 Review
Review

Affiliation:
1Department of Psychology and Neuroscience, Duke University, Durham, NC 27708, USA
2Department of Cell Biology, Duke University School of Medicine, Durham, NC 27710, USA
Email: Moawiah.naffaa@duke.edu
ORCID: https://orcid.org/0000-0003-0451-5901
Affiliation:
3Department of Internal Medicine, Ascension Saint Francis Hospital, Evanston, IL 60202, USA
ORCID: https://orcid.org/0000-0001-6438-7855
Affiliation:
3Department of Internal Medicine, Ascension Saint Francis Hospital, Evanston, IL 60202, USA
ORCID: https://orcid.org/0000-0002-7744-5089
Affiliation:
4Department of Internal Medicine, University of Kansas Medical Center, Kansas City, KS 66103, USA
ORCID: https://orcid.org/0009-0003-0221-589X
Explor Target Antitumor Ther. 2025;6:1002313 DOI: https://doi.org/10.37349/etat.2025.1002313
Received: January 29, 2025 Accepted: April 07, 2025 Published: April 27, 2025
Academic Editor: Weilin Jin, The First Clinical Medical College of Lanzhou University, China
Neoantigen-based immunotherapy has emerged as a transformative approach in cancer treatment, offering precision medicine strategies that target tumor-specific antigens derived from genetic, transcriptomic, and proteomic alterations unique to cancer cells. These neoantigens serve as highly specific targets for personalized therapies, promising more effective and tailored treatments. The aim of this article is to explore the advances in neoantigen-based therapies, highlighting successful treatments such as vaccines, tumor-infiltrating lymphocyte (TIL) therapy, T-cell receptor-engineered T cells therapy (TCR-T), and chimeric antigen receptor T cells therapy (CAR-T), particularly in cancer types like glioblastoma (GBM). Advances in technologies such as next-generation sequencing, RNA-based platforms, and CRISPR gene editing have accelerated the identification and validation of neoantigens, moving them closer to clinical application. Despite promising results, challenges such as tumor heterogeneity, immune evasion, and resistance mechanisms persist. The integration of AI-driven tools and multi-omic data has refined neoantigen discovery, while combination therapies are being developed to address issues like immune suppression and scalability. Additionally, the article discusses the ongoing development of personalized immunotherapies targeting tumor mutations, emphasizing the need for continued collaboration between computational and experimental approaches. Ultimately, the integration of cutting-edge technologies in neoantigen research holds the potential to revolutionize cancer care, offering hope for more effective and targeted treatments.
The discovery of eukaryotic split genes, with non-coding intronic sequences interspersed within protein-coding regions, revolutionized our understanding of gene structure and regulation. This breakthrough illuminated the process of RNA splicing, where introns are excised from pre-messenger RNA (pre-mRNA) by the spliceosome, a ribonucleoprotein complex that ensures the correct ligation of exons to form mature mRNA for translation [1, 2]. Splicing, which occurs in approximately 94% of human genes, is a ubiquitous and essential process in higher eukaryotes, playing a pivotal role in gene expression. Notably, a significant portion of these genes undergoes alternative splicing, generating diverse mRNA isoforms that enable a single gene to produce multiple protein variants [3–5]. This diversification is essential for cellular differentiation, development, and homeostasis. Aberrant splicing, however, can lead to the production of dysfunctional protein isoforms, contributing to various pathological conditions, including cancer and neurodegenerative diseases [6–8]. Furthermore, alternative splicing plays a crucial role in the presentation of neoantigens, which are tumor-specific antigens (TSAs) formed due to genetic mutations and splicing alterations. These neoantigens have emerged as key targets for cancer immunotherapies, highlighting the significance of understanding splicing mechanisms in the development of novel therapeutic strategies.
In cancer biology, the concept of TSAs, or neoantigens, has emerged as a transformative approach in immunotherapy [9, 10]. Neoantigens arise from somatic mutations and are presented on tumor cell surfaces by major histocompatibility complex (MHC) molecules, enabling cytotoxic T lymphocytes (CTLs) to target and eliminate tumor cells [9, 11, 12]. The identification of neoantigens has catalyzed the development of cancer vaccines and adoptive T cell therapies, all of which amplify tumor-specific immune responses [9, 13, 14]. Notably, neoantigens are exclusive to tumor cells, minimizing off-target effects and enabling personalized therapeutic approaches.
While most neoantigen research has focused on mutations, recent studies have highlighted a novel source: aberrant RNA splicing. Dysregulated splicing can produce cryptic exons or exon-skipping events, leading to the generation of tumor-specific splicing isoforms that may serve as immunotherapeutic targets [3]. Advances in next-generation sequencing (NGS) and RNA-seq technologies have facilitated the profiling of cancer-specific splicing landscapes, uncovering new neoepitopes [15, 16]. Additionally, predictive bioinformatics tools that assess MHC-binding affinity and immunogenicity further expand the potential pool of actionable targets [17, 18].
Glioblastomas (GBMs) exemplify the need for innovative therapies. Despite multimodal treatments, including surgery, radiotherapy, and chemotherapy with temozolomide, median survival remains limited to 15 months [19, 20]. FDA-approved therapies such as bevacizumab and tumor-treating fields (TTF) offer minimal survival benefits, and immunotherapeutic agents like nivolumab and EGFRvIII vaccines have failed to achieve efficacy in phase 3 trials. GBM’s inherent heterogeneity, infiltrative growth, and immunosuppressive microenvironment contribute to its resistance to conventional treatments [21, 22].
Neoantigen-based personalized peptide vaccines offer a promising avenue for GBM treatment [9, 23]. By utilizing NGS to identify tumor-specific mutations and splicing variants, these vaccines target unique neoepitopes, generating robust and specific immune responses. Clinical trials targeting the H3K27M mutation in diffuse midline gliomas have demonstrated safety and immunogenicity, underscoring the potential of personalized immunotherapy for GBM management [24, 25]. This strategy represents a critical step in addressing the challenges posed by this devastating malignancy.
This article explores the transformative potential of neoantigen-based immunotherapy, a precision oncology approach that targets TSAs arising from genetic, transcriptomic, and proteomic alterations. It examines various therapeutic modalities, including neoantigen vaccines, tumor-infiltrating lymphocyte (TIL) therapy, T-cell receptor-engineered T cells therapy (TCR-T), and chimeric antigen receptor T cells therapy (CAR-T), all of which show promise in treating GBM. The article also highlights how advances in NGS, RNA-based platforms, and CRISPR gene-editing technologies have accelerated neoantigen discovery, facilitating the development of highly personalized treatments. However, clinical application remains challenging due to factors such as tumor heterogeneity, immune evasion, and therapeutic resistance. To overcome these obstacles, the article underscores the need for an integrated approach that combines multi-omic data analysis, AI-driven predictive modeling, and collaborative efforts between researchers and clinicians to optimize neoantigen-based immunotherapies, paving the way for more effective and durable cancer treatments.
Neoantigen research has significantly advanced our understanding of tumor immunology and has been crucial in the development of targeted cancer therapies. Neoantigens are tumor-specific proteins that arise from somatic mutations in cancer cells, making them highly specific and effective targets for immune system recognition. The identification and validation of these neoantigens have revolutionized cancer immunotherapy by opening up new possibilities for personalized treatments [10, 26].
The exploration of neoantigens dates back to foundational studies in the mid-20th century, which demonstrated the immune system’s ability to recognize tumor cells as foreign [27, 28]. These early insights set the stage for further discoveries, including the recognition of T cell-mediated immune responses against neoantigens in both murine and human tumor models, validating their immunogenic potential [13, 29].
The technological breakthroughs of the early 2000s, such as NGS and flow cytometry, played a pivotal role in advancing neoantigen research. These tools allowed for high-throughput identification and validation of neoantigens, deepening our understanding of tumor immunology and T cell responses [30, 31].
Key milestones in neoantigen research are summarized in Table 1, highlighting how the field has evolved from initial discoveries to the application of personalized neoantigen vaccines and adoptive cell therapies (ACTs). The progress made in identifying and targeting TSAs demonstrates the transformative potential of these.
