25°C for 24 h.
Bone tissues were evaluated for histological characterization of the decellularized bone samples. Initially, tissue samples were fixed in 4% (v/v) neutral buffered formalin for 72 h and then dehydrated using the routine process before embedding in paraffin. 5 μm thick sections were prepared by using a microtome (Histocore Multicut, Leica Biosystems), and hematoxylin eosin (HE) staining was used to evaluate the general morphology of the cells and the ECM matrix of bone tissues. Besides, Masson Trichrome (MT) and Alizarin Red S (ARS) staining were used to observe collagen distribution and mineralization sites, respectively. Untreated native bones were used as controls in all stainings.
The cell nuclei of bone samples were assessed with 4',6-diamidino-2-phenylindole (DAPI, Invitrogen, USA) staining by fluorescence microscopy after the decellularization process. Tissues were fixed in 4% (v/v) formalin and then rinsed with 1× PBS. Cell nuclei were stained with DAPI for observation of the cell nuclei and observed by fluorescence microscopy (Carl Zeiss Observer D1). For the SEM imaging, untreated and decellularized bone samples were fixed with paraformaldehyde for 20 min at room temperature. Following washed with 1× PBS, samples were dehydrated in a graded ethanol series (50%, 70%, 80%, 90% and 100%), and then the samples were observed by SEM (Quanta 450 FEG SEM, Thermo Fisher Scientific).
Total DNA content of untreated and decellularized bone tissues was determined after the decellularization process. Firstly, tissues were homogenized with DNeasy 96 Blood & Tissue Kit (Macherey-Nagel, GmbH&Co KG), following the manufacturer’s protocol, and then DNA content of tissues was measured at 260/280 nm in a Nanodrop spectrophotometer (Nano 2000, Thermo Fisher Scientific).
The total collagen content of the tissues was quantified by hydroxyproline assay. Tissues were hydrolyzed by incubation with 6 M hydrochloric acid at 95°C for 20 h. The sample solutions and test standards were prepared according to the Quickzym (Biosciences) kit manual. The 96-well plate was prepared and incubated at 60°C for 60 min, then the absorbance at 570 nm was measured (Varioskan Flash, Thermo Fisher Scientific). The concentration of collagen was calculated by interpolation from the standard curve.
The GAG content was determined using the Sulfate Glycosaminoglycan Quantification Kit (Amsbio, AMS Biotechnology). After enzymatic digestion with papain at 60°C for 48 h, standard or test solutions were prepared according to the manufacturer’s protocol. The absorbance at 530 nm was measured using a microplate reader (Varioskan Flash, Thermo Fisher Scientific). The resultant concentrations of sulfated sugars representative of GAG were determined by interpolation from the standard curve.
Biomechanical properties of the bones were evaluated with a compression test (Shimadzu Autograph AG-I, Shimadzu Co., Kyoto, Japan) according to ASTM-D 5024-95a standard. Native bones and decellularized bones (n = 6) were used, and tests were carried out with a 1 mm/min crosshead speed up to failure. The sample surface area was measured prior to the mechanical test. Young modulus was determined as the slope in the linear region from stress-strain curves, and maximum stress was calculated based on the strain and stress data.
Mouse preosteoblast MC3T3-E1 cells (Subclone 4 cells, ATCC) were used to examine the cytocompatibility of decellularized bone tissues. Mycoplasma test was routinely monitored using MycoAlert mycoplasma detection kit (Lonza), and all cultures used in experiments tested negative for mycoplasma. The cells were sub-cultured in alpha modified minimum essential medium (α-MEM) containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and L-glutamine at 37°C in a humidified 5% CO2 atmosphere. Prior to cell seeding, the decellularized bone tissues were disinfected using 70% ethanol for an overnight addition to peracetic acid treatment in the decellularization step. Following the disinfection, samples were washed with 1× PBS thrice to remove residual ethanol, and then they were immersed in cell culture medium for 3 h at 37°C for conditioning. MC3T3-E1 cells (1 × 105 cells/scaffolds) were seeded on the decellularized bones (1 × 1 × 0.3 cm3) and cultured for 28 days in α-MEM supplemented with 10% FBS, 1% penicillin/streptomycin, and L-glutamine at 37°C in a humidified atmosphere of 5% CO2 in an incubator. The culture medium was refreshed twice a week during the incubation period. Cells seeded in a tissue culture plate (TCP) were used as a control group.
