 Review
Review

Affiliation:
1INSERM, UMR 1186, 94800 Villejuif, France
2Gustave Roussy Cancer Campus, 94805 Villejuif, France
3University Paris Saclay, Faculty of Medicine, 94270 Le Kremlin Bicêtre, France
Email: jerome.thiery@gustaveroussy.fr
ORCID: https://orcid.org/0000-0002-0998-3627
Explor Target Antitumor Ther. 2022;3:598–629 DOI: https://doi.org/10.37349/etat.2022.00103
Received: June 15, 2022 Accepted: July 21, 2022 Published: October 27, 2022
Academic Editor: Nicola Normanno, Istituto Nazionale Tumori “Fondazione Pascale” Via Mariano Semmola, Italy
The article belongs to the special issue Cancer Immunotherapy and Tumor Microenvironment
Cancer-associated fibroblasts (CAFs) are highly heterogeneous players that shape the tumor microenvironment and influence tumor progression, metastasis formation, and response to conventional therapies. During the past years, some CAFs subsets have also been involved in the modulation of immune cell functions, affecting the efficacy of both innate and adaptive anti-tumor immune responses. Consequently, the implication of these stromal cells in the response to immunotherapeutic strategies raised major concerns. In this review, current knowledge of CAFs origins and heterogeneity in the tumor stroma, as well as their effects on several immune cell populations that explain their immunosuppressive capabilities are summarized. The current development of therapeutic strategies for targeting this population and their implication in the field of cancer immunotherapy is also highlighted.
During the past decades, accumulating evidence has revealed that tumor progression and response to therapies do not only rely on cancer cell genetic or epigenetic alterations but are also controlled by several components of the tumor microenvironment (TME) [1–3]. Indeed, the TME is a complex ecosystem composed of several cell types from endothelial/mesenchymal lineages and of various immune cells embedded in an intricated extracellular matrix (ECM), which enter into a dynamic relationship with tumor cells [2, 4–6]. Of note, over the last years, the TME has emerged also as a crucial regulator that shapes the cellular fate and functions of tumor-infiltrating lymphocytes (TILs), promotes tumor cell evasion from immune cell-mediated cytotoxicity and consequently alters the efficacy of the anti-tumor immune response or potentially immunotherapeutic approaches [7–9]. This last point relies, at least in part, on the ability of tumor cells and the TME components to orchestrate an immunosuppressive landscape, which leads, for example, to the recruitment and differentiation of immunosuppressive cells and ultimately to the inhibition of immune effector/killer cell functions. In particular, within the tumor stroma, fibroblasts that share similarities with fibroblasts activated during tissue injury or by acute or chronic inflammation, also named cancer-associated fibroblasts (CAFs), play a critical role in tumor cell-stroma complex interactions [10–13] and the regulation of the anti-tumor immune response [14–20]. In this review, the cellular, molecular, and biomechanical aspects involved in the immuno-suppressive capabilities of CAFs within the TME are summarized and the latest updates regarding therapeutic targeting of this cell population are highlighted, with potential implications in the field of combined cancer immunotherapies.
In normal tissue, spindle-shaped, interstitial cells lacking epithelial (cytokeratin–, E-cadherin–), endothelial (CD31–), and immune cell (CD45–) markers but from a mesenchymal (vimentin+) lineage are usually identified as resting fibroblasts, which display only negligible metabolic and transcriptional activities [11]. On the opposite, following tissue damages and subsequent repair or acute/chronic inflammation [21, 22], fibroblasts can become activated and exhibit contractile activity, exert physical forces to modify tissue architecture, acquire proliferation and migration properties and become transcriptionally active leading to elevated secretion of cytokines, chemokines and ECM components [21, 23, 24]. This process referred to as “wound healing response” is crucial for normal tissue homeostasis but is hijacked by cancer cells to favor their proliferation, survival, or invasive capabilities [11, 25]. Indeed, several studies have demonstrated that tumor cells can activate resident fibroblasts or promote trans-differentiation of other cell populations within the TME that lead to CAF generation [26, 27], which represent one of the most abundant stromal cell populations of several carcinomas including breast, prostate, pancreatic, esophageal and colon cancers [28]. In the context of cancer, several growth factors and cytokines released by either cancer or infiltrating immune cells are key determinants of CAF generation within the TME. For example, transforming growth factor-β (TGFβ), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), fibroblast growth factor (FGF), reactive oxygen species (ROS), interleukin-1β (IL-1β), and IL-6 or lysophosphatidic acid are important determinants of CAF generation within the TME [29–34]. Interestingly, vitamin A or D deficiency can also promote CAF differentiation under certain circumstances [35–37]. Moreover, it is important to note that CAFs can originate from quiescent resident fibroblasts present within the TME, which is probably the main source of this cell population but can also differentiate from other cell populations (Figure 1A). In particular, endothelial-to-mesenchymal transition (EndMT) has been linked to the trans-differentiation of endothelial cells to CAF-like cells [38, 39]. Similarly, perivascular cells, named pericytes, can also de-differentiate into CAFs [40]. Moreover, in breast cancer, adipocytes were shown to de-differentiate into CAFs [41–43], and in pancreas or liver tumors, stellate cells, involved in fibrosis, are probably an important source of CAFs [44, 45]. Finally, mesenchymal stem cells (MSCs), can be attracted from the bone marrow into the TME before their differentiation into CAFs [42, 46–51]. Together with the diversity of “activation” signals, these various origins undoubtedly represent an important determinant that contributes to the heterogeneity of CAFs, which is also highlighted by the diversity of markers used to identify them. This includes fibroblast-activation protein (FAP), α-smooth muscle actin (αSMA), PDGF receptors (PDGFRs), fibroblast-specific protein-1 (FSP1/S100A4), periostin (POSTN), neuron-glial antigen-2 (NG-2), podoplanin (PDPN), desmin, tenascin-C (TN-C), CD90, integrin β-1 (ITGB1/CD29), discoidin domain-containing receptor 2 (DDR2) or caveolin-1 (CAV1) [25, 28, 52–60]. However, none of these proteins is unequivocally specific for activated fibroblasts and consequently cannot be used as a single marker to distinguish CAFs from normal fibroblasts, or even other cell types. Moreover, these markers show distinct expression profiles between CAFs from different tumor types as well within the same tumors, once again reflecting their high degree of heterogeneity within the TME. In this regard, several studies have defined subtypes of CAFs presents in the TME of breast, ovarian, head, neck, and lung cancers or pancreatic ductal carcinoma (PDAC) [57, 61–63]. For example, based on an integrated flow cytometry analysis of FAP, CD29, αSMA, FSP1, PDGFRβ, and CAV1 expression, four different CAFs subsets (named CAF-S1 to -S4) have been identified in different breast and ovarian tumor subtypes and differentially accumulate within the TME [64] (Figure 1B). In highly aggressive human EGF receptor-2 positive (Her2+) and triple-negative breast tumors, CAF-S1 (FAPHIGH, CD29MED, αSMAMED-HIGH, FSP1MED, PDGFRβMED-HIGH, CAV1LOW) and CAF-S4 (FAPNEG-LOW, CD29HIGH, αSMAHIGH, FSP1LOW-MED, PDGFRβLOW-MED, CAV1LOW) represent the main CAF populations. On the opposite, luminal breast tumors are enriched with CAF-S2 (FAPNEG, CD29LOW, αSMANEG, FSP1NEG-LOW, PDGFRβNEG, CAV1NEG). Finally, CAF-S3 (FAPNEG, CD29MED, αSMANEG, FSP1MED-HIGH, PDGFRβMED, CAV1LOW) appear like normal fibroblasts also found in healthy tissue. Importantly, these 4 CAF subsets have been validated in situ by immunohistochemistry on patient samples [65] and using publicly available single-cell RNASeq (scRNASeq) data, CAF-S1 subtype has been also identified in other tumors including PDAC [66, 67], colorectal [68] or lung cancers [69] and displays inflammation, adhesion and ECM signatures [61] as well as immunosuppressive capabilities. Furthermore, among the CAF-S1 population in PDAC, and more recently in other tumors, two different subsets, αSMALOW CAF [inflammatory CAF (iCAF)] and αSMAHIGH CAF [myofibroblastic CAF (myCAF)] have been identified [44, 61, 70, 71]. The iCAF subpopulation secretes high levels of proinflammatory/immunomodulatory factors and is distant from the neoplastic cells, while the myCAF subset is located in the proximity of tumor cells and secretes ECM components. Moreover, a recent scRNAseq analysis in breast cancer further identified eight different clusters within the CAF-S1 subpopulation [70]. More specifically, within iCAFs subpopulation, IL-iCAF (IL-signaling), interferon-γ (IFNγ)-iCAF (IFNγ-related pathway), and detox-iCAF (detoxification pathway) have been described. Within myCAFs subpopulation, ECM-myCAF (ECM proteins), TGFβ-myCAF (TGFβ-dependent pathway), wound-myCAF (wound-healing signaling), IFNα/β-myCAF (IFNα/β-related pathway), and acto-myCAF (acto-myosin signaling) have been described. Finally, other subsets of CAF have been defined (see [72–74] for review) including a subpopulation of antigen-presenting CAF (apCAF), expressing a high level of major histocompatibility complex (MHC) class II molecules and CD74 [66], which is probably similar to the IFNγ-iCAF subset previously described.