Timeline of key milestones and technological advances in neoantigen-based cancer immunotherapy
| Year | Milestone | Impact on cancer research | Key technologies | Therapeutic applications | References |
|---|---|---|---|---|---|
| 1950s | Immune recognition of carcinogen-induced tumors | Role of immune system in tumor detection | None | Early understanding | [32, 33] |
| 1980s | T cell recognition of neoantigens in mice | Cellular immune mechanisms | Murine models | None | [14, 34] |
| 1990s | Confirmed neoantigen reactivity in mice | Evidence for tumor-specific immune responses | Murine models | None | [35, 36] |
| 1990s | Neoantigen reactivity in human cancers | Broadened immune detection | Human tumor models | None | [13] |
| 2000s | Neoantigen reactivity in CD4+ T cells | Identified immune cell responses | Flow cytometry | None | [37] |
| 2000s | Identified neoantigen-reactive T cells in melanoma patients | Enhanced clinical relevance | Flow cytometry | None | [38, 39] |
| 2000s | Neoantigen reactivity over shared antigens | Clarified neoantigen advantage | Human tumor models | None | [40] |
| 2010s | NGS for immunogenic neoantigen identification | Accelerated antigen discovery | NGS | None | [41, 42] |
| 2010s | NGS for neoantigen-reactive T cells in melanoma | Mapped T cell specificity | NGS, flow cytometry | None | [43, 44] |
| 2010s | Neoantigen-based adoptive cell therapy in GI cancer | First clinical application | NGS, adoptive cell therapy | GI cancer treatment | [45, 46] |
| 2010s | Personalized neoantigen vaccines for melanoma | Pioneered patient-specific vaccines | NGS, peptide synthesis | Melanoma treatment | [47, 48] |
| 2010s | KRAS neoantigen adoptive cell therapy in colorectal cancer | Targeted oncogene mutations | NGS, adoptive cell therapy | Colorectal cancer treatment | [49, 50] |
| 2010s | Personalized neoantigen peptide and RNA vaccines | Advanced specificity and delivery methods | NGS, RNA technology | Multiple tumor types | [51, 52] |
NGS: next-generation sequencing; GI: gastrointestinal
Recent advancements in technologies, such as NGS and RNA-based platforms, have greatly accelerated the clinical translation of neoantigen research, enhancing the specificity and efficacy of therapeutic strategies [31, 53]. Ongoing innovations continue to refine neoantigen-based immunotherapies by improving immune responses while reducing off-target effects.
Looking ahead, the integration of cutting-edge technologies like CRISPR and AI may revolutionize the discovery and validation of neoantigens. These advancements promise to further enhance the effectiveness of cancer treatments, broadening the applicability of neoantigen-based therapies across various cancer types.
Neoantigen generation results from a multifaceted interplay of genetic, transcriptomic, and proteomic mechanisms, all of which contribute to the diversity of the antigenic landscape. These molecular alterations can profoundly affect the immune system’s ability to recognize cancer cells by presenting novel peptide sequences that are not found in normal, healthy tissues [54, 55].
At the genomic level, various mutations, such as single nucleotide variants (SNVs), insertions and deletions (INDELs), and gene fusions, disrupt the normal coding sequences of genes (Table 2). These alterations can lead to the creation of novel open reading frames (ORFs) and chimeric proteins that may possess antigenic properties, making them recognizable by the immune system [56, 57]. Additionally, the integration of viral sequences or the reactivation of endogenous retroviruses (ERVs) further contributes to the diversity of the neoantigen repertoire by introducing foreign protein-coding regions into the genome [58, 59]. These changes can be particularly impactful in cancer, where such genomic alterations can create unique targets for immune recognition.
Classification of neoantigen sources in cancer: genomic, transcriptomic, and proteomic alterations
| Category | Mechanism | Description | Neoantigen impact | References |
|---|---|---|---|---|
| Genomic mutations | Single nucleotide variants (SNVs) | A single-base substitution causes amino acid changes. | Alters the coding sequence, generating novel peptides. | [60] |
| Insertions and deletions (INDELs) | Small insertions or deletions lead to frameshift mutations. | Frameshift mutations alter amino acid sequences and generate novel open reading frames (ORFs). | [61, 62] | |
| Gene fusions | Fusion of two genes creating a chimeric protein. | Generates unique antigenic epitopes due to the formation of hybrid peptides. | [63] | |
| Viral sequences and endogenous retroviruses (ERVs) | Integration/reactivation of viral sequences in the genome. | Introduces foreign protein-coding sequences, producing immunogenic peptides. | [64, 65] | |
| Transcriptomic variants | Constitutive splicing | Standard intron removal and exon joining. | Preserves the normal coding sequence, minimal neoantigen impact. | [66, 67] |
| Exon skipping/inclusion | Variable inclusion or exclusion of exons. | Modifies the protein structure, potentially generating novel peptide regions. | [68, 69] | |
| Alternative 5' and 3' splice sites | Variations in donor and acceptor splice site selection. | Shifts exon boundaries, altering peptide sequence diversity. | [70, 71] | |
| Intron retention | Failure to remove an intron during splicing. | The translation of non-coding regions can introduce premature stop codons and novel antigenic sites. | [72, 73] | |
| Mutually exclusive exons | Expression of one exon while excluding another. | Generates diverse protein isoforms with unique antigenic determinants. | [74, 75] | |
| Exitrons | Hybrid exonic-intronic regions where introns are partially retained. | Alters the final protein product, increasing peptide diversity. | [76, 77] | |
| Adenosine-to-inosine (A-to-I) RNA editing | Post-transcriptional modification converting adenosine to inosine. | Alters codons without changing the underlying genomic sequence, leading to novel peptide generation. | [78, 79] | |
| Proteomic variants | ORFs | Translation initiated from previously untranslated regions. | Produces peptides not expressed under normal conditions, expanding neoantigen diversity. | [80, 81] |
| Coding long non-coding RNAs (lncRNAs) | Some lncRNAs can generate small peptides. | Generates small immunogenic peptides despite being non-coding. | [82, 83] | |
| Defective translation | Translational errors such as frameshifts or premature termination. | Produces aberrant protein fragments, expanding the neoantigen repertoire. | [84] | |
| Alternative start sites | Translation initiated from non-canonical codons (CUG, AGG, AUA). | Generates alternative protein isoforms with novel N-terminal sequences. | [85, 86] | |
| Post-translational modifications | Modifications like phosphorylation, glycosylation, ubiquitination. | Alters peptide structure and may expose hidden epitopes for immune recognition. | [87, 88] |
The transcriptomic landscape also plays a crucial role in expanding neoantigen diversity. Alternative mRNA processing events, such as exon skipping, alternative splice site selection, and intron retention, can modify the protein-coding potential of genes (Table 2). These variations can lead to the translation of modified or truncated protein products, which may differ significantly from the normal protein isoform [89–91]. In addition, mechanisms like mutually exclusive exon expression and the partial retention of exitrons further contribute to the diversity of protein isoforms that can be produced from a single gene [75]. Moreover, post-transcriptional modifications, such as adenosine-to-inosine (A-to-I) RNA editing, introduce codon changes that alter the sequence of the resulting peptides without modifying the underlying DNA sequence. This adds another layer of potential neoantigen generation from modified RNA transcripts [92, 93].
At the proteomic level, the generation of neoantigens is further enhanced by variations in translation and post-translational modifications (Table 2). ORFs can emerge from untranslated regions of the genome, and small peptides derived from long non-coding RNAs (lncRNAs) may also generate unique antigenic peptides [94, 95]. Translation defects, such as premature termination, frameshift mutations, or the use of non-canonical start sites, also contribute to antigenic diversity by producing altered peptide sequences that are not present in healthy tissues [96]. Additionally, post-translational modifications, such as phosphorylation, glycosylation, and ubiquitination, can expose previously hidden epitopes or alter peptide structures in ways that make them more immunologically recognizable [97, 98].
A comprehensive summary of the genetic, transcriptomic, and proteomic mechanisms that contribute to neoantigen diversity is provided in Table 2.
The development of effective cancer immunotherapies relies heavily on the identification and targeting of tumor-specific neoantigens. Neoantigens, which arise from genetic and molecular alterations unique to cancer cells, play a critical role in immune recognition and the design of personalized treatments [26, 42, 99]. Understanding how various therapeutic strategies influence neoantigen generation and presentation is essential for optimizing immunotherapy efficacy.
A comprehensive overview of various treatment modalities and their roles in neoantigen generation within the context of cancer immunotherapy are discussed in Figure 1. Each therapeutic approach affects neoantigen formation through distinct biological mechanisms, with implications for immune response modulation, clinical application, and combination therapy potential.