For the in vitro cytotoxicity assessment, an indirect elution method by water-soluble tetrazolium salt (WST-1, Sigma Aldrich) colorimetric assay was used according to ISO-10993 standard. MC3T3-E1 cells (1 × 105 cells/scaffolds) were seeded on the decellularized bones (1 × 1 × 0.3 cm3, n = 6) and incubated for 72 hours at 37°C in a humidified atmosphere of 5% CO2 in an incubator. The absorbance was measured at 450 nm wavelength, and cell viability was calculated by using the following equation:
where Ax is the average absorbance value of treated samples; A0 is the average absorbance value of control.
Cell proliferation on the decellularized bones was assessed with a WST-1 colorimetric assay by conversion of a water-soluble tetrazolium salt through cellular metabolism into an insoluble formazan. MC3T3-E1 cells (1 × 105 cells/scaffolds) were seeded onto disinfected decellularized bones (n = 6) and incubated for 28 days at 37°C in a humidified atmosphere of 5% CO2. At each time point, cells on the decellularized bones were incubated with cell culture medium containing 1% (v/v) WST-1 solution for 4 hours. After incubation, the absorbance at 450 nm was recorded using a plate reader (Varioskan Flash, Thermo Fisher Scientific).
MC3T3-E1 cells, incubated on dbECM substitutes for 14 days, were observed by fluorescence microscopy. After 14 days of incubation, cells were fixed with 4% (v/v) paraformaldehyde in PBS solution for 20 min at room temperature. Samples were washed with 1× PBS, and 0.1% Triton X-100 was used for permeabilization, then stained with DAPI and Alexa Fluor 555 (Invitrogen, Thermo Fisher Scientific) for observation of the cell nuclei and cytoskeleton, and visualized by fluorescence microscopy (Zeiss Scope A1).
Alkaline phosphatase (ALP) activity of cells on the decellularized bone (n = 6) was quantified at 7, 14, 21, and 28 days. Cells on the decellularized bones were incubated with osteogenic medium (α-MEM containing 10% FBS, 1% penicillin/streptomycin, L-glutamine, 1 μL/mL L-ascorbic acid, 10 μL/mL β-glycerophosphate) for 28 days. Released ALP into the culture medium was measured spectrophotometrically by a plate reader at 405 nm according to the manufacturer’s protocols (Biovision, Inc., USA).
MC3T3-E1 cells were cultured on the decellularized rat and rabbit bone (n = 6) for 28 days of incubation time in osteogenic medium (α-MEM containing 10% FBS, 1% penicillin/streptomycin, L-glutamine, 1 μL/mL L-ascorbic acid, 10 μL/mL β-glycerophosphate). Osteocalcin secretion of cells was determined using the Osteocalcin ELISA Kit (Biovision). Culture media were collected and analyzed for the determination of the osteocalcin concentrations in accordance with the manufacturer’s protocol.
All experiments were repeated thrice, and samples were evaluated in triplicate. The experimental data were represented as mean ± standard deviation (SD). The differences between groups in biochemical, biomechanical, and in vitro biocompatibility tests were analyzed using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. All p-values less than 0.05 were considered to be significant (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
After the decellularization process, bone tissues were stained with HE for general structural evaluation, MT for collagen distribution, and ARS for observation of mineralization sites. HE staining showed that round-shaped osteoblasts were observed in untreated native rat and rabbit bone tissue before decellularization, and cells localized in osteon structure were successfully removed from both rat and rabbit bone tissue after decellularization (Figure 2A, 2B). Figure 2 indicates that cell nuclei in untreated bone tissue before decellularization and cell lacunae were observed to be empty after decellularization. Also, magnified figures (on the right) display that circular osteon structures, which are important for cell localization and provide mechanical support to the bone tissue, were not affected by the decellularization process.
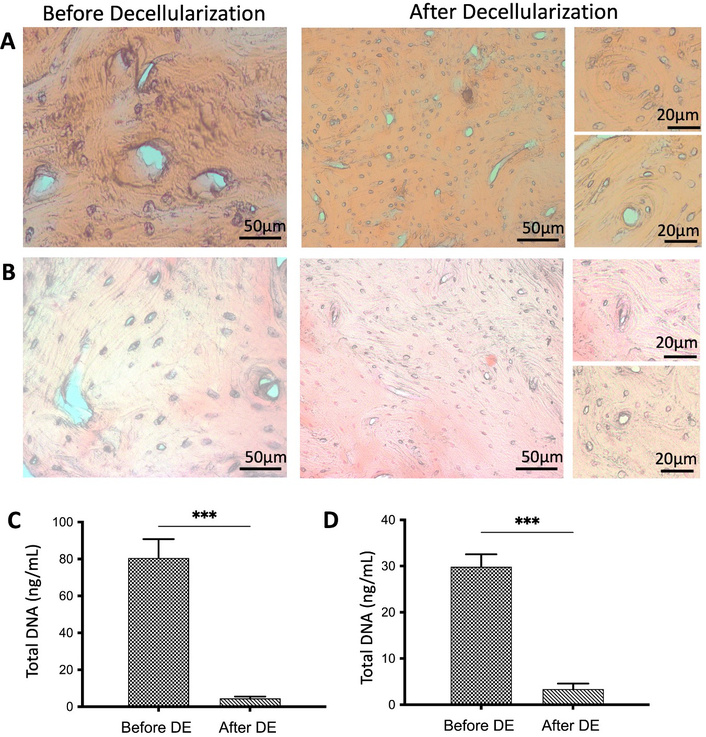
General morphology and DNA determination of the bone tissues. HE staining of rabbit (A) and rat (B) bone tissues before and after decellularization. DNA quantification results of rabbit (C) and rat (D) bone tissues (n = 6) before and after decellularization. Data are presented as mean ± SD (*** p < 0.001). DE: decellularization.