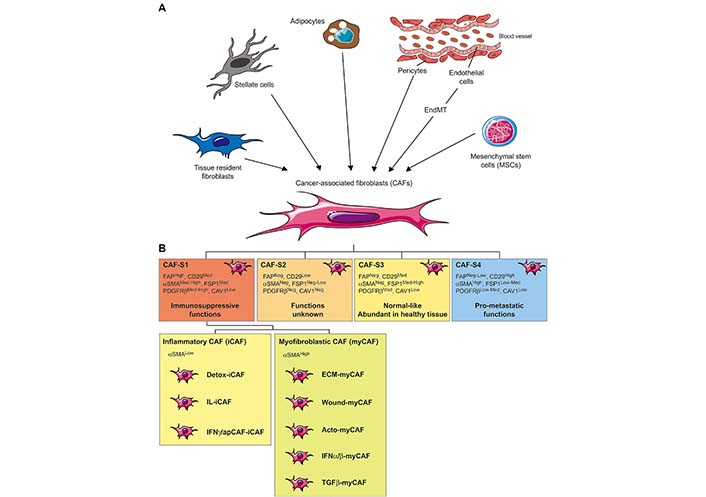
Origins and heterogeneity of CAFs in the TME. A. Schematic representation of CAF origins. CAFs can originate from diverse cell populations through different mechanisms. Local sources of CAFs include activated tissue resident fibroblasts, trans-differentiated endothelial cells resulting from EndMT, and de-differentiated pericytes, adipocytes, or stellate cells. Beyond those local sources, more distant ones can be involved in CAFs recruitment/differentiation in the TME, especially MSCs; B. schematic representation of CAF subsets. Distinct subpopulations of CAFs have been described with the TME. The combined analysis of six CAF markers (FAP, CD29, αSMA, FSP1, PDGFRβ, and CAV1) in breast and ovarian cancer leads to the identification of CAF-S1 to CAF-S4 subtypes. CAF-S1 displays an immune-suppressive function, CAF-S4 promotes invasion and metastasis formation and CAF-S2/-S3 resembles normal fibroblasts. More recently, single-cell RNA sequencing allowed the description of two different subsets of the CAF-S1 population, referred to as myCAF and iCAF. Within these two populations, IL-iCAF (IL-signaling), IFNγ/ap-iCAF (IFNγ-related/antigen presenting pathway), detox-iCAF (detoxification pathway), ECM-myCAF (ECM proteins), TGFβ-myCAF (TGFβ-dependent pathway), wound-myCAF (wound-healing signaling), IFNα/β-myCAF (IFNα/β-related pathway) and acto-myCAF (acto-myosin signaling) have been identified
Note. Adapted from “Alteration of the antitumor immune response by cancer-associated fibroblasts,” by Ziani L, Chouaib S, Thiery J. Front Immunol. 2018;9:414 (https://doi.org/10.3389/fimmu.2018.00414). © 2018 Ziani, Chouaib and Thiery.
In summary, many CAF subsets and clusters have been recently described, with a continuously increasing complexity [75]. Nevertheless, two important points to note are the relative proportion of the CAF-S1 cluster within the sequenced cell from the scRNAseq studies mentioned above and the presence of CAF-S1 within multiple tumor types, confirming the relevance of this subset in the field of immunosuppression with potential implication for immunotherapy.
In the TME, CAFs enter into dynamic crosstalk with tumor cells and/or other TME components and are an important source of several proteins such as ECM components or ECM-remodeling enzymes [e.g., collagens, matrix metallo-proteinases (MMPs)], chemokines [e.g., chemokine C-X-C motif ligand 12 (CXCL12)/stromal cell-derived factor-1 (SDF1)] or chemokine ligands [e.g., C-C motif chemokine ligand 2 (CCL2)/monocyte chemoattractant protein-1 (MCP-1)], angiogenesis-related factors [e.g., vascular endothelial growth factor (VEGF)] and other factors (e.g., TGFβ, EGF, FGF) which are linked to tumor cells proliferation, survival, invasiveness, metabolism reprogramming and stemness [10–13, 25, 28, 76]. Furthermore, and as mentioned above, CAFs have also been involved in the alteration of the anti-tumor immune response by the secretion of several immunomodulators [e.g., TGFβ, IL-1β, IL-6, IL-10, indoleamine-2,3-dioxygenase (IDO), arginase (Arg), CXCL2, CXCL5, CXCL12/SDF1, CCL2/MCP-1, CCL5/regulated upon activation, normal T-cell expressed and secreted (RANTES), VEGF, prostaglandin E2 (PGE2), tumor necrosis factor (TNF) or nitric oxide (NO)], that are key regulators of both innate and adaptive antitumor immune responses [17–19, 77] (Figure 2).
As a key component of the TME, tumor-associated macrophages (TAMs) play critical roles in the regulation of antitumor immune response. TAMs have been sub-classified into two distinct subtypes. Type I macrophages (or M1) secrete important amounts of pro-inflammatory cytokines and ROS and promote a T-helper 1 (Th1) anti-tumor immune response. On the contrary, type II macrophages (or M2) promote tumor progression and are characterized by the secretion of factors with immune-suppressive activity such as TGFβ, IL-10, Arg, and IDO, which particularly affect cytotoxic CD8+ T cell functions [78]. Interestingly, in oral squamous and colorectal cancers, CD163+/DC-SIGN+ M2 macrophages are the most prominent immune cells in the neighborhood of αSMA+, FSP1+, and FAP+ CAF-rich areas, suggesting a close relationship between these two cell populations, with important consequences on the clinical outcome for patients [79, 80]. Further evidence was provided by several studies which have demonstrated that the recruitment of monocytes into the TME and their differentiation toward M2 subtype macrophages are actively promoted by CAFs [81], especially through their secretion of CXCL12/SDF1, macrophage colony-stimulating factor (M-CSF)/CSF-1, IL-6, CCL2/MCP-1 and chitinase-3-like-1 (Chi3L1)/YKL-40 [82–92]. However, and in an intriguing way, CAFs might also alter TAMs infiltration under certain circumstances, by a FAP-mediated modification of the ECM [93]. Finally, it is important to note that reciprocal crosstalk exists between CAFs and TAMs. Indeed, several studies have suggested that M2 macrophages can regulate CAFs generation, for example by enhancing EMT progression through IL-6 and SDF1 [83], or by influencing the trans-differentiation of MSCs into CAFs [94, 95].