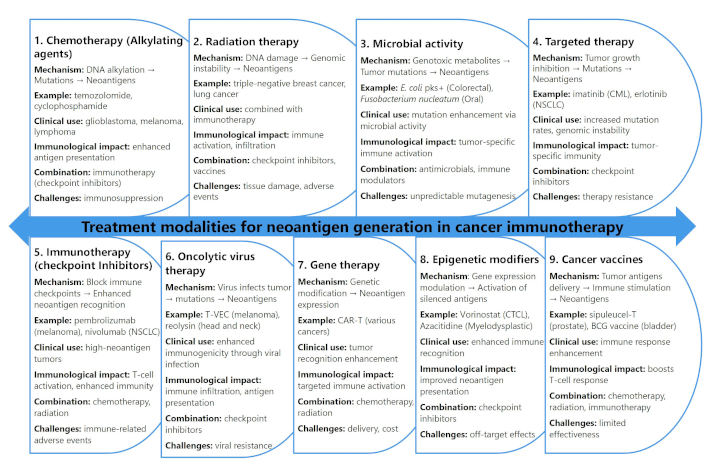
Treatment modalities and their role in neoantigen generation for cancer immunotherapy
Chemotherapy, particularly alkylating agents such as temozolomide and cyclophosphamide, induces DNA alkylation that causes direct strand damage, leading to point mutations and small insertions or deletions [100–102]. These genetic alterations give rise to novel antigenic peptides presented on MHC molecules, making tumor cells more immunogenic. For example, temozolomide treatment has been associated with enhanced neoantigen expression in GBM, particularly when paired with cancer vaccines [23]. However, chemotherapy poses challenges in predicting the extent of neoantigen generation and may cause immunosuppression, complicating its standalone efficacy [103, 104].
Radiation therapy also contributes significantly to neoantigen generation by causing DNA damage through direct ionization and reactive oxygen species (ROS) production. This damage induces genomic instability, leading to the formation of neoantigens from fragmented or mutated tumor DNA [105–107]. Preclinical studies in triple-negative breast cancer and non-small cell lung cancer (NSCLC) have demonstrated that radiation, when combined with immune checkpoint inhibitors such as anti-programmed death-1 (PD-1) inhibitors, enhances antitumor immunity by increasing immune cell infiltration [108, 109]. Nonetheless, radiation therapy carries the risk of localized tissue damage and immune-related adverse effects [110].
Microbial activity, specifically genotoxic microbial metabolites such as colibactin produced by Escherichia coli, can lead to tumor-specific mutations. Strains like pks+ E. coli induce double-strand breaks in host DNA, contributing to neoantigen formation in colorectal cancer [111, 112]. Similarly, Fusobacterium nucleatum and Bacteroides fragilis have been linked to mutagenic effects in colorectal and oral cancers [113, 114]. These microbial-induced neoantigens can promote tumor-specific immune responses, yet challenges remain in targeting specific microbial strains without disrupting the broader microbiome [115].
Targeted therapies, such as tyrosine kinase inhibitors (e.g., imatinib and erlotinib), interfere with signaling pathways involved in tumor growth and survival. These agents can induce mutations or unmask cryptic antigens within the tumor microenvironment (TME) [116, 117]. For instance, imatinib has been associated with increased mutation rates in chronic myelogenous leukemia, contributing to antigenic diversity [118]. However, the emergence of resistance and limited efficacy in tumors with low mutation rates restricts the widespread impact of targeted therapies on neoantigen generation [119].
Immunotherapy, particularly immune checkpoint inhibitors (e.g., pembrolizumab and nivolumab), plays a central role in restoring immune surveillance by blocking inhibitory proteins like PD-1 and CTLA-4 [120, 121]. These inhibitors amplify T-cell activity against tumor-specific neoantigens, especially in tumors with high mutational burdens. However, immune-related adverse events, such as colitis and pneumonitis [11, 122], pose clinical management challenges, necessitating careful patient selection.
Emerging therapeutic modalities, including oncolytic virus therapy and gene therapy, further expand the landscape of neoantigen generation. Oncolytic viruses like T-VEC and Reolysin selectively infect tumor cells, inducing direct lysis and the release of neoantigens [123, 124]. Additionally, viral infection can trigger immunogenic cell death, promoting immune system activation [125]. Gene therapy approaches, such as CAR-T cell therapy and oncolytic virus gene modification, enhance neoantigen presentation through engineered genetic alterations that promote novel antigen expression [126]. However, these advanced therapies face challenges like delivery efficiency, off-target effects, and high costs.
Epigenetic modifiers, including DNA methylation inhibitors and histone deacetylase inhibitors like vorinostat and azacitidine, represent another innovative strategy for neoantigen generation [127, 128]. By altering the epigenetic landscape, these agents can reactivate silenced tumor antigens and promote immune recognition [129, 130]. Nonetheless, concerns regarding off-target epigenetic changes and unpredictable long-term effects remain significant hurdles.
Neoantigen-based immunotherapies represent a promising frontier in cancer treatment, leveraging the body’s immune system to target TSAs [131]. These therapies encompass a variety of approaches, including vaccines, TIL therapy, TCR-T therapy, and CAR-T cells, each designed to stimulate or enhance immune responses against tumor neoantigens [132] (Figure 2). The breadth of neoantigen-targeted strategies underscores the potential for personalized cancer treatments [133, 134], emphasizing the importance of tailoring interventions to the unique antigenic landscape of individual tumors.
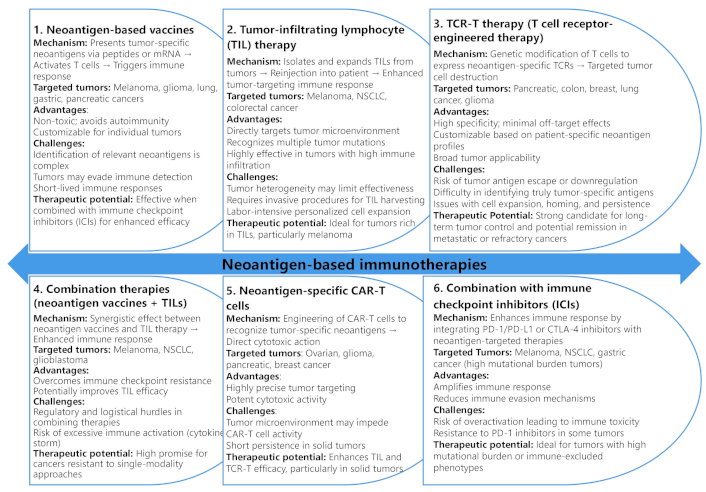
Comparative analysis of neoantigen-based immunotherapies: mechanisms, targeted tumors, and clinical potential
Neoantigen vaccines, for instance, aim to stimulate T cells by presenting neoantigen peptides or mRNA to the immune system, primarily targeting tumors such as melanoma, glioma, and lung cancer [131]. These vaccines offer advantages like non-toxicity and the ability to customize treatment for individual patients. However, challenges remain, including limited neoantigen identification and immune evasion by tumors. Despite these hurdles, studies have demonstrated the high therapeutic potential of neoantigen vaccines, particularly when combined with immune checkpoint inhibitors, leading to improved survival rates and tumor shrinkage [135, 136].
TIL therapy involves isolation, expansion, and reinfusion of TILs from the patient’s tumor to enhance antitumor activity. This approach has shown effectiveness in cancers with high TIL burden, such as melanoma and NSCLC [137, 138]. TIL therapy’s advantages include its ability to target multiple tumor mutations simultaneously and its direct engagement with the TME. However, invasive procedures for TIL harvesting and challenges related to tumor heterogeneity pose significant limitations [138]. Nonetheless, TIL therapy remains a powerful option, especially in immune-permissive tumors, with notable results in metastatic colorectal cancer [139, 140].
Combination therapies, including the use of neoantigen vaccines with TILs or immune checkpoint inhibitors, have gained attention for their synergistic effects in overcoming immune resistance [141]. As shown in Figure 2, combining these therapies can enhance the immune response by targeting multiple pathways simultaneously, particularly in cancers with high mutational burdens like melanoma and NSCLC [142, 143]. However, complex logistics, regulatory hurdles, and the risk of excessive immune activation remain barriers to widespread clinical adoption [144]. Still, the potential for achieving complete remission in refractory cancers underscores the promise of these multimodal strategies in advancing cancer immunotherapy.
Neoantigen-based therapeutic strategies represent a transformative advancement in cancer immunotherapy. These neoantigens are absent in normal tissues, allowing for precise tumor targeting with minimal risk to healthy cells [133, 145]. By focusing on these highly specific antigens, neoantigen therapies aim to harness and amplify the immune system’s natural ability to recognize and destroy malignant cells, thereby reducing the likelihood of off-target cytotoxicity and enhancing the therapeutic index [146, 147]. This precision is particularly advantageous in cancers with high mutational burdens, where the diversity of neoantigens increases the likelihood of effective immune responses [11, 148].
