Review
Review

Affiliation:
1Department of Pharmacology, Luqman College of Pharmacy, Gulbarga 585102, Karnataka, India
Email: Mohsina.patwekar@gmail.com; rohitsharma@bhu.ac.in
ORCID: https://orcid.org/0000-0002-5043-366X
Affiliation:
2Department of Pharmacognosy, Luqman College of Pharmacy, Gulbarga 585102, Karnataka, India
ORCID: https://orcid.org/0000-0001-5737-5290
Affiliation:
1Department of Pharmacology, Luqman College of Pharmacy, Gulbarga 585102, Karnataka, India
ORCID: https://orcid.org/0000-0003-0015-7414
Affiliation:
1Department of Pharmacology, Luqman College of Pharmacy, Gulbarga 585102, Karnataka, India
ORCID: https://orcid.org/0000-0001-6525-5926
Affiliation:
3Institutes for Systems Genetics, Frontiers Science Center for Disease-related Molecular Network, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China
4King Fahd Medical Research Center, King Abdulaziz University, Jeddah 21589, Saudi Arabia
5Department of Pharmacy, Faculty of Allied Health Sciences, Daffodil International University, Dhaka 1207, Bangladesh
6Enzymoics, 7 Peterlee Place, Novel Global Community Educational Foundation, Hebersham NSW 2770, Australia
ORCID: https://orcid.org/0000-0003-0088-0565
Affiliation:
7Department of Biomedical Engineering, Sathyabama Institute of Science & Technology, Chennai 600119, Tamilnadu, India
ORCID: https://orcid.org/0000-0001-7513-8109
Affiliation:
8Department of Rasashastra and Bhaishajya Kalpana, Faculty of Ayurveda, Institute of Medical Sciences, Banaras Hindu University, Varanasi 221005, Uttar Pradesh, India
Email: Mohsina.patwekar@gmail.com; rohitsharma@bhu.ac.in
ORCID: https://orcid.org/0000-0002-3682-3573
Explor Med. 2023;4:637–663 DOI: https://doi.org/10.37349/emed.2023.00166
Received: February 26, 2023 Accepted: April 19, 2023 Published: September 15, 2023
Academic Editor: Haijun Yu, Chinese Academy of Sciences, China
Complex enzyme interactions play a role in the spread of cancer, a process fueled by unregulated cell proliferation. DNA topoisomerases, which are important for fixing DNA topological problems, have drawn a lot of interest as potential targets for anti-cancer medications. Cancer treatment, which includes radiation, surgery, and chemotherapy, tries to control cell survival, demise, and mobility, which are mediated by ion transportation across cell membranes via channels and carriers. The malignant transition is characterised by altered channels and carriers. Chemoresistance, which commonly develops after chemotherapy, denotes decreased therapeutic effectiveness against cancer progression. Chemosensitizers are used in combination with anti-cancer medications to overcome this resistance, particularly against adenosine triphosphate (ATP)-binding cassette (ABC) transporters including P-glycoprotein, multidrug resistance-associated protein 1 (MRP1), breast cancer resistance protein (BCRP). Effective targets for treatment are transcription factors, which play a key role in the development of cancer. With the use of interactions with receptors, enzymes, ion channels, transporters, and TFs, nanotechnology improves the safety of tumour localization, treatment, and diagnostics. As a result of mutations or altered signalling, rat sarcoma (RAS) proteins regulate signalling, which is essential for both healthy growth and the development of cancer. Rational treatments that target RAS pathways have the potential to inhibit the growth and spread of tumours. New treatments are still being developed, and they are showing promise in clinical settings. The roles of receptors on tumour cells, their significance for cancer therapy, and recent advancements in preclinical and clinical research are all included in this overview.
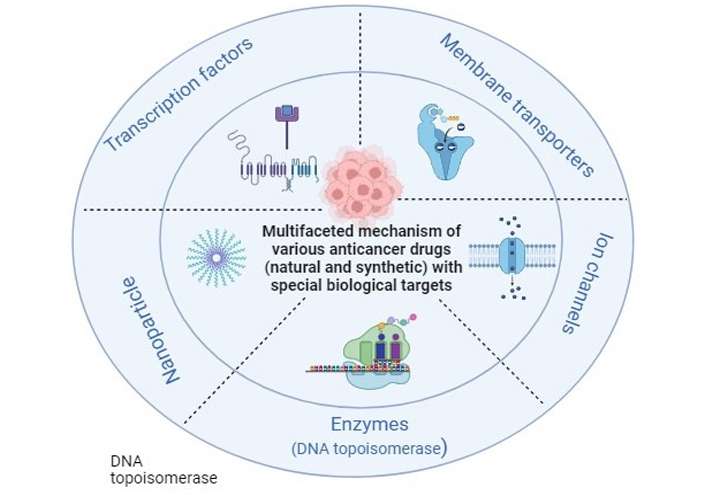
Multifaceted mechanism of various anticancer drugs (natural and synthetic) with special biological targets: enzymes (DNA topoisomerase), ion channels, membrane transporters, transcription factors, nanoparticles
According to the WHO, cancer acquired second place as the most common disease. Uncontrolled division of abnormal cells is a significant cause of cancer. Surgery, chemotherapy, radiation therapy, laser therapies, and a combination of therapies are used for cancer treatment. Despite many promising therapies, there are many failures in chemotherapy treatment because of drug resistance [1]. A high number of patient tumours have developed medication resistance. So, drug conflict is a significant difficulty in cancer management. This review focus on studying different mechanisms to overcome drug resistance and other problems related to chemotherapy. The review highlights many mechanisms involved in cancer treatment (Figure 1). To deal with the topological issues brought on by the double helix, cells contain a group of enzymes generally referred to as DNA topoisomerases [2]. Enzymes called DNA topoisomerases might fix topological issues with DNA that develop throughout biological mechanisms [3]. Finding alternative biological targets which are either exclusive to a specific tumour type or related to a state of cancer growth and have reduced widespread physiological components is one of the main difficulties in oncology. A complex field of study has recently developed around the functional use of ion channels in cancer cell treatment [4]. Various sorts of channels across all documented subfamilies of selective ion channels have been deeply engaged in reproducing cells, especially cancer cells [5]. The research has a significant amount of work on the function of potassium (K+) channels. Among the most effective approaches now seems to be the use of in vivo small interfering RNA (siRNA, interfering RNA) in cancer and other disciplines [6]. Changes in membrane transport are the common mechanism whereby cancerous cells become resistant to chemotherapeutic drugs. Ions and tiny molecules are controlled by membrane transport as they cross biological membranes [7]. Transcription factors (TFs) are essential in chemotherapy, irrespective of the kind of oncogenic variant that arises, equally in vivo and in vitro. Most essential TFs were the subject of extensive research, and human trials of investigational drugs were conducted [8]. Nanotechnology in medicines has considerably grown during the last few decades. The pathogenesis of neoplasm is typically taken into consideration when developing or choosing the size and characteristics of nanoparticles (NPs) used in medicine-targeted delivery [9]. All cancers are brought on by genetic changes. Affected key proteins of particular signalling pathways result from these mutations. The changes that take place in the rat sarcoma (RAS) protein and p53 are two of the most well-known and extensively researched malignant transformation-related changes. The majority of the time, mutations in the genes p53 and RAS cause the appearance of virtually all malignant transformations. Twenty to thirty percent of all human cancers have mutated RAS genes. RAS proteins act as switches that control many processes, including cell division, proliferation, and apoptosis [10]. The “genome guardian,” also known as normal p53 expression, is a crucial molecular regulator of cell proliferation. These proteins’ crucial role in cell growth and death depends on how closely they are related to those processes. Understanding the degree of transformation on RAS and p53, as well as the various biochemical pathways of intracellular signalling that they activate, is crucial for understanding malignant transformation and controlling it, for example by developing new drugs that help eliminate these cells [7].