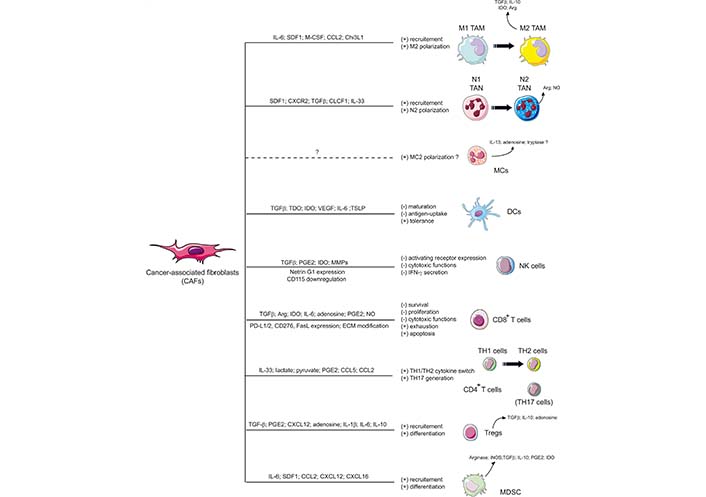
Schematic representation of CAFs-dependent immunosuppression. CAFs shape the tumor immune microenvironment and influence both the innate and adaptive anti-tumor immune response. CAFs are involved in the recruitment of innate immune cells, such as TAMs, and tumor-associated neutrophils (TANs), and promote their acquisition of an immunosuppressive phenotype (M2 and N2 respectively). CAFs also affect the cytotoxic function and cytokine production of natural killer (NK) cells and activate MCs with a potential immunosuppressive phenotype. CAFs also promote the recruitment and differentiation of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) and interfere with the maturation and function or dendritic cells (DCs). CAFs have also the ability to influence CD4+ Th lymphocytes, favoring tumor-promoting Th2 and Th17 responses, and reduce the activation, functions, and survival of CD8+ cytotoxic T cells. MCs: mast cells; CXCR2: C-X-C chemokine receptor 2; CLCF1: cardiotrophin-like cytokine factor 1; TDO: tryptophan 2,3-dioxygenase; TSLP: thymic stromal lymphopoietin; PD-L1: programmed death ligand 1; iNOS: inducible NO synthase; (+): induction; (–): inhibition
Note. Adapted from “Alteration of the antitumor immune response by cancer-associated fibroblasts,” by Ziani L, Chouaib S, Thiery J. Front Immunol. 2018;9:414 (https://doi.org/10.3389/fimmu.2018.00414). © 2018 Ziani, Chouaib and Thiery.
Recent evidence indicates that TANs represent a significant component of the TME [96, 97] and several studies have suggested that TANs can be polarized to an N1 anti-tumoral or N2 pro-tumoral subtype, as observed for TAMs. N1 neutrophils differentiate following TGFβ blockade and express immuno-activating cytokines and chemokines, low levels of Arg 1, and can kill cancer cells. On the opposite, N2 neutrophils are induced following exposure to high TGFβ levels [98], are characterized by expression of CXCR4, VEGF, and MMP9, and can inhibit CD8+ T cell function [99]. Of note, TANs have been linked to a poorer prognosis for patients with renal and pancreatic cancer; gastric, hepatocellular, colorectal, head and neck carcinomas, and melanoma [100, 101]. A few studies have highlighted the crosstalk between CAFs and TANs. For example, CAF-derived CXCL12/SDF1 and CXCR2 are involved in TANs recruitment within the TME and CAF-derived IL-6 stimulates signal transducer and activator of transcription 3 (STAT3) signaling pathway in TANs, potentially inducing immune tolerance through the expression of PD-L1 [102]. CAF-secreted TGFβ can also probably redirect TANs differentiation toward an N2 phenotype [98]. Furthermore, in hepatocellular carcinoma, CAF-derived CLCF1 increases CXCL6 and TGFβ secretion by tumor cells, which subsequently promotes TAN infiltration and polarization [103]. It seems that CAFs can also induce pro-tumorigenic neutrophil extracellular traps (NETs) formation in an amyloid β-dependent manner [104]. In a mouse breast tumor model, it was also shown that CAF-derived IL-33 facilitates lung metastasis by the recruitment of TANs [105]. Interestingly, the N2 polarization is also increased by vascular mimicry between CAF and cancer cells [106]. Finally, reciprocal crosstalk probably exists between CAFs and TANs. For example, neutrophil NETs can promote liver micro-metastasis in pancreatic ductal adenocarcinoma via the activation of CAFs [107] and TANs are capable to promote the differentiation of MSCs into CAFs [108]. It was also shown that the CAF marker PDPN interacts with the neutrophil protein CD177, with possible implications for CAF functions [109].
MCs are tissue-resident sentinel cells that, upon activation, release a wide spectrum of chemokines and cytokines. MCs are mostly known for their role in an allergy but can also modulate tumor initiation and progression. Depending on their localization or cancer type, MCs exert dual effects on tumor progression [110]. As such, it seems that MCs display two subtypes, anti-tumorigenic MC1 and pro-tumorigenic MC2, which produce different mediators with opposite roles in tumorigenesis. In particular, MC1 produces IL-9, and histamine, which induces DC maturation and inhibits tumor growth in murine models. In contrast, MC2 produces a variety of angiogenic and metastatic substances, including VEGF, FGF, MMP9, TGFβ, and cytokines (IL-1β, IL-6, and IL-13) [111, 112]. Importantly, MCs can also alter the anti-tumor immune response. For example, the release of free adenosine [113] or IL-13 by MCs can respectively inhibit T cell function and promotes M2 polarization [114–116]. MCs can also favor the generation of highly suppressive MDSCs and Tregs in the TME [117, 118]. To date, research on the cooperation between MCs and CAFs in tumors is still in its infancy, with only a few studies addressing this question. For example, in odontogenic lesions that affect the jaw or neurofibroma, a large number CAFs and MCs in tumor islets are associated with the aggressiveness of the disease [119, 120]. In pancreatic tumors, stellate cells (a CAF precursor) can activate MCs which in turn enhance CAF proliferation by their secretion of IL-13 and tryptase. This process results in the formation of a fibrotic TME and ultimately suppresses the antitumor immune response [121]. Finally, in an in vitro three-dimensional (3D) microtissue model of prostate cancer, a recent study has revealed cooperation between MCs and CAFs, which enhances the transition from a benign to an anormal epithelia via a tryptase-dependent mechanism [122].