The principal neoantigen-based strategies include TCR-T therapy, CAR-T cell therapy, bispecific antibodies, neoantigen vaccines, TIL therapy, oncolytic virus therapies, cancer peptide vaccines, dendritic cell (DC) vaccines, and personalized multi-epitope vaccines [149]. TCR-T therapy involves the genetic engineering of autologous T cells to express TCRs capable of recognizing neoantigens presented by MHC-I molecules on the tumor cell surface [150]. CAR-T therapy modifies T cells to express synthetic antigen-binding receptors that can directly recognize tumor surface antigens without the need for MHC presentation, broadening their applicability to tumors with low MHC expression [151]. Bispecific antibodies act as immune engagers by simultaneously binding TSAs and CD3 on T cells, physically linking effector cells to tumor cells for targeted cytotoxicity [152, 153]. Neoantigen vaccines, including RNA, DNA, peptide, and DC-based platforms, work by introducing TSAs into the host to prime and expand tumor-reactive T cells [131, 154]. TIL therapy involves isolating and expanding TILs directly from the patient’s tumor, which are then reinfused into the patient to boost anti-tumor immunity [140, 155, 156]. Oncolytic virus therapies use genetically modified viruses that selectively infect and lyse tumor cells while also stimulating a systemic immune response against neoantigens released during tumor cell lysis [126]. Cancer peptide vaccines use synthetic peptides corresponding to TSAs to stimulate T cell responses, while DC vaccines involve loading patient-derived DCs with tumor neoantigens ex vivo before reinfusion [157–159]. Personalized multi-epitope vaccines combine multiple tumor neoantigens tailored to an individual’s tumor mutation profile, maximizing immune coverage [160, 161].
A comparative analysis of these neoantigen-based therapeutic strategies, evaluating critical parameters such as immune response potency, manufacturing complexity, scalability, and antigen breadth, is presented in Figure 3. TCR-T and CAR-T therapies exhibit robust immune responses but face challenges due to high manufacturing complexity and limited scalability, as they require patient-specific modifications [162, 163]. Bispecific antibodies provide a balance of moderate potency and manageable manufacturing demands, making them versatile, though still constrained by limited antigen breadth [164]. Neoantigen vaccines, while highly scalable and capable of broad antigen coverage, often elicit weaker immune responses and struggle to overcome immune suppression in advanced malignancies [146]. TIL therapy offers a potent and personalized response but relies on the availability of tumor-infiltrating cells and involves complex ex vivo expansion protocols [156]. Oncolytic viruses provide a dual mechanism of direct tumor lysis and immune activation but are often limited by pre-existing immunity against the viral vector [165, 166]. Peptide and DC vaccines offer moderate scalability and are less complex to produce but face challenges in generating sustained immune responses [167, 168]. Personalized multi-epitope vaccines maximize antigen breadth but require highly individualized tumor profiling, increasing complexity and cost [134, 141]. This comparative framework aids in selecting the optimal strategy based on clinical context and disease stage.

Comparative evaluation of neoantigen-targeted immunotherapies: key features, advantages, and clinical feasibility
The development of neoantigen-based therapeutic strategies has heralded a transformative era in cancer immunotherapy, capitalizing on TSAs derived from somatic mutations [99, 169]. These neoantigens, exclusive to malignant cells and absent in normal tissues, facilitate precise tumor targeting while minimizing collateral damage to healthy tissues [11, 131, 170]. The convergence of precision medicine and immunotherapeutic innovations positions neoantigen-based therapies as a cornerstone of personalized cancer treatment [103, 169]. Their potential is particularly compelling in patients with high mutational burdens, where the presence of diverse neoantigens drives robust immune responses [171].
Technological advancements have significantly enhanced neoantigen identification and engineering, ensuring higher precision and efficacy in therapeutic development. The integration of AI and high-throughput sequencing has revolutionized neoantigen discovery [172, 173]. AI-driven algorithms analyze extensive genomic and proteomic datasets, expediting the identification of TSAs and enabling tailored treatments for individual tumor profiles [174]. Simultaneously, CRISPR/Cas9 gene-editing technology has expanded the therapeutic horizon by optimizing immune cells to improve neoantigen recognition [175]. The engineering of potent TCR-T and CAR-T cell therapies using CRISPR has been particularly impactful, overcoming challenges such as immune evasion and resistance and broadening the applicability of neoantigen-based strategies across diverse cancer types [175].
A pivotal milestone in advancing neoantigen-based immunotherapies lies in the integration of multi-omic data, including genomics, transcriptomics, proteomics, and metabolomics [176]. This comprehensive approach deepens our understanding of tumor biology, enabling the identification of a personalized, exhaustive set of neoantigens [177, 178]. Multi-omic profiling facilitates the design of immunotherapies that address both well-characterized and novel neoantigens [179, 180]. The advent of personalized multi-epitope vaccines, enriched by insights from multi-omic analyses, has demonstrated enhanced antigen selection and broadened immune coverage [181, 182]. These precision-driven vaccines underscore the transformative potential of combining molecular-level insights with immunotherapeutic strategies.
Despite their transformative promise, neoantigen-based therapies must overcome challenges such as immune evasion and tumor-mediated immunosuppression [147]. TMEs often suppress T-cell activation and proliferation, undermining therapeutic efficacy [183]. Innovative combination therapies that pair neoantigen-targeting strategies with immune checkpoint inhibitors, such as PD-1/PD-L1 or CTLA-4 blockers, show significant promise in mitigating these barriers [120, 184]. These synergistic approaches enhance immune persistence and activity, yielding improved therapeutic outcomes [185, 186]. Additionally, the use of personalized adjuvants targeting immunosuppressive pathways represents a strategic advancement. These adjuvants, when combined with neoantigen vaccines or adoptive T-cell therapies, amplify immune recognition and response, fostering a more effective anti-tumor immunity [134, 173, 187].
Emerging hybrid strategies that integrate multiple therapeutic modalities are gaining traction, addressing the inherent limitations of individual approaches. For example, combining CAR-T cell therapy with bispecific antibody treatments can overcome the antigen breadth constraints of standalone methods, providing broader tumor coverage and enhancing immune activation [188, 189]. Oncolytic viruses deliver dual benefits by directly targeting tumors and augmenting neoantigen presentation through tumor cell death and systemic immune stimulation [126]. Advances in stealth viral vectors and genetically modified oncolytic viruses address pre-existing immunity challenges, ensuring sustained therapeutic efficacy.
The scalability and accessibility of neoantigen-based therapies are critical for their global adoption. Automated, large-scale manufacturing processes are being developed to reduce production costs and streamline therapy distribution [131, 190]. These efforts aim to democratize access to advanced cancer treatments, ensuring that the benefits of precision immunotherapy reach diverse populations worldwide.
The integration of cutting-edge technologies, multi-omic approaches, and scalable production systems is driving the evolution of neoantigen-based cancer therapies [13, 191, 192]. By addressing existing challenges and harnessing emerging opportunities, the field is on the cusp of delivering highly personalized, effective, and accessible cancer treatments. These advancements mark a paradigm shift in cancer immunotherapy, offering renewed hope for improved patient outcomes across a broad spectrum of malignancies.
In personalized cancer immunotherapy, the identification and targeting of neoantigens are critical for developing tailored therapeutic strategies [13, 193]. The process begins with mutation calling, which involves the detection of genetic alterations, such as SNVs, INDELs, gene fusions, and alternative splice variants, all of which are potential sources of neoantigens [84]. Tools like INTEGRATE-neo, PAVIS, and Cancer Genome Interpreter leverage NGS data and machine learning integration to identify these mutations with high sensitivity [194–196] (Figure 4). However, challenges such as false positives from sequencing errors or low variant allele frequencies can complicate mutation detection, highlighting the need for robust validation methods in the neoantigen discovery pipeline [197].

Overview of computational and experimental tools for neoantigen discovery and immunogenicity assessment. HLA: human leukocyte antigen; TCR: T-cell receptor; MHC: major histocompatibility complex
Once mutations are identified, the next crucial step is human leukocyte antigen (HLA) typing, which determines the MHC alleles expressed by the individual. This is vital for predicting the potential presentation of peptides on the surface of tumor cells [198–200]. High-resolution genotyping tools like Polysolver and OptiType use sequence-based typing and Bayesian inference to accurately determine an individual’s HLA haplotype [201–203] (Figure 4). The accurate matching of peptides to specific MHCs is essential for the subsequent prediction of antigen presentation [17, 204, 205]. However, challenges arise in cases of low-coverage sequencing data, where the performance of HLA typing tools may be limited, necessitating further refinement of these methods for clinical applications.