DNA is a polymer made up of a couple of polynucleotide strands that form a double helical construction and carry genetic information [1]. DNA allows genetic material to be reproduced, repaired, and recombined to preserve genetic integrity while allowing for variation in living creatures [11]. The interweaving of the two anti-parallel, complementary strands causes DNA topological stress. The two strands are entangled around one another every 10.4 base pairs under physiological circumstances [2]. Cells have a series of enzymes known collectively as DNA topoisomerases that help them address the topological problems caused by the double helix [3]. DNA topoisomerases are frequently occurring enzymes that may fix topological issues with DNA that develop during biological processes such as replication, recombination, transcription, repair, and chromatin assembly [12]. By tying or untying DNA knots, the topoisomerase enzyme catenates or decatenates circular DNA super-helical strains. Enzymes called DNA topoisomerases are crucial for maintaining the integrity of the genetic material during transcription, replication, and recombination processes. They control DNA topology. Anti-tumour medications that inhibit mammalian enzymes are frequently used. They prevent the DNA relegating step of the catalytic reaction, which stimulates DNA cleavage, to stabilize topoisomerase-DNA cleavable complexes [1]. Prokaryotes and eukaryotes have been shown to have two different types of DNA topoisomerases. They are:
(1) Type I DNA topoisomerases (topo I): mechanism of action by transient double-strand breaking into one strand of DNA [13].
(2) Type II DNA topoisomerases (topo II): mechanism of action by introducing transient single-strand breaking into one strand of DNA [14].
DNA topoisomerases have sparked much attention in recent years as they were discovered to be biological targets for various anticancer medicines. Many of these medications were being used in therapeutic settings prior to their lethal effects being related to topoisomerases. Antibacterial agents such as fluoroquinolones [15] and camptothecin (CPT) [16] are great examples.
Two kinds of topoisomerase targeting anticancer drugs exist, each with a distinct mechanism of action (Table 1) [17].
Categorization of topoisomerase targeting anticancer drugs
| Drug mechanisms and classification | Class-I (topoisomerase poisons) | Class-II (topoisomerase inhibitors) |
|---|---|---|
| Mechanism of drug action | These drugs act as intermediaries in the catalytic cycle of the enzyme, blocking the creation of a covalent topoisomerase-DNA network. | These medications interfere with the enzyme’s catalytic action without trapping the covalent compound. |
| Nomenclature and classification of drug types | Class-I medications are sometimes referred to as “topoisomerase poisons” since they convert the enzyme into a strong cellular toxin. | Class-II drugs are assigned as “topoisomerase inhibitors.” |
| Varieties of topoisomerase medications | Class-I medications contain acridines, alkaloids, anthracyclines, ellipticines, epipodophyllotoxins, actinomycin, quinolones, and isoflavonoids. | Class-II medications are fostriecin analogues and coumarin antibiotics. |
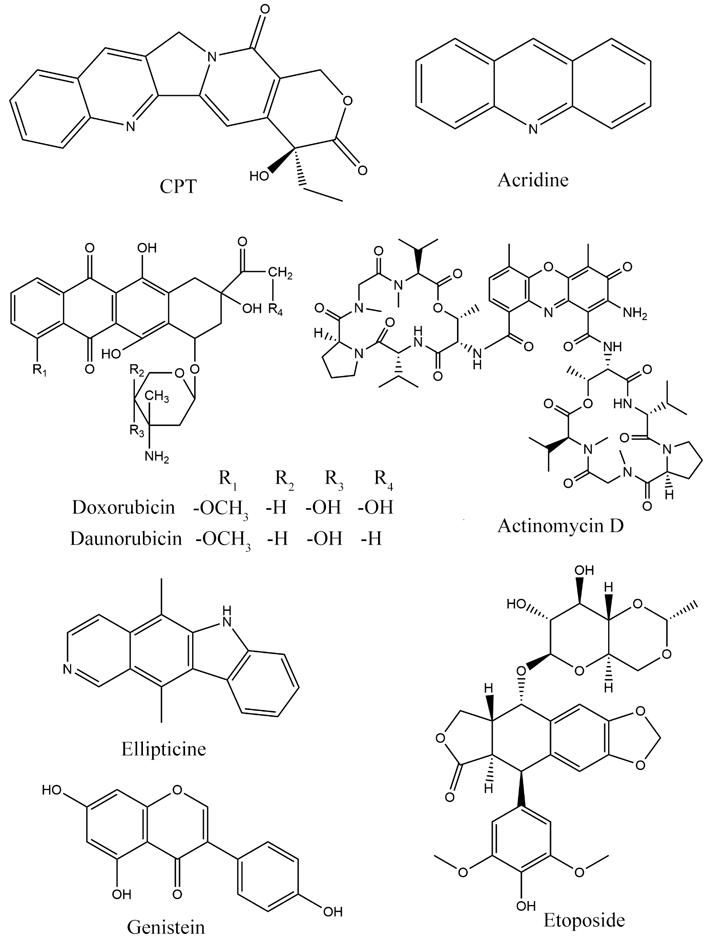
CPT is a cytotoxic alkaloid derived from the Camptotheca acuminata, a Chinese plant that specifically inhibits eukaryotic topo I [18]. In the early 1970s, CPT’s anticancer efficacy was evaluated against leukaemia and lung cancer in clinical studies. It induced myelosuppression and dose-limiting haemorrhagic cystitis [19]. The six-membered lactone rings were essential for anticancer activity and lowered toxicity to increase CPT’s anticancer activity, according to the structural activity investigations of CPT. Several CPT derivatives have been created by altering the parent compound’s A-ring. Topotecan (TPT) [20] and irinotecan (CPT11) [21] are two examples. TPT effectively acts opposite to refractory colorectal cancer, malignant glioma, and head and neck cancers [20]. CPT11 is used to treat breast, and colon gastric cancers, also used against leukaemia and small-cell lung cancer (SCLC) [21].
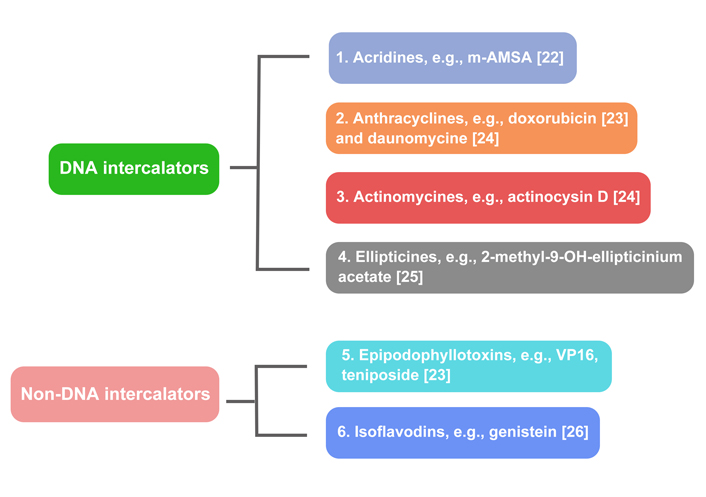
A variety of medications target topo II; they can be distributed into two groups (Figure 2).