DNA content of both rabbit and rat bone tissues was evaluated spectrophotometrically. Total DNA content of rabbit bones was measured as 81.13 ± 12 ng/mL and 8.02 ± 0.4 ng/mL before and after decellularization, respectively (Figure 2C, Table S1). In rat bones, the total amount of DNA was measured as 29.92 ± 8 ng/mL and 4.93 ± 2 ng/mL before and after decellularization, respectively, and statistically significant differences were detected (*** p < 0.001) (Figure 2D, Table S1). After decellularization, 95.88% ± 0.16% for rabbit bone and 92.07% ± 0.45% for rat bone reduction in DNA content were achieved.
MT staining revealed that collagen structures were maintained in both decellularized rabbit and rat bone tissues and were observed in a radial direction around the empty lacuna after decellularization (Figure 3A, 3B). The total collagen content of the bone tissues was determined using a spectrophotometric method based on the amount of hydroxyproline. As shown in Figure 3C, the total collagen amount in rabbit bones decreased slightly from 55.53 ± 2.15 mg/mL to 48.72 ± 1.06 mg/mL after decellularization. In rat bones, it was measured as 37.8 ± 2.68 mg/mL before decellularization and 34.43 ± 4.45 mg/mL after decellularization (Figure 3D). As expected, there was no statistically significant difference observed in either bone sample. GAG amounts in the tissues were evaluated spectrophotometrically. As shown in Figure 3E and 3F, the GAG amounts in rabbit and rat bones were slightly reduced after decellularization, with no statistically significant difference found. According to the biochemical test results, it can be concluded that the DNA content in bone tissues was significantly reduced. However, the total amounts of collagen and GAG in the bone tissues were preserved.
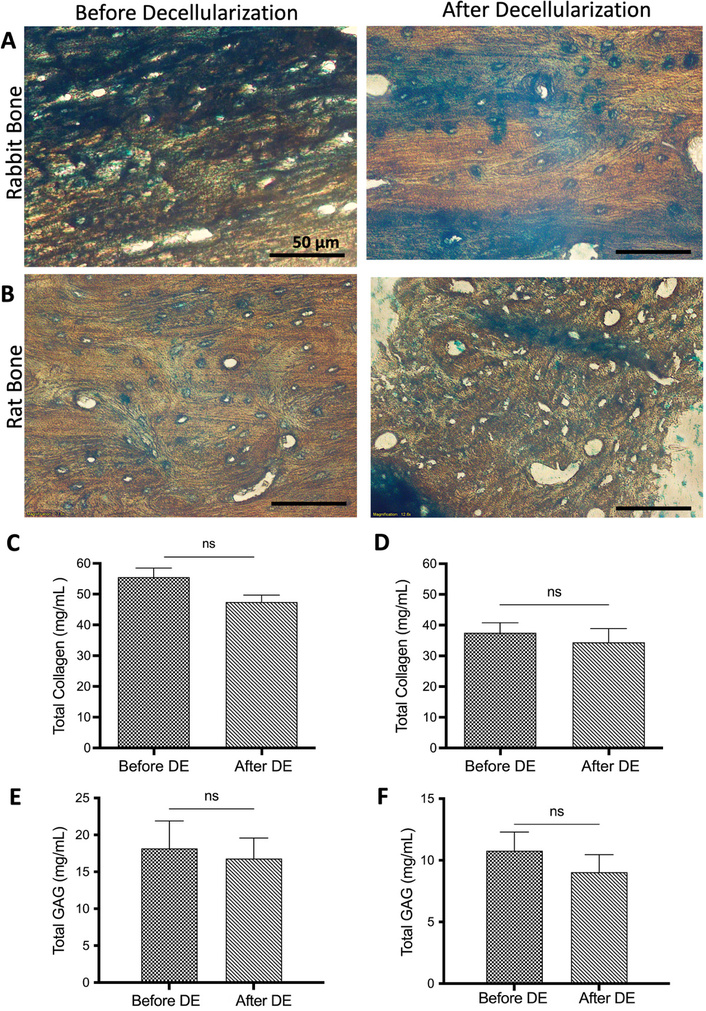
Total collagen and GAG determination of the bone tissues. MT staining of rabbit (A) and rat (B) bone tissues before and after decellularization. Scale bar: 50 µm. Total collagen quantification results of rabbit (C) and rat (D) bone tissues (n = 6). Total GAG amount of rabbit (E) and rat (F) bone tissues (n = 6) before and after decellularization. Data are presented as mean ± SD. DE: decellularization.