HLA binding prediction is the next step, focusing on the prediction of peptide-MHC binding affinity and stability. The ability of peptides derived from mutations to bind to MHC molecules plays a pivotal role in eliciting a T-cell-mediated immune response [206, 207]. Tools such as NetMHCpan and pVAC-Seq employ advanced algorithms, including neural networks and position-specific scoring matrices, to predict the binding of peptides to MHC class I molecules (for CD8+ T cells) and class II molecules (for CD4+ T cells) [57, 172, 208] (Figure 4). These tools provide reliable predictions for peptide-MHC affinity, although they may overlook complex, non-linear binding patterns or peptide stability, which could affect the immune response. As a result, the integration of multiple prediction tools and experimental validation becomes necessary for optimizing neoantigen identification and ensuring effective immunogenicity [209].
To further refine neoantigen candidates, T cell recognition analysis assesses the likelihood of TCR binding to peptide-MHC complexes. Tools such as NetCTL and TCRMatch utilize structural modeling and TCR-peptide docking models to predict TCR interaction and immune activation [210, 211]. Despite their usefulness, these models are often complex and face challenges due to limited experimental validation data. Additionally, neoantigen ranking and prioritization tools like Neopepsee and MuPeXI evaluate immunogenicity and expression levels to prioritize the most promising neoantigens for therapeutic development [172, 212, 213]. Finally, the validation of computational predictions through experimental tools like ELISpot and MHC Tetramer assays is essential to confirm the immunogenicity of neoantigens [41, 214, 215]. While these experimental methods provide the gold standard for validation, they are labor-intensive and require specialized expertise and reagents. The integration of computational tools with experimental validation creates a comprehensive approach to neoantigen prediction, ultimately enabling the development of personalized cancer immunotherapies that target specific tumor mutations.
In the rapidly advancing field of immunogenomics, computational tools have become indispensable for neoantigen identification and immunogenicity prediction. These tools assist in pinpointing potential therapeutic targets, especially in the context of cancer immunotherapy, by predicting the ability of tumor mutations to be presented on MHC molecules and recognized by the immune system [173, 216]. Table 3 compares several prominent computational tools in this domain, each with specific functions, strengths, and limitations. These tools vary in their approach, from mutation calling and HLA typing to peptide binding prediction and T-cell recognition [217]. Despite their distinct methodologies, all these tools aim to provide accurate predictions that can aid in the development of personalized vaccines or other immunotherapies.
Comparison of computational tools for neoantigen identification and immunogenicity prediction
| Tool | Step | Data input | Algorithm type | Key strength | Limitations | References |
|---|---|---|---|---|---|---|
| INTEGRATE-neo | Mutation calling | Whole exome/Genome sequencing | Graph-based detection | High sensitivity for detecting insertions and complex mutations | Limited to specific mutation types; may miss small variants | [218–220] |
| Polysolver | HLA typing | Whole exome sequencing | Bayesian inference | Highly accurate even with low-coverage data | Computationally intensive, requires high processing power | [201–203] |
| NetMHCpan | HLA binding prediction | Peptide sequences | Neural network | Broad coverage of MHC alleles across diverse populations | Sequence length constraints may not predict all peptides | [221–224] |
| NetMHCIIpan | HLA binding prediction | Peptide sequences | Neural network | High sensitivity for MHC class II binding predictions | Limited coverage of MHC class II alleles, not exhaustive | [225, 226] |
| NetCTL | T cell recognition | Peptide-HLA binding data | Machine learning | Specific scoring for CD8+ T cell recognition | Does not model TCR structure or interactions well | [227] |
| MHCflurry | HLA binding prediction | Peptide sequences | Machine learning/Deep learning | Accurate binding affinity predictions for class I peptides | Limited prediction capability for rare MHC alleles | [228, 229] |
| VAXign | Epitope validation | Peptide sequences | Statistical modeling | High throughput capability for large peptide datasets | Requires large amounts of experimental validation data | [230–232] |
| Immune Epitope Database (IEDB) | Epitope validation | Peptide sequences, HLA typing | Database query | Extensive experimental data support, widely recognized database | Limited by available datasets, may lack specificity in certain cases | [233–235] |
| NeoepitopePred | T cell recognition | Peptide-HLA binding data | Hybrid model (SVM, neural networks) | Comprehensive T-cell response prediction model | Requires large training datasets, may overestimate some responses | [172, 236, 237] |
| MHC-I Binding Prediction Tool | HLA binding prediction | Peptide sequences | Weighted scoring system | Effective for a wide range of peptide sequences | Limited by scoring system, may not capture all binding interactions | [238–240] |
HLA: human leukocyte antigen; TCR: T-cell receptor; SVM: support vector machines; MHC: major histocompatibility complex
One of the most prominent tools is INTEGRATE-neo, which focuses on mutation calling through whole exome/genome sequencing. It employs graph-based detection, offering high sensitivity for detecting complex mutations and insertions [219]. However, its major limitation is that it is confined to specific types of mutation and may miss smaller variants. Similarly, Polysolver is a robust tool for HLA typing using Bayesian inference, which is known for its high accuracy, even with low-coverage data [201–203]. Despite its effectiveness, it is computationally intensive and demands significant processing power.
Tools such as NetMHCpan and NetMHCIIpan, which focus on HLA binding prediction, rely on neural network algorithms to predict peptide binding across various MHC alleles. While NetMHCpan offers broad allele coverage, it is constrained by sequence length limitations and may fail to predict certain peptides [221, 222]. Meanwhile, NetMHCIIpan is highly sensitive for MHC class II binding predictions, but its coverage is limited, particularly for rare alleles [241–243].
In addition to mutation calling and peptide binding prediction, several tools aim to predict T-cell recognition. NetCTL, based on machine learning, provides specific scoring for CD8+ T-cell recognition; however, it does not account for TCR structure or interactions, which can be a critical factor in immune response [41, 221, 244]. Similarly, NeoepitopePred integrates a hybrid model using support vector machines (SVM) and neural networks to predict T-cell responses, but it requires large training datasets and may overestimate some immune responses [236, 237, 245]. VAXign, a statistical modeling tool for epitope validation, can handle large peptide datasets efficiently but necessitates substantial experimental validation [230–232]. Meanwhile, the Immune Epitope Database (IEDB) is a widely recognized and extensively supported database query tool for epitope validation, although its performance may be hindered by the availability and specificity of datasets [233–235].
Together, these tools provide complementary insights into the complex process of neoantigen identification, each offering unique advantages but also facing significant challenges in terms of data coverage, computational power, and the need for experimental validation.
GBM is a highly aggressive and treatment-resistant brain cancer, presenting significant challenges in clinical management [246, 247]. Current and emerging therapeutic strategies for GBM encompass a broad spectrum of approaches, each characterized by distinct mechanisms of action, varying levels of efficacy, inherent limitations, and impacts on patient outcomes and quality of life [20, 248, 249]. A thorough evaluation of these therapies highlights the complexity of GBM treatment and underscores the urgent need for innovative and multimodal strategies to achieve meaningful improvements in patient survival and overall well-being.
Conventional treatment options, including surgical resection, temozolomide chemotherapy, and radiation therapy, remain the standard of care for GBM patients. While these modalities collectively offer modest survival benefits, with a median overall survival of approximately 15 months, their limitations are pronounced [250]. Tumor heterogeneity and the emergence of therapeutic resistance contribute to inevitable disease recurrence (Figure 5). Moreover, adverse effects, such as cognitive decline, fatigue, and immunosuppression, significantly compromise the quality of life. These challenges underscore the importance of integrating advanced therapeutic approaches to enhance efficacy and address the long-term limitations of these foundational treatments [251, 252].
Emerging therapies, such as bevacizumab, TTF, immune checkpoint inhibitors, and targeted therapies, provide novel avenues for GBM management (Figure 5) [253, 254]. Bevacizumab, an anti-angiogenic monoclonal antibody, has demonstrated promise in improving progression-free survival (PFS) in recurrent GBM cases but has not shown a significant extension of overall survival, particularly in newly diagnosed patients [255, 256]. Similarly, TTF, a non-invasive therapeutic modality that disrupts mitotic processes, achieves only limited survival benefits [257]. Immune checkpoint inhibitors and targeted therapies face significant challenges, including tumor immune evasion and heterogeneity, which limit their standalone efficacy [258, 259]. However, the potential for combination therapies to overcome these obstacles represents a critical area for further research and optimization.