DNA intercalators: acridines, e.g., 4’-(9-acridinlylamino)methanesulphon-m-anisidide (m-AMSA) [22]; anthracyclines, e.g., doxorubicin and daunomycin [23, 24]; actinomycin, e.g., actinomycin D [24]; ellipticines, e.g., 2-methyl-9-OH-ellipticinium acetate [25].
Non-DNA intercalators: epipodophyllotoxins, e.g., VP16, teniposide [23]; isoflavonoids, e.g., genistein [26].
Acridine derivatives are topo II focusing intercalative medicines utilized to cure several types of leukaemia. Although m-AMSA effectively treats acute myelogenous leukaemia, it is also hazardous [22, 27].
Anthracycline antibiotics are successfully used to counter many tumours, including SCLC, breast, bone marrow, and ovarian malignancies [23, 24, 28].
Actinomycin is, also called dactinomycin, used in prophylaxis of a variety of cancers, including testicular cancer, Wilms tumour, trophoblastic neoplasm, and some ovarian cancer [24, 29].
It is beneficial in the management of mammary gland tumours. However, the side effects include xerostomia, which is linked to weight loss and renal toxicity, and haemolysis [25, 30].
Testicular cancer, central nervous system tumour, malignant lymphoma, SCLC, and Kaposi’s sarcoma are all targets for epipodophyllotoxins. Secondary melanoma, on the other hand, is commonly linked to treatment [23, 31].
The therapeutic drug concentrations at the diseased site are increased by liposomes’ role as nanocarriers, which also reduces toxic effects on healthy tissues. Liposomes deliver the payloads at the target site. The ability of the nanocarrier to target particular tissues has been improved using a number of strategies, including ligand-targeted liposomes and stimuli-responsive liposomes. Due to the targeting ligand attached to their surface, ligand-targeted liposomes have a higher uptake by the target tissue than stimuli-responsive liposomes, which do not release their cargo until they come into contact with an endogenous or exogenous stimulant at the target site. The liposomes that respond to pathologically elevated levels of enzymes at the target site are the main focus of this review [32].
After coming into contact with an enzyme, enzyme-responsive liposomes release their contents through a number of destabilization processes, including: (1) structural changes to the lipid bilayer; (2) removal of a shielding polymer from the surface and increased cellular uptake; (3) cleavage of a lipopeptide or lipopolymer incorporated in the bilayer; (4) activation of a prodrug within the liposomes.
Many enzymes are induced in a variety of pathological situations, including inflammation, malignancy, and infection. This biochemical response can be leveraged to construct nanocarriers that undergo a structural transformation, allowing the contained payload to be released. Different advantages of this technique, liposomes, polymers, quantum dots, and gold have all been used to create enzyme-responsive NPs [32].
sPLA2 raises different atherosclerosis, cancer, and inflammatory diseases. sPLA2 has also been found to be elevated in breast, prostate, and pancreatic cancers [33]. As a result, sPLA2-sensitive liposomes could be promising vehicles for delivering anticancer drugs to tumour tissues. In particular, prostate cancer patients may benefit from sPLA2-responsive liposomes [34].
MMPs are Ca2+ and Zn2+ built upon proteolytic enzymes that break down the extracellular matrix [34]. In several inflammatory diseases, cardiovascular conditions, and malignancies, these enzymes are overexpressed [35]. In addition to their typical functions in tissue remodelling, angiogenesis, and wound healing, causing the overexpression of these enzymes [36]. MMPs are key components in tumour development, invasion, and metastasis [37].
In the various figure of human malignancies, like breast, colon, ovarian, and bladder tumours, the quantity of uPAs are used [38]. Angiogenesis, tumour development, and metastasis are all aided by uPA [39].
Elastase, a protease found in the azurophil granules of activated neutrophils, has a role in arthritis [40], cystic fibrosis, skin, breast cancer, and lung tumours [41].
PSA is a serine protease abundant in prostate tumour tissue. PSA is detected in prostate cancer patients’ serum, although inactive due to enzymatic inactivation [42]. As a result, the extracellular matrix of prostate tissue that is PSA positive is an excellent catalyst for the release of anticancer drugs [43].
Cathepsin B, a papain family lysosomal protease, is overexpressed in a number of clinical conditions and malignancies, including lung, brain, breast, and prostate cancer [44].
Ion channels in the plasma membrane (PM) play a role in almost all fundamental cellular functions as well as the malignant phenotype of cancer cells. The review highlights the function of ion channels in cancer in relation to how they relate to the recognized characteristics of cancer: (1) insensitivity to antigrowth signals; (2) self-sufficiency in growth signals; (3) sustained angiogenesis; (4) infinite capacity for replication; (5) avoidance of programmed cell death (apoptosis); (6) tissue invasion and metastasis. According to recent studies [4, 45], the contribution of particular ion channels to these characteristics varies for various cancer types. Ion channel expression, function, and regulation must therefore be evaluated for each cancer in order to determine the significance of ion channels as targets for cancer diagnosis and treatment. Although knowing this genomic type of cancer can open the path for more tailored therapies, a more traditional chemotherapeutic technique is to uncover biological factors critical to cancer cell physiology. Because the majority of the recognized pharmacological objectives are involved in the cell cycle mechanism, which is effective in every reproducing cell, inhibiting it can have substantial consequences. As a result, finding alternative biological targets that are either exclusive to a particular tumour type or related to a state of cancer growth and have fewer widespread physiological components is one of the main difficulties in oncology. Recently, research on the use of ion channels in cancer cell treatment has developed into a complex field [4]. In neoplastic cells, widespread changes in the functioning of virtually any type of ion channel have been discovered, which simultaneously define particular tumour growth stages and regulate their fate [45]. The main reasons for the extensive range of ion channels that have been linked to tumour growth. To begin with, ion fluxes control the majority of cellular activities adopted in cancer growth, such as cell volume oscillations [46]. Several varieties of ion channels have been linked to cancer’s so-called hallmarks [47]. Over 500 distinct membrane ion channels and transporters may be expressed thanks to instructions included in the human genome. Differential representation of such a vast array of genes is necessary to choose on appropriate control of ionic currents in a variety of physiological settings. Numerous studies have noted the occurrence of odd ion channel expression in developing cells [44–48]. The investigation of the common ion channel patterns in stem cells is ongoing [48]. Much research has already been done on cancer cells, which can have quite a different membrane electrical characteristic from their non-pathological counterparts [5].
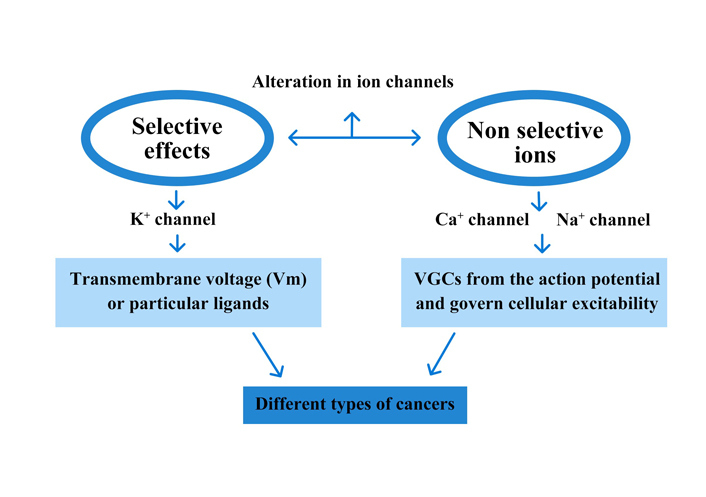
In proliferating cells, particularly cancer cells, different types of channels from all acknowledged subfamilies ion channels are actively involved. The character of K+ channels plays a prominent part in the literature [5]. Different K+ channel subfamilies have been associated with carcinoma. Any type of K+ channel may interfere in some way with cell growth. Additional cell features and external conditions influence specific contributions whenever the membrane voltage reaches a certain point. Excitable tissues that can be triggered by voltage-gated channels (VGCs) include nerve and muscle cells in the heart and other organs [voltage-gated potassium channel (Kv)] (Figure 4) [49]. It is still unknown if Kvs are dispatched just in tumour cells or if they also play an act in normal tissues, at least during some levels of the biological clock of their life process. In reality, no ion channel portrait has been done in the matching of regular original tissues [50, 51]. Prostate cancer has been connected to the non-selective transient receptor potential (TRP) family of ion channels [52]. Other cation channels, such as non-selective cation channels and calcium (Ca2+) and sodium (Na+) channels, have been found in tumour cells. A Ca2+ entry site in cancer cells is T-type Ca2+ channel [53].