In the TME, important antigen-presenting cell subpopulation, known as DCs, have a pivotal role in the activation of T cell-mediated, adaptive, anti-tumor immunity [123] and their global biology can be affected by the CAFs, even if in-depth mechanisms remain poorly understood. As a major source of TGFβ in the TME, CAFs can probably affect DC functions, in particular through the inhibition of MHC class II molecules, co-stimulatory molecules (CD40, CD80, and CD86), and cytokines (TNF-α, IFNγ, and IL-12) expression/secretion [124], which alter CD8+ cytotoxic T cell activation and Th1 polarization of CD4+ Th cell populations, and also promote the formation of CD4+ forkhead box protein P3 (FoxP3)+ Treg cells that potently inhibit the function of other T cells [125, 126]. Similarly, in hepatocellular carcinoma, CAFs have been described as a major source of IL-6 that affects DC functions through the activation of the STAT3 pathway leading to the generation of regulatory DCs, characterized by low expression of costimulatory molecules and high secretion of immune-suppressive cytokines, which impair T-cell proliferation and promote Tregs expansion [127]. Furthermore, CAF-produced IL-6 can also favor the emergence of pro-tumorigenic TAMs from monocytes at the expense of DCs [82]. Interestingly, in lung tumors, galectin1-driven secretion of TDO2 and IDO by CAFs promotes tryptophan degradation in kynurenines that inhibits DCs differentiation and functions [128]. In pancreatic tumors, the secretion of TNF-α and IL-1β by tumor cells promotes CAFs activation and their secretion of TSLP, which favor the generation of DCs with Th2-polarizing capabilities, associated with reduced patient survival [129]. In mouse esophageal squamous cell carcinoma, CAFs-secreted Wnt family member (WNT2) has been linked to suppression of the DC-initiated antitumor T-cell response via the suppressor of cytokine signaling 3 (SOCS3)/phosphorylated Janus kinase 2 (p-JAK2)/phosphorylated STAT3 (p-STAT3) signaling pathway. On the opposite, anti-WNT2 monoclonal antibodies (mAbs) can significantly restore T-cell responses and enhance the efficacy of anti-programmed cell death 1 (PD-1) therapy by increasing active DCs [130]. Furthermore, in ovarian cancers, CAFs can secrete wingless-type mouse mammary tumor virus integration site 16B (WNT16B) in response to DNA damage-associated treatment, which promotes the secretion of IL-10 and TGFβ by DCs [131]. Finally, as a major source of VEGF, CAFs might inhibit DC generation, maturation, and functions through this pathway [132].
CAFs can also alter the activity of NK cells, which are a major participant in the early immune response through their cytotoxic functions, and contribute to the adaptive immune response through their secretion of cytokines and the promotion of DC maturation. The detailed mechanisms of the complex relationship between CAFs and NK cells are still emerging and most likely involve multiple molecules. As such, TGFβ released in the TME by CAFs most likely plays an important role in the alteration of NK cell activation and cytotoxic activity [133], for example by reducing NK-activating receptor expression [134–136]. Furthermore, more direct evidence of the effect of CAFs on NK cells has been provided during the past few years. Independent studies involving melanoma, colorectal, and hepatocellular carcinoma-derived fibroblasts have shown that CAFs, through the secretion of PGE2 and IDO, can decrease the expression of several natural cytotoxicity receptors [NCRs, e.g., NKp30, NKp44 and NK receptor DNAX accessory molecule (DNAM)] at the NK cell surface, as well as perforin and granzyme B [137–139], leading to attenuated cytotoxic capabilities of NK cells. We also demonstrated that melanoma-associated CAFs decrease the sensitivity of melanoma tumor cells to NK cell-mediated killing through the secretion of MMPs which cleave MHC class I-related chain (MIC)-A and MIC-B [two ligands of NK group 2D (NKG2D)], at the surface of the tumor cells and consequently decrease both NKG2D-dependent cytotoxic activity of NK and their secretion of IFNγ [140]. In pancreatic ductal models, the high expression of the glutamatergic pre-synaptic protein netrin G1 (NetG1) in CAFs is also linked to their ability to inhibit NK cell-mediated killing of tumor cells [141]. Furthermore, in endometrial cancer, CAFs can decrease NK cells’ lytic potential through their downregulation of poliovirus receptor (PVR/CD155), a ligand of the activating DNAM-1/CD226 [142]. Finally, in the context of radiotherapy, CAFs isolated from non-small cell lung cancer inhibit NK cell activation and cytotoxic functions [143].
In summary, due to their secretion of cytokines, chemokines or other soluble factors, and possibly other mechanisms, CAFs shape the TME and favor the recruitment of innate immune cells and their acquisition of an immunosuppressive phenotype like M2 macrophages, N2 neutrophils, possibly MC2, but also affect DC functions or cytotoxic potential and cytokine production of NK cells.
CAFs also hamper the adaptive anti-tumor immune response at different levels, ultimately leading to the alteration of effector T cell functions in the TME (Figure 2). Of note, among FAPHIGH CAF, the recent single cell analyses revealing the heterogeneity within this population mentioned earlier in this review have also strongly suggested that specific clusters, in particular those characterized by wound-healing signature, ECM accumulation, and TGFβ-signaling, are particularly associated with an immunosuppressive environment, at least in some tumor types [70, 144].
As mentioned above, CAFs are an important source of TGFβ in the TME which acts on both CD8+ and CD4+ T cells [124, 126] and consequently hamper the antitumor T cell-dependent immune response and the response to immunotherapies. For example, in breast cancer, one of the cellular clusters identified among FAPHIGH CAFs is characterized by TGFβ signaling and is linked to immunosuppression and resistance to immunotherapy [70]. Similarly, poor response to immunotherapies in the metastatic urothelial, lung, and colon cancer and melanoma have been linked to TGFβ signature in CAFs [145, 146]. Furthermore, in an ovarian cancer cohort, it has been shown that the key determinant of T cell exclusion is the up-regulation of TGFβ in the activated stromal compartment [147]. Mechanistically, TGFβ is known to have pleiotropic “bad” effects on the T cell-dependent immune response. This includes the alteration of effector CD8+ T cell survival through the inhibition of the pro-survival protein B cell lymphoma-2 (Bcl-2) expression [148], the reduction of CD8+ T cell cytolytic functions through the reduction of perforin, granzymes A and B, Fas ligand (CD95L) and IFNγ expression [149, 150], the reduction of CD8+ T cells infiltration [145], the alteration of the acquisition of effector function by memory CD8+ T cells [149, 151] or the promotion of Tregs recruitment and differentiation [152]. As such, TGFβ-secreting myCAF is very abundant in immune-excluded ovarian tumors [153], and αSMA+FAP+ CAFs from head and neck tumors have been shown to inhibit CD8+ T cell proliferation and to promote the recruitment of Tregs in a TGFβ-dependent manner [154]. Of note, it has been suggested that CAFs and Tregs enter into a reciprocal cross-talk via their mutual expression of TGFβ, increasing in parallel CAFs activation and Tregs activity [155].
Furthermore, CAFs are also an important source of cytokines and chemokines in the TME, with once again a potential pleiotropic effect on T cells. For example, in αSMA+FAP+ CAFs from head and neck tumors mentioned above, IL-6 secretion cooperates with TGFβ to inhibit CD8+ T cell proliferation and promote the recruitment of Tregs [154]. Similarly, in murine PDAC models, IL-6 depletion specifically in αSMA+ CAFs synergizes with anti-PD-1 immunotherapy to significantly improve the survival of tumor-bearing mice [156]. In breast cancer, CAF-derived IL-33 has been identified as a driver of the Th2-polarized immune response [25]. Furthermore, in lung and pancreatic tumors, the secretion of CXCL12/SDF1 by CAFs contributes to the exclusion of T cells from the cancer cell proximity [157, 158]. Similarly, in high-grade serous ovarian cancers, CAF-S1 increases the attraction, survival, and differentiation of Tregs via microRNA-141/200a (miR-141/200a)-dependent secretion of CXCL12β [61]. Similarly, recent scRNAseq analysis in breast cancer has also demonstrated that CXCL12 is highly secreted by iCAFs [70]. In the TME, CAFs-secreted CCL2, CCL5, CCL17, IL-1, IL-6, IL-13, and IL-26 can also promote a Th2 and Th17 CD4+ polarization, at the expense of anti-tumor Th1 response [33, 159–161]. Consequently, in vivo elimination of CAFs in a murine model of breast cancer using a vaccine targeting FAP can shift CD4+ T cell polarization from a Th2 to a Th1, increase expression of IL-2, increase CD8+ T cell functions, and hamper Tregs recruitment [161].