Personalized neoantigen-derived peptide vaccines represent a promising advancement in precision medicine for GBM (Figure 5) [23, 260]. These vaccines leverage patient-specific tumor antigens to elicit robust T-cell responses, offering the potential for extended survival and improved quality of life [23]. Despite their promise, challenges such as the time-intensive production process and the immunosuppressive TME hinder their broad applicability [41, 103]. When combined with complementary therapies like checkpoint inhibitors, these vaccines demonstrate synergistic potential, paving the way for transformative developments in GBM treatment [261, 262].
By critically analyzing the strengths and limitations of these therapeutic modalities, researchers and clinicians can navigate the evolving landscape of GBM management more effectively and identify innovative strategies to enhance patient outcomes. The next section will explore the role of personalized neoantigen-derived peptide vaccines in greater detail.
Neoantigens have catalyzed the development of advanced immunotherapeutic strategies for GBM. These strategies encompass a spectrum of approaches aimed at enhancing the immune system’s ability to recognize and destroy tumor cells. Personalized cancer vaccines, for instance, utilize patient-specific neoantigens to activate CD8+ and helper T cells, initiating a robust immune response [141]. However, tumor heterogeneity and immune evasion mechanisms remain significant barriers to efficacy (Table 4). To address these challenges, research has integrated vaccines with checkpoint inhibitors, demonstrating improved immune activation in GBM [263]. Future directions involve expanding vaccine applications to other tumor types and refining neoantigen identification processes to enhance immunogenicity.
Neoantigen-driven immunotherapeutic strategies for glioblastoma
| Therapeutic strategy | Mechanism | Application in GBM | Key challenges | Recent advances | Future directions | References |
|---|---|---|---|---|---|---|
| Personalized cancer vaccines | Prime the immune system with patient-specific neoantigens. | Induces tumor-specific immune responses by presenting identified neoantigens, activating both CD8+ T cells and helper T cells. | Limited immune response due to tumor heterogeneity and immune evasion mechanisms (e.g., checkpoint inhibition). | Development of combinatory approaches with checkpoint inhibitors to boost efficacy in GBM. | Expansion to other tumor types beyond GBM and improvement in neoantigen identification for higher immunogenicity. | [134] |
| Adoptive T cell therapies | Infuse patients with expanded T cells specifically targeting neoantigens. | Increases the quantity of tumor-specific T cells, improving tumor cell recognition and destruction through cytotoxic mechanisms. | Difficulty in maintaining T cell persistence and activity within the immunosuppressive GBM tumor microenvironment. | Use of IL-2 or other cytokines to support T cell expansion and persistence in vivo. | Optimizing T cell expansion methods and improving the persistence of T cells within the GBM microenvironment. | [264] |
| TCR-engineered lymphocytes | Modify T cells ex vivo to express receptors targeting specific neoantigens on tumor cells, then reinfuse them into the patient. | Improves T cell specificity and efficacy in targeting neoantigen-expressing tumor cells, enhancing tumor cell recognition and killing. | Off-target effects and T cell exhaustion in the hostile tumor environment can reduce efficacy. | Advances in TCR optimization to avoid off-target effects and increase the recognition of a broader range of neoantigens in GBM. | Developing next-generation TCR engineering techniques for targeting neoantigens more effectively, along with immune checkpoint blockers. | [149] |
| Immunotherapy combination therapy | Combine personalized vaccines, adoptive T cells, or TCR-engineered lymphocytes with other immunomodulatory agents (e.g., checkpoint inhibitors). | Provides synergistic effects, improving immune response and overcoming GBM’s immune evasion mechanisms (e.g., PD-1/PD-L1 axis). | Combination therapy may lead to increased toxicity, requiring careful patient monitoring and dosing. | Promising results combining anti-PD-1/PD-L1 with TCR-engineered T cells or vaccines to overcome immune suppression. | Expanding personalized combination therapies to include other immune modulators and identifying the most effective pairings. | [261] |
| Chimeric antigen receptor (CAR)-T cells | Engineer T cells with a CAR that targets a specific antigen on GBM cells, enhancing their ability to recognize and attack tumors. | Enhances tumor cell recognition and cytotoxicity against neoantigen-expressing GBM cells. | Limited by antigen escape, GBM’s heterogeneous nature, and CAR-T cell exhaustion in the tumor microenvironment. | Development of CAR-T cells targeting novel neoantigens and improving persistence through advanced cytokine support and genetic engineering. | Exploring multi-antigen CAR T cells targeting diverse neoantigens in GBM, along with methods to sustain CAR-T cell activity. | [265] |
| Oncolytic virus therapy | Use modified viruses that selectively infect and kill tumor cells while stimulating anti-tumor immune responses. | Induces tumor cell death and enhances immune responses to tumor neoantigens, potentially increasing efficacy of vaccines or T cell therapies. | Potential for oncolytic virus resistance and insufficient targeting specificity in the heterogeneous GBM tumor environment. | Oncolytic virus therapies combined with checkpoint inhibitors have shown early promise in preclinical models. | Investigating improved viral vectors that can better target GBM cells and enhance immune activation. | [123] |
| CRISPR-Cas9 gene editing | Modify the genome of T cells or tumor cells to enhance neoantigen targeting or create new therapeutic pathways for immune evasion. | Potential to enhance the precision and efficiency of T cell therapies or alter GBM’s immune evasion mechanisms to increase treatment efficacy. | Risks of off-target mutations, ethical concerns with gene-editing, and long-term effects of genetic modifications. | Successful use of CRISPR-Cas9 to engineer T cells for better targeting of neoantigens, with promising results in early-phase trials. | Expansion of CRISPR-based approaches to more effectively engineer immune cells and GBM cells for personalized therapies. | [175] |
| Peptide-based immunotherapies | Use synthetic peptides derived from identified neoantigens to activate immune cells in the tumor microenvironment. | Can specifically activate immune responses targeting neoantigens in GBM, enhancing tumor-specific immunity. | Limited by peptide delivery and the complexity of ensuring effective immune activation against heterogenous tumor populations. | Nanoparticle-mediated peptide delivery systems have enhanced peptide efficacy in preclinical models. | Optimizing delivery methods for peptide-based therapies to ensure efficient and targeted immune activation within GBM tumors. | [52] |
| Neoantigen-based biomarkers | Utilize identified neoantigens as biomarkers to predict treatment response and monitor therapy efficacy. | Can guide personalized therapy choices, monitor immune response, and track disease progression in GBM patients. | The complexity of tracking neoantigen responses in vivo, and the need for accurate biomarkers to predict treatment outcomes. | Development of liquid biopsy approaches using neoantigen biomarkers for non-invasive tracking of tumor progression and therapy response. | Continued refinement of non-invasive biomarkers for GBM treatment monitoring, including early detection of resistance. | [266] |
TCR: T-cell receptor; GBM: glioblastoma
Adoptive T cell therapies and TCR-engineered lymphocytes represent another pivotal class of neoantigen-focused therapies. Adoptive T cell therapies bolster tumor-specific cytotoxicity by infusing patients with expanded T cells targeting GBM-specific neoantigens (Table 4) [149]. However, sustaining T cell activity in GBM’s immunosuppressive environment is challenging [267]. Advances in cytokine supplementation, such as interleukin-2 (IL-2), have shown promise in enhancing T cell persistence and efficacy [268, 269]. Similarly, TCR-engineered lymphocytes improve tumor targeting by expressing receptors specific to neoantigens. Innovations in TCR optimization have minimized off-target effects and expanded the range of neoantigens targeted in GBM [150]. Emerging techniques aim to refine TCR engineering, leveraging combination therapies to further enhance treatment efficacy [270, 271].
CAR-T cells and oncolytic virus therapies highlight the potential of engineered and biologically driven approaches to target neoantigens (Table 4) [272]. CAR-T cells are equipped with receptors that specifically recognize GBM antigens, significantly enhancing tumor cell recognition and destruction [273, 274]. However, antigen escape and tumor heterogeneity present significant obstacles. Advanced CAR designs targeting multiple neoantigens and incorporating genetic modifications to sustain T cell activity are under investigation [275, 276]. Oncolytic virus therapies, meanwhile, employ modified viruses to selectively kill tumor cells and stimulate immune responses [123, 165]. Recent advancements in viral engineering and combinatory use with checkpoint inhibitors have demonstrated enhanced efficacy, particularly in preclinical models, underscoring the potential of synergistic therapeutic combinations [277, 278].