Mineralization sites of the bone tissues were observed as red/brown color in ARS staining (Figure 4A, 4B). In rabbit bone tissue, mineralization sites surrounding the osteon structures were clearly observed before and after decellularization, revealing no changes in the structures throughout the process (Figure 4A). In contrast, rat bone tissue displayed partially irregular structures after decellularization; however, the mineralization sites, indicated by a brown color, remained unchanged (Figure 4B). The tissue volume and density of rabbit and rat bones show differences as reported previously [42]. Also, due to the smaller size of rat bones, it might be possible to observe damage than in rabbit bone. This might be the reason that we found partial damage in the rat tissue. Moreover, the FTIR spectrum showed the inorganic and organic compounds of both rabbit and rat bones preserved after decellularization (Figure S1).
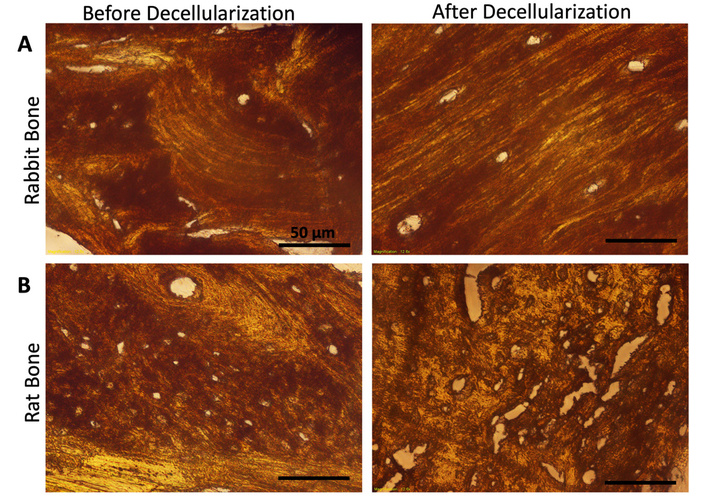
Evaluation of the mineralization site of the bone tissues. ARS staining of rabbit (A) and rat (B) bone tissues before and after decellularization. Scale bar: 50 µm.
DAPI staining was used to observe the cells in the bone tissues for fluorescence imaging. Cell distribution was observed homogenously on the untreated rabbit and rat bone tissues as control groups (Figure 5A). Especially, cells were localized around round-shaped osteon structures as seen in bright blue color. After the decellularization process, no cell nuclei in the decellularized bone tissues were found as expected. Similar to histological results, fluorescence imaging indicated the removal of the cellular components from the bone tissues successfully with the decellularization process.
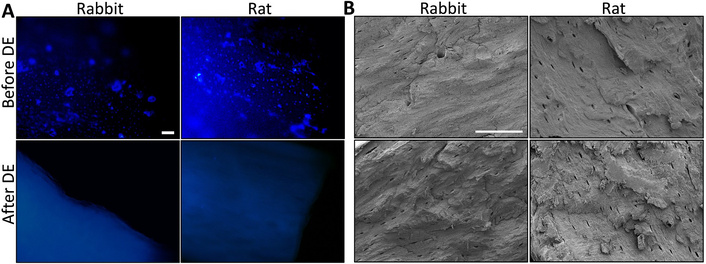
General morphology and nuclei evaluation of the bone tissues. Fluorescence (A) and SEM (B) imaging of the rabbit and rat bone tissue before and after decellularization. Scale bars: 100 µm (A), 100 µm (B). DE: decellularization.
SEM analysis revealed the surface morphology of rabbit and rat bones, both before and after decellularization. Figure 5B illustrates the overall bone structure, including the Haversian canal centrally located within the osteons and the canaliculi present in both rabbit and rat samples, suggesting that the micro-histoarchitecture of the bone tissue remained intact following decellularization.