Of note, and as mentioned earlier in this review, the presence of CAFs in the TME profoundly affects the ECM through the deposition of several components (e.g., fibronectin or type I collagen) and proteolytic degradation of normal ECM structure in an MMPs-dependent manner. This remodeling has important consequences on both tumor behavior [23, 162–165] and the efficacy of the antitumor immune response [166]. Indeed, this modified ECM is presumed to restrict access of immune cells to cancer cells, serving as a physical barrier [166, 167]. The perfect example is PDAC, where fibrosis is extensive and the “scar-like” ECM acts as a barrier for cytotoxic T cell infiltration into tumor cell areas [168, 169]. This also occurs in other cancer types such as lung tumors, where T cells poorly migrate in dense ECM areas [158, 170]. Similarly, the presence of FAPHIGH ECM-secreting CAFs has been linked to the exclusion of CD8+ T cells from the tumor and their accumulation in the collagen-rich peritumoral stroma [70]. Furthermore, in tumors with the accumulation of matrix proteins in the ECM, tumor tissues are often poorly oxygenated, resulting in the presence of areas with a low oxygen pressure called “hypoxic zones” [16, 171, 172]. Interestingly, several studies indicated that hypoxia is involved in the process of CAFs activation and in their functionality within the TME [173–177]. In parallel, in melanoma, our group recently provided evidence that hypoxia increases CAFs TGFβ, IL-6, IL-10, VEGF, and PD-L1 expression and/or secretion and demonstrates that hypoxic CAF exerts a more profound effect on T cell-mediated cytotoxicity than their normoxic counterpart [178].
In addition, CAFs can also impair T cell proliferation and effector functions through the metabolic reprogramming of the TME. In particular, the secretion by CAFs of IDO1 [179, 180], an immuno-regulatory enzyme that catabolizes tryptophan degradation [181, 182], or Arg 2, an enzyme involved in the deprivation of arginine in the TME [183], have a potentially important effect on T cells. In this regard, a poor clinical outcome for PDAC patients has been linked to the presence of CAFs expressing Arg 2 in hypoxic zones [184]. In addition, CAFs can use aerobic glycolysis as a source of energy, which results in the production of pyruvate and lactate that switch T cell polarization, reducing the percentage of Th1 CD4+ T cells and increasing Treg recruitment [185–187]. Furthermore, stromal cells from cervical tumors express high levels of CD39 and CD73, two molecules known to hydrolyze ATP, generating free adenosine, which possesses important immunosuppressive properties [188]. Similarly, FAPHIGH CAFs from breast, colorectal and ovarian cancer express high levels of CD73, which potentially promotes immunosuppression in, at least in part, a Tregs-dependent manner [61, 189, 190]. Interestingly, it was also recently shown that CAFs upregulate CD39 expression on T cells, and in turn, T cells upregulate CD73 expression on CAFs [191]. CAFs highly express cyclooxygenase-2 (COX2) and are consequently a major source of PGE2 [192, 193], with important implications in the field of immunosuppression [194], especially by shifting the balance between Th1 and Th2 responses, by suppressing CD8+ T cell-mediated cytotoxic activity and by promoting Treg recruitment. As such, PDAC-derived CAFs strongly inhibited T-cell proliferation in a PGE2-dependent manner, and its inhibition by indomethacin, a non-steroidal anti-inflammatory molecule, partially restored the proliferative capacities of both CD4+ and CD8+ T-cells [195]. Finally, in breast cancer, NO secretion by FAPHIGH PDPN+ CAFs can suppress T cell proliferation [196].
Finally, CAF can hamper T-cell-mediated immune response through many other miscellaneous mechanisms, many of them being under investigation or probably not yet elucidated. For example, it was suggested that CAFs can trigger cytotoxic T cell apoptosis via their expression of FasL/CD95L [197] and that CAFs secretion of galectins that have a high affinity for β-galactosides [198, 199], alters T cell functions [200–202].
CAFs in the TME can also interfere with the adaptive immune response by facilitating the infiltration and generation of MDSCs, involved in the direct or indirect alteration of the T cell-mediated immune response through their secretion of several factors including Arg, iNOS, TGFβ, IL-10, PGE2 and IDO [203, 204]. In this regard, in pancreatic tumors, CAF-secreted IL-6 favors monocyte precursors differentiation towards an MDSC phenotype, in a STAT3-dependent manner [85, 205]. Similar results were obtained in esophageal squamous cell carcinoma, where CAF-secreted IL-6 and CAF-derived, exosome-packed, miR-21 promote MDSC differentiation via STAT3 signaling [206]. Furthermore, secretion of CXCL12/SDF1 by hepatic carcinoma-derived CAFs attracts monocytes into the tumor stroma and engages their differentiation into MDSCs in an IL-6- and STAT3-dependent manner [207]. MDSC-promoting factors (e.g., IL-6, VEGF, M-CSF, CXCL12, CCL2) can also be produced by pancreatic stellate cells, described as CAFs precursors [85]. Similar results were also obtained in murine liver tumor models, where FAP+ CAFs are a major source of CCL2 that enhances the recruitment of MDSCs and predicts poor prognosis of patients with intrahepatic cholangiocarcinoma [208]. In lung squamous cell carcinoma, CAFs have also been reported to promote peripheral CCR2+ monocyte migration via CCL2 and their reprogramming into MDSCs [209]. Another study described similar effects of CAF-secreted CXCL16 on monocytes in triple-negative breast cancers [210].
Immune checkpoint receptors and ligands respectively expressed at the surface of T-cells and tumor cells, have clearly emerged as one of the main contributors to T-cell dysfunction within the TME. In particular, PD-L1 and PD-L2, two members of the B7 family of co-stimulatory/co-inhibitory molecules expressed by a large variety of cancer cells, engage their receptor PD-1 expressed on T-cells. This interaction strongly counteracts T cell receptor (TCR) signaling and CD28-co-stimulation [211], which result in the inhibition of T cell activation, proliferation, and functions. As such, PD-L1/PD-1 blocking antibodies now receive great attention in the field of tumor immunotherapies, especially in melanoma, lung, and renal cell carcinomas [212].