Emerging technologies such as CRISPR-Cas9 gene editing and peptide-based immunotherapies provide further avenues for innovation in neoantigen-targeted strategies [175, 279]. CRISPR-Cas9 has enabled precise modifications in T cells, enhancing neoantigen recognition and therapeutic outcomes in early-phase studies (Table 4) [175, 280]. Future research aims to expand its applications while addressing concerns about off-target effects and long-term safety. Peptide-based immunotherapies utilize synthetic peptides to activate immune cells against GBM-specific neoantigens. However, effective delivery remains a challenge [281, 282]. Nanoparticle-mediated delivery systems have improved peptide stability and target preclinical models, paving the way for clinical translation. Additionally, neoantigen-based biomarkers emerge as tools for monitoring therapy efficacy and guiding personalized treatments (Table 4) [134]. Non-invasive liquid biopsy techniques show potential for tracking tumor progression and detecting resistance, signaling a paradigm shift in GBM management [283].
Neoantigen-based immunotherapies represent a promising frontier in the treatment of GBM, but their clinical application faces several significant challenges [131, 261]. Among the most formidable obstacles is tumor heterogeneity, a hallmark of GBM that manifests as profound genetic and phenotypic diversity [284, 285]. This heterogeneity enables tumors to adapt swiftly to therapeutic pressures, often leading to immune escape [286]. For instance, tumor cells may lose the expression of targeted neoantigens or develop resistance mechanisms that diminish immune surveillance, ultimately limiting the efficacy of neoantigen-targeted treatments [11, 133]. Such adaptability underscores the need for strategies that can address the dynamic and heterogeneous nature of GBM [287].
The immunosuppressive TME further complicates the application of neoantigen-based immunotherapies [11, 149]. Regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and immunosuppressive cytokines like TGF-β collectively create a microenvironment that inhibits immune cell activity [288, 289]. This suppression prevents immune cells from fully engaging with and eliminating tumor cells, thereby significantly reducing the effectiveness of therapeutic interventions [290, 291]. In addition, immune evasion mechanisms, such as those mediated by the PD-1/PD-L1 and CTLA-4 pathways, further enable tumors to escape immune detection and suppression [292, 293], presenting yet another barrier to successful treatment.
To overcome these challenges, researchers are exploring combination therapies designed to enhance the efficacy of neoantigen-based immunotherapies [294]. One promising approach involves the use of immune checkpoint inhibitors, such as anti-PD-1 or anti-CTLA-4 antibodies, which block tumor-mediated immune suppression [120, 295]. These inhibitors enhance the immune system’s ability to recognize and target neoantigen-expressing cells, thereby improving therapeutic outcomes [11, 296]. Concurrently, efforts to reprogram the immunosuppressive TME by targeting Tregs, MDSCs, and immunosuppressive cytokines aim to create a more supportive environment for immune-mediated tumor destruction [288, 297].
Personalized neoantigen vaccines have emerged as a particularly compelling therapeutic strategy. These vaccines, which focus on tumor-specific neoantigens, have demonstrated the ability to elicit robust T cell responses in GBM patients, providing evidence of their potential to stimulate tailored immune responses while sparing healthy tissues [131, 161, 261]. However, several critical aspects must be optimized to fully realize their therapeutic potential, including the selection of immunogenic neoantigens, the development of efficient vaccine delivery platforms, and the refinement of dosing strategies [173]. Ensuring that the selected neoantigens are both specific to the patient’s tumor and capable of eliciting strong immune responses is paramount for the success of these therapies.
TCR-engineered lymphocytes offer another promising avenue for advancing neoantigen-based immunotherapy. These modified T cells are engineered to exhibit enhanced specificity and potency, allowing them to selectively target and eliminate neoantigen-expressing GBM cells [149]. To further enhance their effectiveness, researchers are investigating the combination of TCR-engineered lymphocytes with immune checkpoint inhibitors [270, 298]. By blocking tumor-induced immune suppression, these combinations may sustain immune responses and reduce the likelihood of tumor immune escape [299, 300].
Clinical trials investigating neoantigen-targeted therapies in GBM have provided encouraging, though preliminary, results [260, 261]. Personalized neoantigen vaccines have demonstrated their ability to activate immune responses, offering valuable insights into their therapeutic potential [134, 136, 301]. However, long-term clinical outcomes, such as survival benefits, remain to be thoroughly evaluated. Early successes in integrating neoantigen vaccines with other immunotherapeutic approaches highlight the potential of combination strategies, but additional research is needed to refine these approaches and address the immunosuppressive elements of the TME [103, 302]. Overcoming the inhibitory effects of Tregs, MDSCs, and immunosuppressive cytokines like TGF-β will be critical for improving the efficacy of neoantigen-based therapies [303, 304].
By addressing these challenges and leveraging innovative combination strategies, neoantigen-based immunotherapies have the potential to transform GBM treatment and improve patient outcomes. Continued research and clinical investigation will be essential to unlock the full therapeutic promise of these approaches.
Neoantigen-based immunotherapies have emerged as a promising approach for personalized cancer treatment, leveraging TSAs to provoke robust immune responses. Currently, a variety of therapeutic strategies—such as cancer vaccines, ACT, CAR-T therapy, and immune checkpoint blockade (ICB)—are undergoing clinical evaluation (Table 5). These therapies are being assessed for their ability to enhance immune responses, reduce tumor burden, and improve patient outcomes. Clinical trials are closely monitoring the safety, efficacy, and overall clinical impact of these therapies across various cancer types, highlighting the ongoing evolution of neoantigen-targeted immunotherapies (Table 5).
Clinical trials investigating neoantigen-based immunotherapies
| Therapy type | Cancer type(s) | Trial identifier(s) | Therapy combinations | Key outcomes/endpoints |
|---|---|---|---|---|
| Neoantigen-directed cancer vaccines | ||||
| Dendritic cell (DC) vaccine | Melanoma | NCT00683670 | Induction of neoantigen-specific T cells | |
| Synthetic long peptide (SLP) vaccine | Melanoma | NCT01970358, NCT02035956 | No evidence of disease (NED) in some patients, recurrence in others; response to PD-1 therapy | |
| SLP vaccine (GAPVAC) | Glioblastoma | NCT02149225, NCT02287428 | Increased neoantigen-specific T cell response | |
| Ribonucleic acid (RNA) vaccine (IVAC MUTANOME) | Melanoma | NCT02035956 | Induction of neoantigen-specific T cells | |
| SLP vaccine (NEO-PV-01) | NSCLC | NCT03380871 | Anti-PD-1 & Chemotherapy | Induction of neoantigen-specific T cells |
| Adenoviral & self-amplifying messenger RNA (samRNA) vectors | Microsatellite-Stable colorectal cancer (MSS-CRC), gastroesophageal adenocarcinoma (GEAC), NSCLC | NCT03639714 | Anti-PD-1 | One complete response (CR) (GEAC); neoantigen-specific T cell induction |
| Neoantigen-directed adoptive cell transfer (ACT) | ||||
| Tumor-infiltrating lymphocyte adoptive cell transfer (TIL-ACT) | Cholangiocarcinoma | NCT01174121 | Anti-PD-1 | Durable response (~9 years) after retreatment with TIL-ACT |
| MSS-CRC | NCT01174121 | Regression of all metastases | ||
| Breast cancer | NCT01174121 | One CR (>5 years), partial response (PR) in others | ||
| T cell receptor-engineered adoptive cell transfer (TCR-ACT) | Pancreatic cancer | NCT04146298 | PR, metastasis regression at 6 months | |
| T cell receptor-engineered T cell therapy (TCR-T) | ||||
| TCR-T | Multiple solid tumors | NCT05194735, NCT05105815, NCT04625205, NCT03171220, NCT05020119 | Safety, overall response rate (ORR), disease-free survival (DFS) endpoints | |
| Various cancers | NCT04102436, NCT04596033, NCT02280811, NCT02858310 | Chemotherapy | Response rate, adverse events (AEs), dose-limiting toxicities (DLTs) | |
| Solid tumors | NCT05349890, NCT04520711, NCT03970382 | Immune checkpoint blockade (ICB) | Safety, tolerability, DLTs | |
| Multiple tumors | NCT03412877, NCT04536922 | ICB & Chemotherapy | Response rate, treatment effect | |
| Hepatocellular carcinoma | NCT03199807 | Radiotherapy | AEs | |
| Tumor-infiltrating lymphocytes (TILs) | ||||
| TIL therapy | Solid tumors | NCT05141474 | AEs, serious adverse events (SAEs), treatment-limiting toxicity (TLT) | |
| TILs | Gastrointestinal (GI) & pancreatic cancers | NCT04426669, NCT03658785, NCT02959905 | Chemotherapy | Maximum tolerated dose (MTD), ORR, AEs |
| TILs | Melanoma, NSCLC | NCT03997474, NCT04032847 | ICB | AEs |
| TILs | NSCLC, squamous cell carcinoma (SCC), adenosquamous carcinoma | NCT03215810 | ICB & chemotherapy | DLTs |
| Chimeric antigen receptor t cell therapy (CAR-T) | ||||
| CAR-T | Glioblastoma multiforme | NCT02844062 | Chemotherapy | Safety |
| Immune checkpoint blockade (ICB) therapy | ||||
| ICB monotherapy | Various cancers | NCT03600155, NCT02553642, NCT03827044, NCT03718767, NCT03925246, NCT03082534, NCT03357757, NCT03813394, NCT02437279, NCT04825990, NCT03130764, NCT03653052, NCT02113657, NCT03040791, NCT04019964, NCT04293419, NCT04262089 | Clinical response, safety, DFS, ORR, progression-free survival (PFS) | |
| ICB | Various cancers | NCT04214249, NCT03978624, NCT02990845, NCT02453620, NCT03409198, NCT05456165, NCT05201612, NCT03832621, NCT03186326, NCT04659382, NCT04262687, NCT05141721, NCT04014530, NCT03918499, NCT03655002, NCT03126812, NCT03554317, NCT04336943, NCT04068194, NCT05317000, NCT02883062 | Chemotherapy | Clinical response, MTD, AEs, PFS, ORR |
| ICB | Cutaneous T-cell lymphoma | NCT03385226 | Radiotherapy | ORR |
| ICB | Colorectal cancer, meningioma, rectal cancer | NCT03854799, NCT03604978, NCT04340401 | Chemotherapy & radiotherapy | Pathologic response, MTD, ORR, AEs |
The clinical trials focus on five primary therapeutic categories: neoantigen-directed cancer vaccines, adoptive cell transfer, TIL therapy, CAR-T therapy, and ICB therapy. Each approach seeks to harness the body’s immune system to more effectively target cancer, either by stimulating immune responses against neoantigens or by genetically engineering immune cells to recognize and eliminate cancer cells (Table 5). For example, vaccines utilizing DCs, synthetic long peptides (SLPs), and RNA aim to prime the immune system to identify and attack tumors expressing neoantigens [305, 306]. ACT and TCR-engineered adoptive cell transfer (TCR-ACT) approaches involve modifying and expanding T cells to enhance their ability to recognize and kill cancer cells [150]. CAR-T therapy involves engineering T cells to express chimeric receptors that specifically target cancer cells [307], while ICB therapy aims to lift immune system inhibitory signals, enabling immune cells to attack tumor cells more effectively [308].