Ion channels are sheath proteins that transition between nonconductive (‘closed’) and conductive (‘open’) conformational categories to govern passive ion fluxes. Although alternative mechanisms are possible, Vm or particular ligands are frequently used to induce a ‘gating’ process (activation). There are many different forms of ion channels, each including a particular degree of ion collection and kinetic lineaments, allowing for a wide range of functional flexibility. VGCs form the action potential and govern cellular excitability. They can also stimulate exocytosis and muscle contraction by regulating transmembrane Ca2+ fluxes. Ligand-gated channels usually exert synaptic functions. Cell volume, transepithelial ion outflow, sensory transduction, synaptic action, and many more physiological functions are controlled by multiple channel types’ combined action. Channels close when the stimulus is interrupted (deactivation). Otherwise, they frequently transition to a non-conductive phase through a measure known as an adolescent for VGCs and desensitization for alternative channel categories. Cells can use allosteric regulators or phosphorylation to control the energy distinction among states and the speed of transition processes. Ion channels are frequently formulated from multiple subunits or domains that envelop low or high conduction pathways from a structural standpoint. The VGC family, which includes most of the ion channels described, is briefly discussed [54, 55]. Communication with other subunits or partner proteins is controlled by the N-terminus, while the intracellular regions provide consensus sequences for phosphorylation. The quadruplicate elements that isolate the pore in Na+ and Ca2+ channels follow the same structure, except that the four elements are repeating domains of a continuous polypeptide rather than independent subunits. A K+ channel subunit is homologous to each domain. VGCs are intrinsically voltage-dependent, meaning that Vm controls their conformation, whereas auxiliary proteins (subunits) control accessory characteristics. Many genes code for mammalian VGCs and a consistent nomenclature has been assigned. Classic names, which identify large functional properties less frequent to various gene substances, are still utilized in describing cellular currents. For example, abounding K+ Channels of the Kv 1 and Kv 10 molecular tribe produce late outward rectifying currents that stimulate depolarization and usually control the action potential’s repolarisation state. KDR is the aggregate name for several channels.
The inward rectifying K+ [inwardly rectifying potassium (IRK)] channels have an evolutionary connection to the VGC clan. They’re similarly composed of four subunits, in which every individual has only a pair of transmembrane domains (M1 and M2), which are connected by a pore loop and are comparable to the S5 and S6 of VGCs [56]. Finally, the TRP gene branch is connected to the VGCs, keeping the above-mentioned structural characteristics. The homo- or heterotetrametric channels are formed by the subunits. TRP canonical (TRPC), TRP vanilloid (TRPV), TRP melastatin (TRPM), TRP ankyrin (TRPA), TRP polycystin (TRPP), and TRP mucolipin (TRPML) are six TRP families with permeability ratios that differ from zero to very extreme between Ca2+ and monovalent cations. These proteins are abundant in mammalian tissues and have been associated with a variety of human illnesses, along with cancer. The physiological significance of changed TRP channel expression in tumours is currently being researched [57].
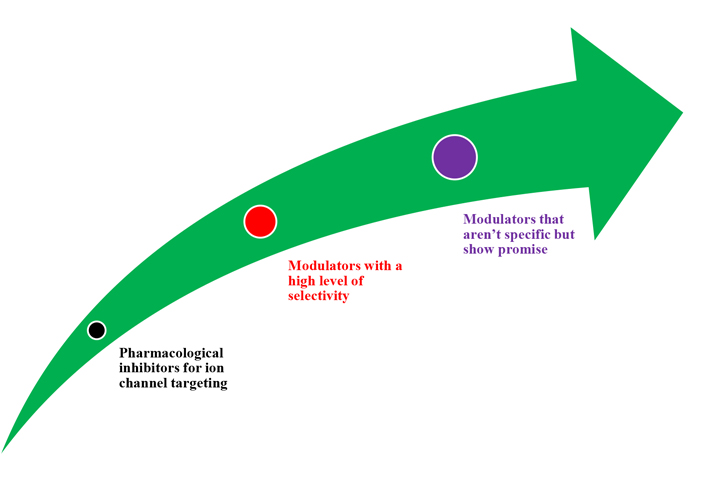
Depending on the pathophysiological role a specific channel plays in a tumour, ion channel targeting should include both inhibition and activation techniques (Figure 5). Peptide toxins, small chemicals, and antibodies are among the many agents available today that modulate ion channel activity. Many of these compounds can be active anticancer medicines. There are not many cancer growth inhibitors that are safe and effective enough to get into clinical trials. Like some other medicine, channel modulators might have adverse side effects. Human ether-a-go-go-related gene (hERG) K+ channel barricade, for example, has been demonstrated in many cases to produce cardiac arrhythmia (long-QT syndrome) [58, 59].
New highly selective chemicals would be needed to modulate ion channel activity and reduce side effects [6]. Aside from recent small compounds and antibodies, siRNA could soon provide a choosy gadget for modulating ion channel action and many other proteins. In vivo siRNA application in cancer and other fields appears to be one of the most promising techniques nowadays [60].
Old modulators with low specificity should not be completely ruled out of clinical trials. Many of them have shown anti-tumour action in vitro. For example, neuroblastoma cells have been demonstrated to be inhibited by dendrotoxins and quercetin [61, 62]. In addition to stimulating the growth-inhibitory action of the cytostatic medication, tamoxifen, dequalinium, and amiodarone have shown substantial hindrance action on human prostate cancer, breast cells, and colon cancer cell development [63]. Other natural antidepressant compounds, such as those found in Hypericum perforatum L. extracts, also have anticancer effects, owing to ion channel modulation [64]. The long-time practice of Ca2+ channel blockers such as verapamil, an antihypertensive medicine, has already yielded important data. Prostate cancer benefit has a convincingly inverse connection, according to a thorough prospective investigation of the long-term risk of prostate cancer in male Ca2+ channel blocker users. At least in persons with a specific genetic background, able to decrease the risk of prostate cancer [65, 66].
The most prevalent method by which tumour cells turn resistant to antineoplastic medicines is a change in membrane transport, which is one of the most significant difficulties in current cancer treatment. An emphasis on understanding how medications are delivered into cells has begun, and this should be recently been placed, and this should continue to aid efforts to solve these issues [7].