Very interestingly, several studies have now demonstrated that CAFs can express some of these immune checkpoint molecules. For example, CAFs from renal, colon, and lung cancers or melanoma can express programmed PD-L1, PD-L2, or CD276 (also known as B7-H3) [178, 197, 213–216], with potential participation in T cell exhaustion. Interestingly, the expression of some immune checkpoint ligands, especially OX40 ligand (OX40L)/CD242 and PD-L2, by FAP+ CAFs also allows their long-term interaction with Tregs, at least in vitro [61]. In parallel, CAFs can contribute to the expression of these immune checkpoints by other cell populations present within the TME, through their various production of cytokines or exosomes. As such, in pancreatic cancer, CAFs have been reported to increase the expression of PD-1, cytotoxic lymphocyte-associated antigen-4 (CTLA-4), lymphocyte-activation gene-3 (LAG-3), and T cell immunoglobulin and mucin-domain containing-3 (TIM-3), on both CD4+ and CD8+ T cell [195]. Similarly, in breast cancer, FAPHIGH ECM-myCAF can recruit Tregs and increase PD-1 and CTLA-4 expression at their surface [70]. Moreover, CAF-secreted IL-6 can induce PD-L1 expression on TANs in a STAT3-dependent manner [102]. Importantly, CAF can also promote the expression of immune checkpoint ligands by tumor cells. For example, CAF-secreted CXCL5 was involved in the expression of PD-L1 on mouse melanoma and colorectal tumor cells in a phosphatidylinositol 3-kinase (PI3K)/AKT-dependent manner [217]. Similarly, in lung adenocarcinoma, CXCL2 secretion by αSMA+ CAFs increases PD-L1 expression by tumor cells [218] and αSMA+ CXCL5-secreting CAFs are positively correlated to PD-L1 expression by melanoma and colorectal carcinoma tumor cells [217]. Additionally, in human breast cancer, CAF-derived exosomes containing miR-92 decrease the expression of large tumor suppressor 2 (LATS2), and secondarily promotes the nuclear translocation of yes-associated protein 1 (YAP1) and its binding to the enhancer region of PD-L1 to promote its transcription within tumor cells [219]. Nevertheless, it is crucial to emphasize that further studies are clearly needed to clarify both the mechanisms of CAF-induced immune checkpoint expression by the diverse population present in the TME and the influence of immune checkpoint ligand expression by CAF on the T cell-mediated anti-tumor immune response.
In summary, CAF can shape the adaptive antitumor immune response by switching CD4+ Th lymphocytes polarization from a Th1 to a Th2 phenotype, affecting Tregs and Th17 cells generation, affecting CD8+ T cell functions, modifying the ECM and T cell migration, by affecting MDSCs generation or through their effect on immune checkpoint receptors/ligands expression.
Based on the capacities of CAFs to impair the anti-tumor immunity, and more generally exert pro-tumorigenic effects, the development of therapeutic strategies to target these cells in the TME is very seductive to improve the antitumor immune response and more generally may represent a great therapeutic advance in the fight against cancer. Several strategies are thus being explored in preclinical and/or clinical studies [220, 221] and mainly rely on depletion of CAFs, targeting of CAFs surface markers, restoration of their quiescent phenotype, targeting of CAFs-effector molecules, or targeting of CAFs-associated ECM remodeling [23] (Figure 3 and Table 1). Of note, it is also important to consider that the specificity of these therapeutic strategies is a real challenge. In other words, challenging research is needed for the development of anti-CAF therapies capable of specifically modulating CAFs activity without side effects on their normal counterparts, as normal fibroblasts can also be considered, under certain circumstances, as factors that limit tumor growth and invasiveness.
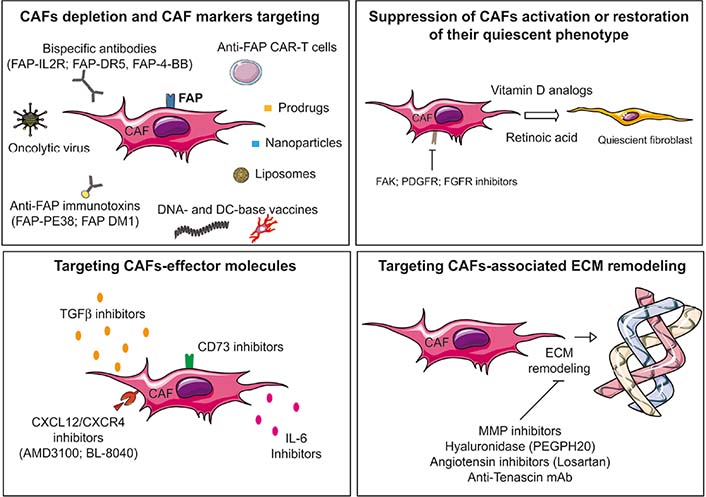
Schematic representation of the current main strategies to target CAFs. Several strategies are being explored in preclinical and/or clinical studies to target CAF-associated immunosuppression such as depletion of CAFs, restoration of their quiescent phenotype, targeting of CAFs-effector molecules or targeting of CAFs-associated ECM remodeling. FAK: focal adhesion kinase; FGFR: FGF receptor
Examples of clinical trials targeting CAFs
| Strategy | Approach | Indications | Combination | Trial ID |
|---|---|---|---|---|
| CAF depletion | Anti Nectin-4 and FAP CAR T cells | Nectin4-positive advanced malignant solid tumors | - | NCT03932565 |
| FAP-IL-2R (R06874281) | Advanced or metastatic melanoma | Anti-PD-1 | NCT03875079 | |
| Unresectable advanced and/or metastatic renal cell carcinoma | Anti-PD-L1 ± anti-VEGF | NCT03063762 | ||
| Metastatic pancreatic ductal adenocarcinoma | Chemotherapy or anti-PD-L1 | NCT03193190 | ||
| Breast cancer | Anti-Her2; anti-EGFR | NCT02627274 | ||
| FAP inhibitor (talabostat/BXCL701) | Advanced solid cancers | Anti-PD-1 | NCT04171219 | |
| Suppression of CAF activation | Vitamin D | Cervical/uterine cancer | Radiation or anti-PD-1 | NCT03192059 |
| Metastatic pancreatic ductal adenocarcinoma | Chemotherapy or anti-PD-1 | NCT02754726 | ||
| Targeting CAF-effector molecules | CXCR4 antagonist motixafortide (BL-8040) | Metastatic pancreatic cancer | Anti-PD-1 | NCT02826486 |
| CD73 blockade | Advanced solid tumors | Anti-PD-1 | NCT02754141 | |
| Targeting CAF-induced ECM remodeling | Pegylated recombinant hyaluronidase (PEGPH20) | Pancreatic cancer | Chemotherapy | NCT01839487 |
| Gastric, gastroesophageal, or esophageal cancer | Chemotherapy or anti-PD-L1 | NCT03281369 |
CAR: chimeric antigen-receptor; EGFR: EGF receptor
To date, CAF-depleting therapies have been mainly focused on strategies targeting cell surface markers. Based on a pioneer study demonstrating that FAP genetic depletion in mouse models causes rapid necrosis of both Lewis lung tumor cells and stromal cells in an IFNγ, TNF-α and CD8+ T cells-dependent manner [222, 223], many direct CAFs depletion strategies have been developed to target this marker [224, 225], such as vaccination approaches or CAR T cells. For example, an oral DNA vaccine targeting FAP has successfully demonstrated its ability to induce CD8+ T cell-mediated killing of CAF and to suppress primary tumor cell growth and metastasis of colon and breast murine carcinoma [226]. Murine LL2 (lung cancer), B16F10 (melanoma), and CT26 (colon cancer) tumor cells modified to express FAP, used as a whole-tumor cell vaccine, can induce antitumor immunity against both tumor cells and CAFs, with a notable enhancement of CD8+ T lymphocytes infiltration and a decrease of immunosuppressive cell accumulation within the TME [227]. Similarly, in murine melanoma models, the vaccination-based depletion of FAP+ stromal cells has been linked to the reduction of immunosuppressive cell frequencies and functions, resulting in a robust CD8+ T cell response and prolonged survival of melanoma-bearing mice [223]. More recently, a synthetic consensus sequence approach to provide MHC class II help was used to develop a FAP DNA vaccine, which was shown to synergize with other tumor antigen-specific DNA vaccines to enhance CD8+ and CD4+ antitumor immunity [228]. A FAP vaccine using a modified vaccinia Ankara vector combined with cyclophosphamide also significantly enhanced anti-tumor immune response decreased Tregs infiltration, and prolonged the survival of 4T1 tumor-bearing mice [229]. Furthermore, a DC vaccine that encodes an A20-specific short hairpin RNA (shRNA) to enhance DC function, targets FAP and the tumor antigen tyrosinase-related protein 2 (TRP-2), has demonstrated its ability to enhance tumor infiltration of CD8+ T cells and to induce robust FAP- and TRP-2-specific T-cell responses in a B16 melanoma model [230]. More innovative approaches such as the fusion of DCs with FAP+ CAFs have been developed, for example in H22 mouse hepatoma models, and can efficiently stimulate T cell-mediated immune response in vitro and inhibit the growth of H22 xenografts in vivo [231]. Similarly, in