Untreated and decellularized bone tissues were tested under compression to determine their compressive properties. The maximum compression force that could be applied to untreated bone tissue was determined as 162.02 ± 11.81 N for rabbit bone and 49.78 ± 8.72 N for rat bone before decellularization. These values slightly decreased to 153.83 ± 10.57 N and 42.72 ± 9.53 N after decellularization (Figure 6). No statistically significant differences were found before and after decellularization. The compression strength of untreated rabbit and rat bone was determined to be 17.95 ± 2.05 MPa and 11.69 ± 1.21 MPa, respectively. After decellularization, the compression strength of the bone tissues did not change significantly, and there were no statistical differences before and after decellularization (Figure 6). Additionally, the Young’s modulus of the bones was determined before and after decellularization. The Young’s modulus of both bone tissues did not change much after decellularization. Furthermore, no statistically significant difference was found for Young’s modulus, confirming that the decellularization did not affect the biomechanical properties of the bones (Figure 6).
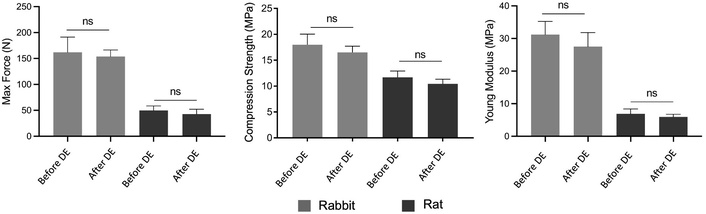
Biomechanical test results of rabbit and rat bone tissues before and after decellularization. The tests were performed using six replicates (n = 6) and data represented as mean ± SD. DE: decellularization.
To evaluate the cytocompatibility of the dbECM, MC3T3-E1 cells were incubated for a 28-day culture period. Figure 7 depicts the fluorescence images of the cells covered on the decellularized substitutes on day 14 of the culture; the cell nuclei and cytoskeleton are shown in blue and green, respectively. The cells were spread and distributed homogeneously on the surface of the dbECM substitutes. The finding of the indirect cytotoxicity test showed that cells maintained their viability, and the decellularization process and dbECM substitutes had no influence on growing MC3T3-E1 cells. There were statistically significant differences found at 24, 48, and 72 h for both rat and rabbit dbECM; however, there was no statistically significant difference detected between rat and rabbit dbECM after 72 h incubation. The proliferation assay results showed that cells proliferated with an increasing trend for both rat and rabbit dbECM. It was determined that the proliferation capacity of the cells on dbECM substitutes was higher in comparison to the TCP control, also cell proliferation for rabbit dbECM was higher than rat dbECM. The significant differences were found on days 21 and 28 of culture between TCP and dbECM groups. Thus, it is estimated that the dbECM substrates would increase the cellular activity in a natural microenvironment as a natural substrate. The viability and proliferation results indicate that the dbECM substrates could be accepted as biocompatible and have the potential for cell growth. Although MC3T3-E1 cells are of murine origin, they exhibited high proliferation capacity on both decellularized rabbit bone extracellular matrix (dRbECM) and decellularized rat bone extracellular matrix (dRaECM), indicating that the decellularization process was effective in minimizing immunological responses, which was one of the main aspects of the decellularization of the tissues. Also, cells showed better growth on dRbECM when compared to the dRaECM substrates. The implications of this result in selecting optimal substrates for bone tissue engineering, suggesting that rabbit dbECM may provide a more favorable microenvironment for osteoblast proliferation and thus could be considered a more suitable substrate. Furthermore, ALP activity was also assessed on dbECM substitutes. ALP release by the cells was stable on days 7 and 14 for each group; however, it was increased on day 21 of culture, and statistically significant differences were found between TCP and both dRbECM and dRaECM on the 21st-day incubation. Also, both rat and rabbit dbECM reached the maximum ALP release on the 28th day of culture, and there were statistical differences compared to the TCP control group. On the other hand, secretion of osteocalcin was determined, and since the MC3T3-E1 pre-osteoblast cells had differentiation capacity, the higher osteocalcin secretion was detected in both rat and rabbit recellularized bones on day 28 of the culture period (Figure 7F). Cell differentiation studies revealed that both dRaECM and dRbECM induce higher secretion of ALP and osteocalcin by MC3T3-E1 cells compared to TCP control (Figure 7E, 7F). The results of these two markers confirmed the osteogenic effectiveness of decellularized bone substitutes due to HAp content.