Membrane transport is a phrase utilized in cellular biology to characterize a group of systems that control the movement of ions and tiny molecules across biological membranes; these membranes are composed of lipid bilayers that include proteins. Selective membrane permeability, a characteristic of biological membranes that enables them to segregate components of various chemical natures, controls passage through the membrane. In other words, certain chemicals can travel through them but not others [67]. In the dynamic connection between cellular integrity, localization, the removal of toxic metabolites, or the admission of essential nutrients, the expression of drug transporters and these processes have become obvious [68]. Furthermore, representative of the adenosine triphosphate (ATP)-binding cassette (ABC) [69] and solute carrier (SLC) transporter [70] superfamilies have been identified as significant predictors of many medicines’ therapeutic responses.
Chemotherapy is the preferred treatment for many forms of cancer. Chemotherapeutic medicines can lose their effectiveness over time due to the development of resistance involving a group of membrane proteins. These multidrug transporters keep intracellular drug concentrations below the cell-killing threshold by removing harmful compounds from the cell. These transporters belong to the superfamily of ABC proteins, which are found in all organisms, from bacteria to humans. According to clinical studies, the overexpression of particular ABC transporters, recognized as multidrug resistance (MDR) proteins, is linked to the MDR phenotype in malignancies [69].
MDR is the opposite impact of a wide range of structurally and mechanistically unrelated medicines across the membrane. Because cancer can reject and reduce intracellular drug concentrations chemotherapy drugs and diminish intracellular drug altitude, this process is the most frequent cause of chemotherapy for cancer failure. It has been determined that members of the ABC family are involved in this procedure [71]. According to numerous clinical studies, the overexpression of particular ABC transporters is well-known as MDR resistance phenotype in malignancies [72]. P-glycoprotein [P-gp, MDR protein 1 (MDR1), ABC sub-family B member 1 (ABCB1)] is the first known ABC transporter [73], and it is identified to be answerable for the most common mechanism in MDR [74]. Following the cloning and characterization of MDR1, it became clear that other efflux pumps are tangled in transport-related drug resistance as well. MDR-associated protein 1 (MRP1), ABC sub-family C member 1 (ABCC1), or the MDR protein [75] and mitoxantrone resistance protein [multixenobiotic resistance (MXR)/breast cancer resistance protein (BCRP), ABC sub-family G member 2 (ABCG2)] [76] are two additional ABC transporters that have been firmly linked to tumour MDR. The review examines ABCB1, ABCC1, and ABCG2, the three most therapeutically significant ABC transporters, which produce MDR during cancer treatment.
The first human ABC transporter found was called P-gp, and it was discovered to be a glycoprotein that modulates drug permeability [73]. By MDR, the gene encoding P-gp is located on chromosome 7 at q21 and contains 28 exons, and is 170 kDa protein [77]. P-gp exports xenobiotics and hydrophobic compounds that are neutral or positively charged in order to protect cells from cytotoxicity [78]. It was shown how important P-gp is for maintaining the blood-brain barrier (BBB) [10]. Chemo-resistance is a result of the ability of glycoprotein to efflux chemotherapeutic medicines and reduce intracellular drug altitude [79]. Furthermore, poorly differentiated and invasive cells are more likely to express P-gp [80]. P-gp is expressed most strongly in the most aggressive soft tissue sarcomas [81].
Alkaloids have a heterocyclic ring and their main structural component is one or more basic nitrogen atoms. P-gp inhibitory activity requires such a nitrogen atom [82]. (1) Quinine is effective in reversing doxorubicin resistance [83]; (2) berberine is a Ca2+ channel blocker [84]; (3) P-gp mRNA is able to be downregulated by berberine [85].
Depending on how many isoprene units their parent structure contains, terpenoids are divided into a variety of types. To be effective P-gp inhibitors, terpenoids must include carbonyl groups at C2, C3, and C8 as well as lipophilic compound substituents at C6 [86]. Sooneuphanone D, a P-gp inhibitor isolated from Euphorbia soongarica, has a substantially more impressive MDR cancellation effect than verapamil [87].
According to several classifications of terpenoids acute lymphoblastic leukaemia (ALL), retinal progenitors, fibrosarcoma, hepatic metastases, glioblastoma, gastric carcinoma, non-SCLC, and myeloma are among the multidrug-resistant malignant cell disorders associated with ABCG2 [87]. MDR results from co-expression, which necessitates the use of multi-transporter inhibitors to improve clinical outcomes [88]. Even though the MDR pathways involving ABCG2 are understood mainly, therapeutic trials, including ABCG2 inhibitors, have yielded mixed outcomes [89].
Naringenin and its derivatives were tested in cancer cells to see if they could reverse MDR [90]. Chrysin has an inhibiting effect on ABCG2 [91].
Guajadial is a natural meroterpenoid derived from the Psidium guajava’s leaves that have been shown to have anti-tumour action, particularly in breast cancer cell lines [92].
Tariquidar belongs to the tertian generation of P-gp inhibitor capable of inverse doxorubicin and vinblastine resistance in breast cancer patients [93].
ABCC1 was discovered in 1992 in human cell lines of SCLC that exhibited drug resistance without overexpression of P-gp [94]. Endometriosis, lymphoblastic leukaemia, acute myeloblastic leukaemia, head tumour and neck cancer, melanoma, neuroblastoma, breast, renal, prostate, and thyroid cancer are linked to ABCC1 overexpression [95].
ABCC1 was discovered to be inhibited by triple flavanol stilbenes isolated from Sophora alopecuroides L [96]. Timosaponin A-III (TAIII), a saponin obtained from the rhizome of Anemarrhena asphodeloides, reversed the twain P-gp and ABCC1-induced drug resistance [97]. Compared to chrysin, 3-methoxy-chrysin, and 5,7-dihydroxy-4’-fluoro-flavone, verapamil is less efficient and risky [98].
The effect of SLC, which are often influx or bi-directional transporters, on cancer treatment is not thoroughly studied. Ion channel transporters, passive transporters, and exchangers are exposed in intracellular organelles or on the PM, among which around 400 SLC transporter genes are recognized and distributed in 55 families (e.g., mitochondrial or vesicular transporters) [99]. The SLC superfamily regulates the shipping of a broad range of compounds and various supplements and medicines [100]. To improve cancer treatment outcomes, researchers must comprehensively define the action of SLC drug carriers in organs crucial for anticancer medication disposition, like the liver, intestine, and kidney. Furthermore, through a better understanding of the changing phase of SLC transporters in diverse cancerous cells, we can create new cancer-treatment techniques. Down-regulation of specific critical transporters for cancerous cell existence, for example, could be used to produce transporter-targeted chemotherapy. The following are the primary subfamilies of SLC members of studied cancer therapy to varying degrees (Figure 6) [101–103].

Primary subfamilies of SLC members about cancer therapy to varying degrees. OCT: organic cation transporters; OAT: organic anion transporters; OATPs: OAT polypeptides; MATEs: multidrug and toxin extrusion proteins
The molecular foundation of neoplastic transformation has been a critical and fruitful topic of research for many years, regardless of what kind of oncogenic mutation occurs. According to core ideas which have evolved, TFs perform a very vital part in the transformation. As a result, significant effort has been made to therapeutically inactivate certain TFs in neoplastic cells, both in vitro and in vivo. A handful of these TFs have undergone extensive investigation and human testing. In other instances, the data is recent but expanding fast. TFs include, but are not limited to, the activator protein-1 (AP-1) family, the myc family, and the myb proteins. The nuclear dimeric proteins known as AP-1 TFs are composed of the Fos, Jun, musculoaponeurotic fibrosarcoma oncogene homolog (MAF), and activating TF (ATF) subunits. The activity of AP-1 is controlled by a variety of stimuli, and hormones, including growth factors, and environmental variables. AP-1 affects cellular processes including apoptosis and cell cycle progression [8, 104].