These clinical trials encompass a wide range of cancer types, including melanoma, GBM, NSCLC, and colorectal cancer, among others (Table 5). The studies also explore various combinations of these therapies with other treatments, such as PD-1 inhibitors, chemotherapy, radiotherapy, and ICB, to further enhance therapeutic efficacy. Clinical outcomes, such as overall response rate (ORR), disease-free survival (DFS), and PFS, are being used to evaluate the success of these therapies.
The increasing focus on neoantigen-based cancer immunotherapies—encompassing vaccines, ACTs, and immune checkpoint inhibitors—highlights their transformative potential in revolutionizing cancer treatment [10, 103]. Neoantigens, by being tumor-specific, allow for precise targeting of malignancies while sparing healthy tissues, offering a more personalized alternative to traditional therapies [133, 145, 301]. However, while promising, several obstacles need to be addressed to maximize the efficacy and applicability of these treatments.
One primary challenge lies in the identification of immunogenic neoantigens and their corresponding TCRs [30, 309]. Current methods, such as NGS and immunopeptidomics, have made strides in identifying neoantigens [9]. Despite these advancements, the accuracy of epitope prediction remains suboptimal, necessitating the refinement of computational workflows [36, 41]. The experimental validation of these antigens is resource-intensive and time-consuming, further complicating progress. Emerging technologies, such as T-Scan, which assess T cell-mediated killing, hold promise for scaling these processes, but a more efficient approach to TCR mapping, high-throughput platforms, and enhanced algorithms is vital for advancing personalized therapies [310, 311].
The financial and time-related constraints associated with personalized neoantigen therapies represent another significant hurdle. While these therapies promise tailored treatments, the high costs and lengthy timelines associated with producing custom vaccines and identifying TCRs hinder their widespread implementation. A promising alternative lies in the development of off-the-shelf therapies targeting public neoantigens, which arise from common mutations across different tumor types [84, 312]. Although such therapies could alleviate cost and time challenges, targeting shared neoantigens across diverse patient populations and creating effective vaccines for these antigens remains a major scientific hurdle [131, 147]. Future research should focus on the development of public neoantigen-based vaccines, bispecific antibodies, and TCR-based therapies to overcome these limitations.
Combination therapies, particularly pairing neoantigen-based vaccines with ICIs, are emerging as a potential solution to improve treatment efficacy. These combination strategies, which may also include chemotherapy or radiation to boost neoantigen presentation, have shown promise in overcoming immune evasion in solid tumors, especially epithelial cancers [12, 131, 145, 313]. However, combining therapies introduces its own set of challenges, particularly with respect to balancing immune activation and managing immune-related adverse effects [122, 314, 315]. Further exploration is needed into immune-modulating agents such as interferons and TLR agonists to determine optimal dosing schedules and improve their therapeutic impact.
Despite the challenges, the future of neoantigen-based immunotherapies is promising. Future research must focus on optimizing the screening and production processes to enhance scalability and reduce costs. Advances in bioinformatics, machine learning, and AI could significantly improve the accuracy of neoantigen prediction and streamline the process of identifying optimal therapeutic targets. Moreover, novel delivery systems, such as nanoparticles and tumor-derived extracellular vesicles (EVs), hold great potential in augmenting immune responses and improving overall therapeutic outcomes [316, 317]. As clinical trials continue, the integration of neoantigen-based therapies into personalized treatment regimens for specific patient populations, such as the elderly, could further improve treatment efficacy, considering distinct disease progression patterns in these individuals.
In conclusion, neoantigen-based immunotherapies hold significant promise as a frontier in cancer treatment, yet several challenges must be addressed to fully realize their potential. Key obstacles include the efficient identification of neoantigens, cost reduction, and the scalability of therapies. Overcoming these hurdles will require continued advancements in bioinformatics, high-throughput technologies, and combination therapeutic strategies. While the future of neoantigen-based cancer immunotherapy appears promising, achieving its full impact will require overcoming technical, logistical, and financial barriers.
To unlock the potential of personalized therapies, substantial investments in infrastructure, computational tools, and collaborative efforts across disciplines are essential. Collaboration between industry partners, academic institutions, and regulatory bodies will play a pivotal role in translating research findings into clinical applications. The challenge of ensuring cost-effectiveness remains critical to providing equitable access to these therapies for all patients.
Looking forward, enhancing the precision of neoantigen identification through improved bioinformatics models and high-throughput technologies represents a promising direction. Additionally, exploring the use of combination therapies—integrating immune checkpoint inhibitors with other immune modulators—could offer a more comprehensive approach to overcoming tumor resistance mechanisms. Expanding the focus to investigate common neoantigens across various cancers could also lead to the development of more broadly applicable therapies. However, challenges related to computational prediction, resource constraints, and the complexity of immune responses must be addressed through continuous innovation and rigorous clinical testing. Ultimately, while substantial work remains, the future of neoantigen-based immunotherapy is bright, with the potential to revolutionize cancer treatment, particularly in the realm of personalized medicine.
ACTs: adoptive cell therapies
CAR-T: chimeric antigen receptor T cells therapy
CTLs: cytotoxic T lymphocytes
DC: dendritic cell
GBMs: glioblastomas
HLA: human leukocyte antigen
ICB: immune checkpoint blockade
INDELs: insertions and deletions
MDSCs: myeloid-derived suppressor cells
MHC: major histocompatibility complex
NGS: next-generation sequencing
NSCLC: non-small cell lung cancer
ORFs: novel open reading frames
PD-1: programmed death-1
PFS: progression-free survival
SNVs: single nucleotide variants
TCR-T: T-cell receptor-engineered T cells therapy
TIL: tumor-infiltrating lymphocyte
TME: tumor microenvironment
Tregs: regulatory T cells
TSAs: tumor-specific antigens
TTF: tumor-treating fields
MMN: Conceptualization, Writing—original draft, Writing—review & editing. OAA: Writing—original draft, Writing—review & editing. SG: Writing—original draft. VB: Writing—review & editing. All authors read and approved the submitted version.
The authors declare there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.