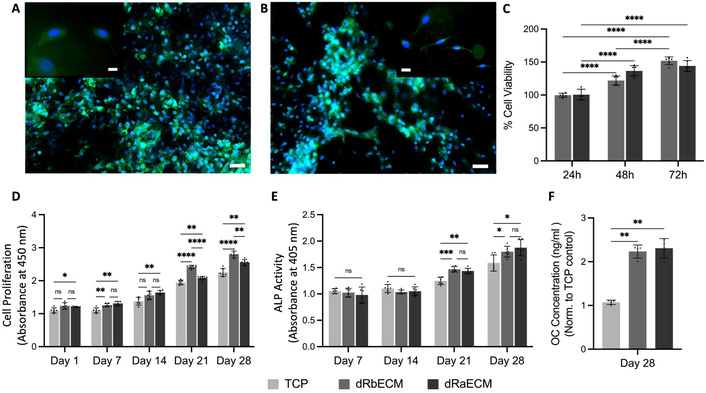
Recellularization study of decellularized rabbit dbECM (dRbECM) and rat dbECM (dRaECM). Fluorescence microscopy images of the cell-seeded dRbECM (A) and dRaECM (B) on 14 days of the culture period. Scale bars: 50 µm, 20 µm (inlets). Cytotoxicity of the decellularized bones according to cell viability (%) of the elution test (C). Proliferation (D), ALP activity (E), and osteocalcin (OC) secretion (F) of the cells on dbECM during 28 days of incubation. All assays were performed using six replicates (n = 6), and data are shown as mean ± SD (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001).
The decellularized scaffold utilizes the natural three-dimensional architecture of tissues to create scaffolding materials suitable for tissue engineering and regenerative medicine. Physical decellularization methods, including repeated freeze-thaw cycles and high-pressure treatments, often fail to completely eliminate cellular debris, potentially triggering immunogenic reactions. To address these limitations, various techniques have been developed [43–45]. Among them, a combination of Triton X-100 and SDS has proven most effective in cellular component removal; however, its impact on the ECM components has been noted [26]. Additionally, SDS, Triton X-100, and TnBP are commonly used to remove cells and cellular debris from bone and other tissues [46, 47]. SDS, in particular, demonstrates superior efficiency in cellular component removal compared to Triton X-100 and TnBP [48]. Despite its effectiveness, chemical decellularization, though yielding the best results among available methods, has been observed to alter the biomechanical properties of tissues due to ECM degradation [49, 50]. Such biomechanical changes can negatively impact cellular activity and the regenerative capacity of the tissue. The development of scaffolds using decellularized bone has been motivated by the need to enhance the biocompatibility of allograft bone while preserving its native structure.
Bone tissue has a unique composition, including osteon structures, Haversian and Volkmann’s canals, and specialized cell types. The osteon, the fundamental unit of cortical bone, consists of concentric lamellae composed of osteoblasts and osteocytes surrounding a central Haversian canal containing blood vessels [51]. Due to this compact, layered structure, removing cellular components from bone proves more challenging than from other tissues. A combination of treatments can enhance decellularization efficiency while maintaining the natural ECM. In this study, a novel decellularization method incorporating physical, chemical, and enzymatic treatments successfully cleared cellular components from rat and rabbit bone tissue. Histological HE staining confirmed cellular component removal while preserving the circular osteon structures critical for cell localization (Figure 2). Furthermore, DNA assays validated the effective removal of DNA from both rabbit and rat bone tissue (Figure 2C, 2D). After decellularization, DNA content was measured at 8.02 ± 0.4 ng/mL and 4.93 ± 2 ng/mL, respectively. These values are significantly below the recommended upper limit of 50 ng DNA for complete decellularization [18]. Laker et al. [47] found that using combined detergents enhances decellularization effectiveness compared to SDS alone, as excessive SDS exposure can damage ECM integrity. However, in this study, an optimized SDS concentration of 0.1% enabled effective decellularization while minimizing ECM degradation. This approach achieved a 95% and 92% reduction in DNA content for rabbit and rat bone, respectively, aligning with DNA removal efficiencies reported by Yates et al. [52] and Ibrahim et al. [53]. Unlike previous methods relying solely on washing steps that degrade proteins and disrupt ECM structure, the present decellularization process integrates physical, chemical, and enzymatic treatments, achieving high DNA removal without significant ECM protein loss.