As a result, similarly, the myc and myb TF families control cellular proliferation and abnormal activity has been seen in a variety of human malignancies [105].
Steroid receptors, more likely retinoid receptors, are some other types of nuclear TFs involved in various cancers. The stimulation of retinoid receptors can cause cellular differentiation, which is used to treat cancers such as ALL and neuroblastoma. The use of pharmacological agents to target these receptors for cancer therapy as well as prohibition is still being researched [106–108]. There is a large corpus of knowledge about translocations involving TFs. More of these fusion proteins have an abnormal transactivating function, which contributes to malignant phenotypes [109, 110].
Signal transducer and activator of transcription (STAT) 3, STAT5, nuclear factor kappa B (NF-kB), and hypoxia-inducible factor 1 (HIF-1) are the TFs in question. All four proteins are found in the cytoplasm in a dormant state until they are triggered by particular stimuli and translocated to the nucleus, where they are attached to DNA and also manage gene expression. New cancer medicines that target these proteins have much promise. When selecting a suitable molecular target against which to direct innovative treatments, there must be clear evidence that the putative target’s activity is necessary for ontogenesis [111].
Inhibiting the functional domains of TFs and the post-translational changes that affect their action are two techniques for identifying and removing TFs. In contrast to kinases, which have the deepest connecting compartments for tiny fragments, TFs bear ample surface space for protein-DNA and protein-protein communication, making them challenging to target. One more difficulty with suppressing TFs is that they are primarily found in the nucleus, making rehabilitative procedures less available. Regardless of these challenges, important growth has been made in the creation of more potent medicines and improved delivery systems that significantly target tumour cells [112].
STAT are TFs that are cytoplasmic until tyrosine phosphorylation is applied. After activation, STATs balance genes that control cell growth, differentiation, and even existence by designing dimers in the nucleus. Inhibitory molecules such as protein tyrosine phosphatases, protein suppressors of effective STAT proteins, and inhibitors of cytokine signalling proteins maintain STAT signalling transient and tightly controlled in normal cells. Nevertheless, tumour cells usually acquire enduring STAT activation due to upstream tyrosine kinase hyper-activation or overexpression, or failure of action of negative regulators. Arresting STAT3 function may be a helpful antineoplastic treatment because stimulated STATs, especially STAT3, stimulate the production of genes essential in cell multiplication but also in survival [113]. Reduced STAT3 expression by RNA interference (RNAi), which reduces STAT3-specific gene expression and lowers tumour development in vivo, is one technique for inhibiting STAT3 activity [114, 115]. NPs, which have reduced toxicity, better stability, and higher tumour accumulation, have been used in current delivery systems. STAT3 siRNA NPs reduce STAT3 expression and trigger apoptosis, which makes lung neoplastic tissues more sensitive to antineoplastic drugs [116]. Another is the block of STAT3 DNA binding via decoying oligodeoxynucleotides (ODNs). In a phase 0 clinical trial involving head and neck neoplastic patients, a STAT3 ODN effectively reduced STAT3-specific gene expression [117].
A protein complex called NF-kB serves as a TF in cancer. Five proteins, reticuloendotheliosis viral oncogene homolog (Rel, c-Rel), RelA (p65), RelB, NF-kB1 (p50), and NF-kB2 (p52), each contribute a Rel homology domain and, through homo- and heterodimerization, form functional TFs. In unstimulated cells, NF-kB is kept inactive in the cytoplasm by inhibitors of NF-kB (IkB). The cytokines [tumour necrosis factor (TNF), interleukin (IL)-1b], and also pathogen-associated molecular model cause IkB to be degraded by proteasomes, allowing NF-kB to be released and translocated to the nucleus, where it manages gene transcription which affects cell multiplication, survival, metastasis, and angiogenesis. Monocyte chemotactic protein-1 (MCP-1), IL-6, IL-8, IL-1b, and TNF-a are proinflammatory genes that NF-kB controls. Many malignancies contain persistent NF-kB activation, which causes an inflammatory action that may contribute to cancer formation. As a result, inhibiting the NF-kB chain has therapeutical value. Direct suppression of NF-kB DNA binding is one technique for targeting the NF-kB pathway [118]. The contribution of NF-kB signalling to tumour breakout from the immune regulation also consequent tolerance to chemotherapy may be a critical activity of NF-kB signalling in cancer. The activity of the TF NF-kB p50 in tumour cells aims for the recruitment of anti-inflammatory M2 macrophages, which leads to tumour tolerance in radiation treatment. Radiation treatment is most efficient in mice that are deficient in specific proteins. NF-kB p50 is a protein found in the NF-kB gene [119]. As a result of their capacity to directly inhibit NF-kB, drugs are generally utilized in the treatment of some other diseases and are prescribed as prospective antineoplastic therapy. Bindarit is a non-steroidal anti-inflammatory drug [120–122]. In prostate and breast cancer animal tests, it inhibits carcinogenesis and metastasis. Bindarit reduces tumour-associated macrophage (TAM) and myeloid-derived suppressor cells (MDSC) tumour invasion by increasing IkB expression, suppressing NF-kB nuclear translocation but also MCP-1 transcription [123], FTY720, also known as fingolimod, functions as a prodrug that obstructs downstream signalling involving NF-kB/IL-6/STAT3, along with the expression of receptors linked to sphingosine 1-phosphate (S1P). This suggests its potential utility in addressing malignancies associated with persistent inflammation. The activation of M2 macrophages through prostaglandin E2 (PGE2), linked to NF-kB-mediated release of IL-6 and PGE2, is thought to contribute to resistance against platinum-based cancer therapies. By utilizing a specific inhibitor targeting IkB phosphorylation, the capacity of cervical and ovarian cancer cells to generate IL-6, PGE2, and STAT3, as well as the induction of M2 macrophages, appears to be diminished. By inhibiting M2 macrophage induction, the cyclooxygenase inhibitor indomethacin, and the anti-IL-6R antibody tocilizumab can enhance the effects of platinum-based chemotherapy [124].
Finding novel and creative cancer curing methods is a big demand around the globe. The efficacy of various malignant cancer treatments has grown in tandem with the development of novel cancer therapies and the philosophy of individualized care. In recent years, nanotechnology has been used increasingly often in medication, with implementations that are safer and more effective for tumour localization and therapy, including diagnostics. In cancer chemotherapy, the benefits of NP-based drugs have been extensively studied. These benefits include good pharmacokinetics, precise tumour cell specificity, low toxicity, and increased drug tolerance. The size and features of NPs utilized in medicine delivery systems are generally developed or selected depending on the pathophysiology of malignancies [9]. Nanocarriers are used in treating cancer to focus tumour cells mechanically after being absorbed. This is accomplished by the carrier activity of NPs and the localizing function of the desired substance. They then provide the medicine to tumour cells in order to destroy them. The inside of the nanocarriers contains conventional chemotherapeutic medicines, including nucleic acids, suggesting that they might be used for both cytotoxic and gene therapy. Additionally, NPs offer a substrate for encapsulation and transport into circulation for some medications that are poorly soluble. Drugs with nanocarriers have a longer half-life and are more likely to accumulate in tumour tissues [125].