colon, melanoma, lung, and breast cancer models, exosome-like nanovesicles derived from FAP-engineered tumor cells have been used as a vaccine that inhibits tumor growth by a cytotoxic T lymphocyte (CTL)-mediated immune response against both tumor cells and FAP+ CAFs [232]. Furthermore, the development of CAR T cells targeting FAP has also shown promising results in murine models [233–235] and in malignant pleural mesothelioma patient-derived xenograft (PDX) models [236] and are now in clinical trials (see [237] for review). For example, a clinical trial using a fourth-generation CAR T targeting Nectin-4 and FAP in advanced malignant solid tumors (NCT03932565) is ongoing. Furthermore, recent studies also investigated the use of a bispecific antibody (R06874281/Simlukafusp alfa) which binds to FAP on CAF and IL-2 receptor on immune cells [238]. This approach was designed to stimulate antibody-dependent or T cell-dependent cellular cytotoxicity against CAFs, increase the pool of CD8+ T and NK cells immune effectors, and reduce Tregs activity [238]. Of note, given its promising preclinical results, several clinical trials are ongoing using R06874281 (e.g., NCT03875079; NCT03193190; NCT02627274; NCT03063762). Similarly, an optimized tetravalent FAP-DR5 bispecific antibody (RG7386) was developed [239], as well as a bispecific antibody targeting FAP and 4-1BB/CD137 [240, 241]. Finally, other FAP-targeting approaches have been developed. For example, the FAP inhibitor talabostat/BXCL701 [242] has been used in a phase II trial as a single agent for patients with metastatic colorectal cancer [242] or in association with cisplatin in melanoma [243] and is currently tested in association with anti-PD-1 therapy in advance solid cancers (NCT04171219). Finally, CAF depleting strategies also include immunotoxin targeting FAP, such as FAP-PE38 [244] or FAP-DM1 [245], FAP targeting oncolytic adenovirus [234, 246], liposomes [247], prodrugs [248, 249], nanoparticles [250]; nanocarriers [251], light-activated nanohyperthermia [252, 253], small molecules such as ABT-263 [254] or anti-FAP antibodies labeled with 131Iodine [255].
In summary, CAFs depleting strategies have been mainly focused on FAP, even if a clinical trial targeting PDGFR, another CAF marker, is ongoing using dasatinib [256]. Nevertheless, it should be noted that, in addition to CAFs, FAP can be expressed by cells present in several tissues, including multipotent bone marrow stem cells or skeletal muscles, with potential side effects of the strategies targeting FAP, as suggested [257], highlighting caution against its use as a universal target. This last point also suggests that more highly selective markers are probably required to improve the precision of CAF depletion-based therapies. In this regard, targeting CD10 and G protein-coupled receptor 77 (GPR77), two markers for a specific CAF subset that correlates with chemoresistance and poor survival in multiple cohorts of breast and lung cancer patients, could be an effective therapeutic strategy, as suggested [63].
Another strategy to restrain CAFs function within the TME relies on the normalization of their quiescent state. To date, this approach mainly uses retinoic acid (a metabolite of vitamin A) or vitamin D [37], even if other approaches exist or will certainly emerge. Indeed, as already mentioned, vitamin A or D deficiency can promote CAF activation [35–37]. Consequently, it was hypothesized that targeting this pathway may enable CAFs conversion back to the normal quiescent state. In this regard, in 2D and 3D PDAC models, all-trans retinoic acid (ATRA) treatment reverts CAFs to a quiescent state together with a reduction of proliferation and increased apoptosis of surrounding pancreatic cancer cells [36]. Similarly, treatment with calcipotriol, a vitamin D receptor ligand, reprogram the tumor stroma to a more quiescent state, which improves gemcitabine delivery in PDAC tumors and ultimately enhances antitumor activity compared to chemotherapy alone [258]. Consequently, several clinical trials are ongoing to evaluate the clinical efficacy of vitamin D analogs, in combination with other treatments, especially immunotherapies. For example, a phase II study is ongoing to evaluate the combination of vitamin D with PD-1 inhibitors and radiation in a patient with advanced and refractory cervical cancer, endometrial carcinoma, or uterine sarcoma (NCT03192059), and treatment with vitamin D in association with chemo- or anti-PD-1-therapies are currently evaluated for patients with pancreatic cancer (NCT02754726). Nevertheless, a recent study in PDAC demonstrated that calcipotriol, a vitamin D3 analog, reduces the tumor supportive activity of CAFs, but at the same time decreases T cell effector functions, which highlights the needed caution with this approach [259]. Finally, targeted therapies that could modulate pathways involved in CAF activation have been developed. For example, the FAK pathway is potentially an important target since it promotes the emergence of a fibrotic and inflammatory TME and is essential for CAF development. As such, FAK inhibitors have demonstrated a synergistic effect with PD-1 inhibitors in PDAC models [260]. Similarly, targeting the Hedgehog signaling [261] pathway has been also considered [262]. Several indirect potential targets are also currently explored. For example, pharmacological inhibition of nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) using GKT137831, a small organic molecule of the pyrazolopyridine dione chemical class, prevents and reverses ROS-dependent myofibroblast activation [263]. Other potential targets are for example PDGFR or FGFR [264].
Because depleting CAFs from the TME is still challenging, targeting the CAF secretome to attenuate their immunosuppressive role in the TME is also an interesting strategy. However, it is important to note that this approach is less specific since the immunomodulatory factors expressed and secreted by CAFs are also expressed and secreted by other cell populations within the TME and by the tumor cells. The importance of TGFβ in the activation of CAFs and its crucial role in their immunosuppressive capabilities makes this cytokine an obvious target. For example, artemisinin inactivates CAFs by the suppression of TGFβ signaling in breast cancer [265] and tranilast (Rizaben), a known suppressor of fibroblast proliferation and TGFβ secretion, has demonstrated a synergistic effect with a DC-based vaccine in C57BL/6 mice bearing syngeneic E-G7 lymphoma, LLC1 Lewis lung cancer or B16F1 melanoma [266]. Consequently, multiple preclinical and clinical trials using TGFβ-targeting drugs (including neutralizing antibodies, ligand traps, small-molecule kinase inhibitors, or antisense oligonucleotides) alone or in combination with immunotherapies or other treatments are ongoing, even if the current results are, at least partly, disappointing [267, 268]. Another potential target is CXCL12. In this regard, a crucial study has demonstrated that targeting CXCL12 from FAP+ CAFs with plerixafor (AMD3100) synergizes with anti-PD-L1 treatment in pancreatic cancer [157]. The immunosuppressive axis driven by CAFs in a CXCL12-CXCR4-dependent manner is also targeted by the CXCR4 antagonist motixafortide (BL-8040) in combination with anti-PD-1 antibodies in phase II clinical trial for patients with pancreatic cancer (NCT02826486). Since CAFs also secrete high levels of IL-6, which negatively affect NK and T cell functions, IL-6 or IL-6 receptor (IL-6R)-targeting agents [269] could also be useful to interfere with CAFs immunosuppressive activity. Finally, as CAFs have been identified as CD73 highly expressing cells [189], blocking this ectonucleotidase is probably a way of choice to hamper CAFs immunosuppressive effects, in synergy with anti-PD-1 or anti-CTLA-4 antibodies [270–272], cancer vaccines [273] or CAR T cells [274]. Accordingly, several clinical trials using CD73 blocking are ongoing in combination with immune checkpoint blockade, targeted- or chemo-therapies (e.g., NCT02754141).