Preservation of ECM proteins is another critical factor in evaluating decellularization effectiveness. Collagen and GAG content remained largely intact following the decellularization process, as confirmed by histological and biochemical analyses. Collagen, the primary ECM structural component, provides tensile strength, supports cell adhesion, and facilitates cell proliferation [54, 55]. Studies indicate that SDS and Triton X-100 have milder effects on ECM integrity than trypsin, which leads to substantial ECM component loss [56, 57]. MT staining confirmed the retention of collagen structures after decellularization (Figure 3), while SEM further demonstrated the preservation of collagen fibers, indicating that bone tissue microarchitecture remained unaltered (Figure 5). Additionally, biochemical assays verified that total collagen and GAG content were preserved after decellularization (Figure 3C–3F). Literature reports highlight varying ECM molecule preservation across different decellularization methods, with SDS and Triton X-100 treatments often resulting in GAG reduction [28, 58, 59]. Xu et al. [60] demonstrated that among different detergents tested on annulus fibrosus tissue, trypsin caused the greatest GAG loss, followed by SDS and Triton X-100, with the latter achieving successful decellularization while maintaining macroscopic structure. The present method effectively preserved collagen and GAG content, representing a key achievement in maintaining bone ECM integrity.
Bone mineralization, a defining feature of bone tissue, has often been overlooked in decellularization studies [32, 34]. On the other hand, there are several studies that combine decellularization and demineralization processes in the literature [3, 38, 39, 61]. However, removing inorganic components, which are essential for osteoinductivity, can hinder cellular function in bone regeneration. This study evaluated bone mineralization sites post-decellularization to assess osteogenic activity and osteoinductivity. Histological staining confirmed that mineralization sites remained intact in both rat and rabbit bone samples (Figure 4), further supporting the bioactivity retention of the decellularized bone matrix.
Biomechanical properties are crucial for structural stability and scaffold functionality. Decellularized scaffolds provide a complex biochemical and mechanical environment that guides cell adhesion, proliferation, and differentiation. Bracey et al. [4] achieved 98% DNA content reduction in decellularized bone scaffolds using 0.05% trypsin and 2% Triton X-100; however, mechanical properties declined, resulting in reduced Young’s modulus and stiffness [4]. In contrast, the present method maintained both maximum force and Young’s modulus of rat and rabbit bones post-decellularization, with no statistically significant differences compared to untreated samples after 0.1% SDS treatment. These findings confirm that the decellularization process preserved bone histoarchitecture and mechanical strength (Figures 5 and 6).
Recellularization approach of the decellularized tissue is a critical step for the clinical usage of the decellularized ECM. Biocompatibility of the decellularized scaffolds, such as adipose tissue, skin, cartilage, meniscus, myocardium, pancreas, etc. has been investigated for the tissue regeneration capacity [30, 62–67]. As is known, the bioactive components induce the cellular activity, the preserved ECM components (i.e., collagen, proteoglycans) of the bone after the decellularization process have been shown to allow proliferation and differentiation of the osteoblasts [68]. In our recellularization study, fluorescence images confirmed that dbECM could support cell attachment as the cells attached, elongated, and spread on dRaECM and dRbECM surfaces (Figure 7A, 7B). Also, preosteoblasts proliferated on the dbECM and did not show any cytotoxicity (Figure 7C, 7D). Furthermore, ALP activity, known as an important osteogenic differentiation marker at early stages of bone regeneration, was also assessed on dbECM substitutes [69, 70]. The main structure of bone preserved after decellularization induced osteoblast differentiation by promoting cell attachment and proliferation as well as ALP secretion (Figure 7E). On the other hand, secretion of osteocalcin, which is the osteogenic marker for bone tissue, was determined [71, 72]. Since the MC3T3-E1 pre-osteoblast cells have differentiation capacity, the higher OC secretion was detected in both rat and rabbit recellularized bones on day 28 of the culture period (Figure 7F). Besides the cell differentiation capacity, it was estimated that HAp content of the decellularized bones also induced the osteocalcin secretion and osteogenic differentiation. The increasing ALP activity and the osteocalcin secretion of MC3T3-E1 pre-osteoblasts on dbECM substrates may be attributed to collagen fibers and HAp content, as well as residual growth factors preserved after the decellularization process. Previously reported studies showed that dbECM has the ability to retain biochemical signaling molecules such as growth factors, cytokines, and also induces cell differentiation [73, 74]. Cell differentiation studies revealed that both dRaECM and dRbECM induce higher secretion of ALP and osteocalcin by MC3T3-E1 cells compared to TCP control (Figure 7E, 7F). Although ALP is typically considered an early marker of osteogenic differentiation, we observed a high level of ALP activity on day 28, which aligns with elevated osteocalcin levels. This result may be attributed to the osteoinductive nature of the decellularized bone ECM, which can provide sustained biochemical cues that prolong or modulate the temporal pattern of osteogenic marker expression. Previous studies have reported similar trends in ECM-based scaffolds, where ALP activity remained high or peaked at later stages of differentiation [75–77]. These findings suggest that the bone-derived ECM not only supports early osteogenic commitment but may also enhance later stages of maturation and mineralization. The results of these two markers confirmed the osteogenic effectiveness of decellularized bone substitutes due to HAp content. Mattioli-Belmonte et al. [37] reported successful cellular removal from the tissue with decellularization and demineralization methods. It was determined a significant increase in collagen and osteonectin, as well as a significant decrease in ALP and osteocalcin after 14 days of cell culture [37]. Unlike our method, the decellularization process, including demineralization or decalcification of the tissues, might affect the recellularization process in accordance with changing the biomineralization feature of the tissue; thus, this could lead to a decrease in the secretion of osteogenic markers.