These characteristics have a substantial impact on the efficiency of nano-drug transport and, consequently, therapeutic efficacy control. NPs used in therapeutic care often have certain sizes, shapes, and surface qualities. Because they can transport drugs well and have an enhanced permeability and retention (EPR) impact, NPs with a diameter of 10 mm to 100 nm are frequently utilized in cancer therapy [126]. The surface characteristics of NPs have an impact on their bioavailability and half-life. Hydrophilic coatings on NPs, such as polyethylene glycol (PEG), for instance, diminish opsonization and hence hinder immune system clearance. In order to increase medication penetration and accumulation in tumours, NPs are frequently altered to become hydrophilic [127].
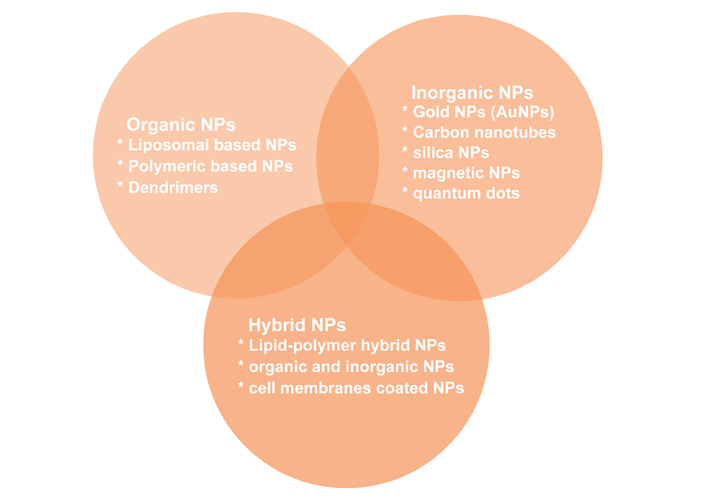
Types of NPs used in cancer treatment: organic NPs, inorganic NPs, and hybrid NPs (Figure 7) [128].
Organic NPs are studied for years and they have a diverse range of components. Liposomes, the first nano-scale medicine to be licensed for clinical use, are made up of an outer lipid layer and a core that contains either a hydrophobic or hydrophilic chemical. By changing the lipid layer structure, liposomes can perform a variety of roles, including mimicking the biophysical features of living cells, which can aid in the administration of more efficient medicinal drugs [129]. Liposomes have gone through multiple waves of development thanks to decades of research. Liposomes are a useful platform for in vivo administration of anti-tumour medications like doxorubicin and paclitaxel, among cancer treating substances, and nucleic acids in cancer therapy [130].
The surface area to volume ratio of inorganic NPs is higher than that of organic NPs. Although they are easy to produce and have a flexible surface conjugation chemistry, this usually comes at the expense of biocompatibility and biodegradability. Among the inorganic NPs studied are carbon nanotubes, quantum dots, magnetic NPs, silica NPs, and AuNPs [single-nucleotide polymorphism (SNPs)]. One of the extensively studied inorganic NPs (AuNPs) is a mixture of monolayer-protected clusters with a gold core that may be used to deliver drugs [131, 132].
Since both organic and inorganic NPs have advantages and disadvantages, combining them into a single hybrid drug delivery system improves the biological features of the multi-effective carrier, enhancing therapeutic activity and reducing drug resistance. Lipid-polymer hybrid NPs, which comprise an inner polymeric core and a lipid shell, have been demonstrated to be a promising drug delivery platform in the treatment of pancreatic cancer [133], breast cancer [134], and metastatic prostate cancer [135]. This type of hybrid NP allows for the encapsulation of both hydrophilic and hydrophobic medications for a more potent therapeutic effect since it combines the excellent biocompatibility of lipids with the structural stability of polymer NPs [136].
Cancer cell targeting is an important feature of nanocarriers for drug delivery because it improves therapeutic effectiveness while preserving normal cells from damage. Numerous investigations on the targeted design of NP-based medications have been conducted. It is critical to figure out tumour characteristics also the interaction among nanocarriers and tumour cells in order to properly handle the issues of tumour localizing and nano carrier system design. There are two types of targeting mechanisms: passive targeting and active targeting.
The distinct properties of tumour and normal tissue are used in passive targeting. The medications are favourably transported to the target place to have a therapeutical action in passive targeting. High cancer cell proliferation causes neovascularization, and big holes in the vascular wall cause tumour vessels to have more selectivity than normal vessels [137]. Macromolecules, counting NPs, leak from blood arteries that supply the tumour and accumulate inside the tumour tissue due to fast and faulty angiogenesis. Meanwhile, cancer patients’ poor lymphatic outflow promotes NP retention, allowing nanocarriers to transfer their contents to tumour cells. One of the driving forces of passive targeting is the EPR effect [138], which is caused because of these processes. The size of NPs affects the EPR effect since much research has shown that small NPs contain superior penetration ability but do not leak into regular capillaries. Larger particles, on the other side, are most likely to be eliminated by the immune system [139].
Active targeting specifically targets cancer cells through interactions between ligands and receptors. To distinguish between cancer cells and healthy cells, the ligands on the surface of NPs are selected to target molecules that are overexpressed on the surfaces of cancer cells [140]. When ligands on NPs interact with receptors on the surface of cancer cells, a process known as receptor-mediated endocytosis, it enables internalized NPs to release therapeutic drugs in a potent manner. In light of this, active targeting is especially well suited for the delivery of macromolecular medicines like proteins and siRNAs [141]. Examples of targeting moieties include monoclonal antibodies, peptides, amino acids, vitamins, and carbohydrates. The transferrin receptor, folate receptor, glycoproteins, and the epidermal growth factor (EGF) receptor are among the most researched of these ligands that connect to receptors on targeted cells [142].
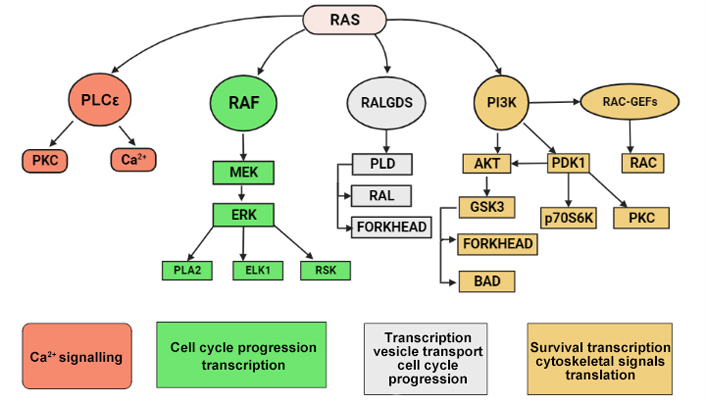
Once in its active, guanosine triphosphate (GTP)-bound state, RAS will interact with several families of effector proteins, resulting in stimulation of their catalytic activity. The main effectors are shown here. Rapidly accelerated fibrosarcoma (RAF) protein kinases initiate the mitogen-activated protein (MAP) kinase cascade, which leads to extracellular signal-regulated kinase (ERK) activation. This kinase has numerous substrates both in the cytoplasm and in the nucleus, including E26 transformation-specific (ETS) family TFs such as ETS like TF 1 (ELK1) that regulate cell cycle progression. Phospholipase Cε (PLCε) catalyses the hydrolysis of phosphatidylinositol 4,5-bisphosphate to diacylglycerol and inositol trisphosphate, resulting in protein kinase C (PKC) activation and Ca2+ mobilization from intracellular stores. ERK, glycogen synthase kinase 3 (GSK3), mitogen-activated kinase/ERK kinase (MEK), p70 ribosomal protein S6 kinase (p70S6K), 3-phosphoinositide dependent protein kinase-1 (PDK1), phospholipase A2 (PLA2), phospholipase D (PLD), p90 ribosomal protein S6 kinase (RSK). RalGDS proteins are guanine nucleotide exchange factors (GEFs) for RAS-like (RAL), a RAS-related protein. Downstream targets include forkhead TFs. Type I phosphoinositide 3-kinases (PI3Ks) generate second-messenger lipids, such as phosphatidylinositol-3,4,5-trisphosphate, which activate numerous target proteins including the survival signalling kinase protein kinase B (AKT/PKB). RAC is a member of the Rho family of small GTPases, which act as molecular switches to control a wide array of cellular functions. In particular, RAC signalling has been implicated in the control of cell-cell adhesions, cell-matrix adhesions, cell migration, cell cycle progression, and cellular transformation [143].