As mentioned earlier, CAF-induced ECM remodeling is an important future that affects immune effector cell recruitment to cancer cell areas [166, 167]. Consequently, targeting the ECM remodeling is a potential therapeutic option to hamper CAFs-mediated immunosuppression. One potential CAF-produced target currently explored is hyaluronan (HA). HA is a large aminoglycan and a key ECM component involved in stromal fibrosis [275]. Mechanistically, HA-enriched TME promotes tumor vasculature compression in a collagen-dependent manner, resulting in tumor hypoxia, and also blocks the delivery of peripheral immune cells or drugs from blood vessels to tumors [276]. Consequently, HA-targeting approaches have been developed, such as PEGPH20, a pegylated recombinant hyaluronidase. PEGPH20 facilitates tumor reoxygenation and the intra-tumoral penetration of chemotherapeutic agents in preclinical models [276–278] and displays therapeutic benefit in association with gemcitabine for patients with advanced pancreatic cancer (NCT01839487) [279]. However, more recent data have demonstrated the poor clinical benefit of this treatment in association with paclitaxel/gemcitabine in patients with HAHIGH metastatic pancreatic ductal adenocarcinoma [280]. PEGPH20 is also currently tested for gastric or esophageal cancers (NCT03281369). Moreover, the angiotensin II inhibitor losartan also displays the ability to decrease collagen and HA production by inhibiting TGFβ, connective tissue growth factor (CTGF), and endothelin-1 (ET-1) profibrotic signals, and consequently improves drug and oxygen delivery to tumors, thereby potentiating chemotherapy and reducing hypoxia in breast and pancreatic cancer models [281]. Nevertheless, to date, the effect of PEGPH20 or Losartan on immune effector cell infiltration within tumors has never been addressed.
TN-C, a glycoprotein expressed in the ECM of several tissues and overexpressed in a variety of cancer tissues is also a potential target [282]. Indeed, recent studies have demonstrated that CAFs express TN-C in many tumors [283] and several antibodies have been engineered to target this protein. For example, F16 and P12 antibodies specific to the alternatively spliced domains of the large isoform of TN-C [284], have been fused with IL-2 to promote CD45+ immune cell recruitment and tested in a xenograft model of human breast cancer [285]. Interestingly, it was also shown that antibody-based inhibition of TN-C in autophagy-deficient breast cancer cells improves their CTL-mediated killing and the efficacy of a single anti-PD-1/PD-L1 treatment [286].
Other potential CAF-produced targets are also actually explored such as MMPs, which greatly influence ECM degradation. However, despite promising preclinical results supporting the use of MMPs inhibitor for cancer treatment, the obtained clinical data have been disappointing [286]. Nevertheless, as more specific MMPs inhibitors are now developed, MMPs targeting will be probably reconsidered, especially to target CAF-secreted MMPs to improve immune responses. In this regard, we have shown in vitro that the inhibition of MMPs secreted by melanoma-associated CAFs improves the NK-mediated killing of melanoma tumor cells [140].
Despite their abundance in the TME, fibroblasts have been ignored over decades, but are now considered a major player in tumor initiation and progression. Meanwhile, an increasing amount of research has revealed their heterogeneity in terms of origins and subsets, which also reflects their pleiotropic functions in tumor growth and the variety of chemokines, cytokines, and other factors secreted within the TME. Additionally, their function in the alteration of the antitumor immune response is now widely recognized, thanks to the extensive efforts which made it possible to grasp their secretome and its complex immunosuppressive network that affect both innate and adaptive immune system. Furthermore, CAFs are now considered targets that can be manipulated through therapeutic intervention, as demonstrated by the numerous clinical trials involving CAF-targeting agents used as monotherapy or in combination with existing treatments. These approaches are also expected to enhance immune effector cell infiltration and cytotoxic functions within the tumor, and to enhance the efficacy of current immunotherapy approaches. Nevertheless, multiple challenges are still ahead, such as the definition of CAFs more specific markers, the precise definition of the different CAF subpopulation functions and their localization during tumor progression, and finally the development of targeting agents that are specific enough to spare normal stromal cells in healthy tissues. Furthermore, it is also important to note that some CAFs subsets exert tumor-inhibiting effects, it is therefore conceivable that, under certain circumstances or tumor tissues, these cells may act as both heroes and villains [287], making this field even more challenging.
3D: three-dimensional
Arg: arginase
CAFs: cancer-associated fibroblasts
CAR: chimeric antigen-receptor
CAV1: caveolin-1
CCL2: C-C motif chemokine ligand 2
Chi3L1: chitinase-3-like-1
CLCF1: cardiotrophin-like cytokine factor 1
CTLA-4: cytotoxic lymphocyte-associated antigen-4
CXCL12: chemokine C-X-C motif ligand 12
CXCR2: C-X-C chemokine receptor 2
DCs: dendritic cells
ECM: extracellular matrix
FAK: focal adhesion kinase
FAP: fibroblast-activation protein
FGF: fibroblast growth factor
FGFR: fibroblast growth factor receptor
FSP1: fibroblast-specific protein-1
HA: hyaluronan
iCAF: inflammatory cancer-associated fibroblast
IDO: indoleamine-2,3-dioxygenase
IFNγ: interferon-γ
IL-1β: interleukin-1β
iNOS: inducible nitric oxide synthase
MCP-1: monocyte chemoattractant protein-1
MCs: mast cells
M-CSF: macrophage colony-stimulating factor
MDSCs: myeloid-derived suppressor cells
MHC: major histocompatibility complex
miR-141/200a: microRNA-141/200a
MMPs: matrix metallo-proteinases
MSCs: mesenchymal stem cells
myCAF: myofibroblastic cancer-associated fibroblast
NK: natural killer
NO: nitric oxide
PD-1: programmed cell death 1
PDAC: pancreatic ductal carcinoma
PDGFRs: platelet-derived growth factor receptors
PD-L1: programmed death ligand 1
PDPN: podoplanin
PGE2: prostaglandin E2
ROS: reactive oxygen species
SDF1: stromal cell-derived factor-1
STAT3: signal transducer and activator of transcription 3
TAMs: tumor-associated macrophages
TANs: tumor-associated neutrophils
TGFβ: transforming growth factor-β
Th: T-helper
TME: tumor microenvironment
TN-C: tenascin-C
TNF: tumor necrosis factor
Tregs: regulatory T cells
VEGF: vascular endothelial growth factor
αSMA: α-smooth muscle actin
JT acknowledges further work that was done by our colleagues in the fields of CAF-dependent immunosuppression and apologizes for the missing works/citations due to space limitations.
The author contributed solely to the work.
The author declares no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by INSERM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Mohammad A. Al-Mterin, Eyad Elkord
Maria Teresa Palano ... Lorenzo Mortara
Mohamad Omar Ashi ... Stéphanie Corgnac
Pengkun Yuan ... Bin Ma
Flora Doffe ... Pierre Savagner
Manisha Singh ... Rachana
Vanessa C. Talayero, Miguel Vicente-Manzanares
Matthew Moghaddam ... Benjamin Bonavida
Soumaya Kouidhi ... Amel Ben Ammar El Gaaied
Max Kam-Kwan Chan ... Patrick Ming-Kuen Tang
Ai Tsuji ... Satoru Matsuda
Sayuri Yoshikawa ... Satoru Matsuda