Considering all these results, the presented study demonstrated that cellular components were removed from the bone tissue, and ECM proteins and inorganic components were preserved without any significant change in the mechanical properties. Therefore, the method developed in the present study is favorable to the development of biological bone scaffolds, may be a promising candidate for use in bone regeneration. Decellularized bone scaffolds possess biochemical and mechanical properties and have the potential for clinical use in the future.
Moreover, the biological activity of dbECM is a promising biomaterial for bone tissue engineering due to its native bioactivity, osteoinductivity, and structural support for cell attachment and differentiation. It can be used as a substrate for bone regeneration, a reinforcement in bioinks for 3D bioprinting, and as a physiologically relevant substrate for in vitro models. Future developments aim to standardize production, enhance mechanical and biological performance through hybrid materials, and enable personalized therapies. Ongoing research also focuses on optimizing immune compatibility and translating these materials into clinical applications.
Decellularization is a highly effective method for the development of natural biological ECM for tissue regeneration. Specifically, decellularized bone tissue serves as an ideal scaffold for bone tissue regeneration due to its excellent structural and mechanical properties and lack of immunogenicity. We developed a novel and efficient decellularization process to generate biological bone ECM for bone regeneration. Our method preserves both the organic and inorganic components of bone tissue. To the best of our knowledge, this is the first study to assess both the inorganic and organic content of decellularized bone, demonstrating the ability to obtain biocompatible bone scaffolds with minimal chemical exposure and short incubation times compared to existing methods. Our results confirm the successful removal of cellular components while maintaining key ECM elements, including collagen, GAG, and mineralization sites. Notably, the mechanical strength of the bone remained unchanged after decellularization. Biocompatibility assays further revealed that cells proliferated on the decellularized bone and differentiated into osteoblasts over a 28-day culture period, indicating that the scaffold supports osteogenic differentiation in vitro. Decellularized ECM scaffolds derived from rabbit and rat bones demonstrated strong potential for bone regeneration due to the inductive effects of ECM-derived molecules on cellular adhesion, proliferation, and differentiation. Additionally, our findings highlight the importance of preserving bone mineralization as a key factor in osteogenic activity. Overall, our method offers a promising approach for developing biological bone substitutes, which may serve as effective bone graft alternatives for clinical applications in the future.
ARS: Alizarin Red S
DAPI: 4',6-diamidino-2-phenylindole
dbECM: decellularized bone extracellular matrix
dRaECM: decellularized rat bone extracellular matrix
dRbECM: decellularized rabbit bone extracellular matrix
ECM: extracellular matrix
FBS: fetal bovine serum
GAG: glycosaminoglycan
HAp: hydroxyapatite
HE: hematoxylin eosin
MT: Masson Trichrome
OC: osteocalcin
TCP: tissue culture plate
TnBP: tri-n-butyl phosphate
α-MEM: alpha modified minimum essential medium
The supplementary figure and table for this article are available at: https://www.explorationpub.com/uploads/Article/file/101345_sup_1.pdf.
The authors are grateful to the Izmir Institute of Technology Biotechnology and Bioengineering Research and Application Center (IZTECH BIOMER) and Center for Materials Research (IZTECH MAM) for fluorescence analysis, microtome sectioning, histological imaging, and SEM imaging.
AKÖ: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. HH: Conceptualization, Validation, Writing—review & editing. FT: Conceptualization, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
The study was approved by the Animal Experiments Ethical Committee of the Dokuz Eylul University School of Medicine, Izmir, Turkey (Protocol No: 44/2019). The study complies with the Guide for the Care and Use of Laboratory Animals.
Not applicable.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
This research is supported by the Izmir Institute of Technology Scientific Research Project [2020-İYTE-0018]. Kara Özenler A. would like to thank Council of Higher Education (CoHE) (YÖK) for the scholarship YÖK 100/2000 PhD Biomaterials and Tissue Engineering. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Damla Arslantunali Sahin ... Vasif Hasirci
Momoko Ebisawa ... Yutaka Inoue
Athira S. Dev ... Renu Mohan
Viktor Borysiuk ... Serhii Nedilko
Andrew Linz ... Yannick J Bomble