AKT/PKB is a group of three serine/threonine protein kinases with close genetic resemblances that bind to PI3K lipid products at their amino termini. They become active in response to PI3K stimulation and send the cell anti-apoptotic signals. B cell lymphoma-2-like 11 (BIM) is a member of the B-cell lymphoma 2 (BCL-2) family of apoptosis regulatory proteins, which induces apoptosis by acting at mitochondria to promote the release of cytochrome c into the cytosol. Fas cell surface death receptor ligand is an extracellular protein that binds and activates the death receptor Fas, also known as CD95 and APO1, which results in the beginning of apoptosis (Figure 8) [143].
There is a tremendous amount of molecular information about the RAS proteins’ molecular mechanisms in the nearly 20 years since it was discovered that RAS was an activated oncogene in human tumours. They have control over a complex signalling network that has numerous cross-talk, bifurcation, and feedback points. Although there is undoubtedly still much to learn and we may be fooling ourselves if we think we have a good understanding of this system, there are clearly a lot of leads worth following in the search for cancer treatments. Specificity questions will continue to be crucial: How specific should a drug be for its intended target, and can it be too specific without also inhibiting related molecules that could act as a substitute for the intended target? More importantly, it’s still unclear how wide of a therapeutic window many of these drugs with rational design will likely offer. The RAS signalling pathways are essential for the existence of both normal and tumour cells, even though tumour cells may be more dependent on them. They share this trait with many other signalling pathways that are under scrutiny by the pharmaceutical industry. Given the intense selective pressure placed on the genetically unstable cancer cells, even if these medications and their replacements are successful, the problem of resistance will inevitably arise quite quickly. In the end, assuming we ever have access to such an arsenal, this is most likely to be overcome by taking a cue from the fight against infectious diseases and using numerous effective drugs concurrently. Clinicians may need to change the endpoints used in clinical trial designs to assess drug efficacy because if reduction of tumour mass by single agents is the only metric used, many effective drugs may be discarded before they have a chance to demonstrate their value in combination therapies. Even if these medications and their replacements are successful, resistance will unavoidably develop quite quickly. In this regard, the creation of suitable surrogate markers to track drug action will be essential. If drugs that are effective on relatively uncommon tumour types are to be identified, tumour stratification will also be crucial in the selection of patients on whom to test these inhibitors. There is reason to be cautiously optimistic that some of the drugs discussed here, or more likely their derivatives, will eventually serve as the foundation for truly effective treatments for many common cancers, even though we are still only near the beginning of a very long and labour-intensive process [144].
Cancer is one of the most harmful diseases. Many chemotherapies are used to treat cancer. Other than medication, radiotherapy, laser treatment, immunotherapy, surgery, and hormone therapy are used for cancer. Resistance to the medication is a significant reason for drug investigation and structural modification. Many potent and less toxic anticancer drugs by changing their mechanism of action. Monitoring and timely management of adverse effects are critical to minimise toxicities and optimise efficacy from these classes of therapeutics. Future directions include optimising efficacy with novel combinations to overcome resistance mechanisms, as well as further development of predictive biomarkers for better selection of patients who will benefit from the therapies. Enzyme improves tumour tolerance and prevents healthy adult cells from becoming cancerous. Ion channels place an actual performance in the management of tumours. Membrane transporters are rulers of critical function in tumour cell chemosensitivity and resistance. TFs are a prominent class of cancer cell dependencies that are frequently altered in the aetiology of human cancer. NPs have been used for the past few years because of their safety and effectiveness. Delivering the medication to the targeted site is done by nanoencapsulation.
Many different tumour types are activated by RAS proteins, their regulators, and the downstream enzymes that are controlled by a variety of mechanisms, including oncogenic mutation of RAS genes. (1) They play a key role in mediating a number of the malignant traits of transformed cells, making them excellent candidates for tumour therapy. To be biologically active, RAS proteins need to undergo farnesylation post-translational modification. Although farnesyl transferase inhibitors appear to act on targets other than RAS, they do have some anti-tumour activity in the clinic. (2) Kinase inhibitors that work downstream of RAS in the RAF/MAP kinase pathway and inhibit either RAF or MAP kinase have been created and have shown promise in preliminary clinical studies. (3) Researchers have created inhibitors that target the human EGF receptor 2 (ERBB2) upstream activators of RAS and the EGF receptor. Small-molecule EGF receptor inhibitors have shown promise in clinical trials against lung cancer, whereas antibodies against ERBB2 have been approved for the treatment of breast cancer. (4) Inhibitors of the AKT/PKB kinase activity, which is triggered by the RAS oncogenic mutation and the loss of the tumour-suppressor gene phosphatase and tensin homolog (PTEN), are among the RAS-related treatments currently under development.
These data suggest novel potential pharmacological targets for the treatment of human malignancies associated with oncogenic activation.
ABC: adenosine triphosphate-binding cassette
ABCB1: adenosine triphosphate-binding cassette sub-family B member 1
ABCC1: adenosine triphosphate-binding cassette sub-family C member 1
ABCG2: adenosine triphosphate-binding cassette sub-family G member 2
AKT/PKB: protein kinase B
AP-1: activator protein-1
AuNPs: gold nanoparticles
CPT: camptothecin
EPR: enhanced permeability and retention
ERK: extracellular signal-regulated kinase
IkB: inhibitors of nuclear factor kappa B
IL: interleukin
Kv: voltage-gated potassium channel
MAP: mitogen-activated protein
MDR: multidrug resistance
MDR1: multidrug resistance protein 1
MMPs: matric metalloproteinases
NF-kB: nuclear factor kappa B
NPs: nanoparticles
PGE2: prostaglandin E2
P-gp: P-glycoprotein
PI3Ks: phosphoinositide 3-kinases
PSA: prostate-specific antigen
RAF: rapidly accelerated fibrosarcoma
RAS: rat sarcoma
Rel: reticuloendotheliosis viral oncogene homolog
SCLC: small-cell lung cancer
siRNA: small interfering RNA
SLC: solute carrier
sPLA2: secreted phospholipase A2
STAT: signal transducer and activator of transcription
TFs: transcription factors
topo II: type II DNA topoisomerases
TRP: transient receptor potential
uPA: urokinase plasminogen activator
VGCs: voltage-gated channels
Vm: transmembrane voltage
Authors would like to offer special thanks to Luqman College of pharmacy for allowing to carry out this work and other research projects. Advice given by Mr. Faheem IP and Dr. Mohammad Kamal has been a great help in our work. Authors particularly grateful for the assistance, and coordination support provided by Anuradha Madikeri, Shaikh Daniyal, and Gulzar Ahmed Rather.
MP, FP, AM, SD, MAK, GAR, and RS equally contributed to: Writing—original draft, Writing—review & editing, Conceptualization. All authors read and approved the final manuscript.
The authors declare no conflict of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.