 Review
Review

Affiliation:
1Department of Gastroenterology and Hepatology, University Medical Center Groningen, University of Groningen, 9700 RB Groningen, The Netherlands
2Department of Pathology and Medical Biology, University Medical Center Groningen, University of Groningen, 9700 RB Groningen, The Netherlands
†
ORCID: https://orcid.org/0000-0003-4605-2423
Affiliation:
3Department of Sociology, Faculty of Behavioral and Social Sciences, University of Groningen, 9700 RB Groningen, The Netherlands
†
ORCID: https://orcid.org/0009-0006-4902-5294
Affiliation:
2Department of Pathology and Medical Biology, University Medical Center Groningen, University of Groningen, 9700 RB Groningen, The Netherlands
Email: m.c.harmsen@umcg.nl
ORCID: https://orcid.org/0000-0002-7128-2741
Affiliation:
1Department of Gastroenterology and Hepatology, University Medical Center Groningen, University of Groningen, 9700 RB Groningen, The Netherlands
Email: a.j.moshage@umcg.nl
ORCID: https://orcid.org/0000-0002-4764-0246
Explor Dig Dis. 2025;4:100580 DOI: https://doi.org/10.37349/edd.2025.100580
Received: March 27, 2025 Accepted: June 06, 2025 Published: July 08, 2025
Academic Editor: Ina Bergheim, University of Vienna, Austria
The liver operates as a highly coordinated microsystem, where various liver cell types engage in dynamic interactions to maintain homeostasis. This intercellular cooperation resembles sociological models of sustainable cooperation, encompassing mechanisms such as resource sharing, communication networks, and conflict resolution. However, both in biology and sociology, cooperation can break down due to external pressures and self-serving behaviors. In metabolic dysfunction-associated steatotic liver disease (MASLD), chronic metabolic stress disrupts this equilibrium, leading to endothelial dysfunction, immune overactivation, and fibrosis—akin to sociological models of systemic collapse. A common model in sociology, Hardin’s Tragedy of the Commons, describes how individuals overexploit shared resources when acting in self-interest, ultimately leading to resource depletion. Similarly, under metabolic stress, hepatic cells prioritize short-term survival by increasing lipid storage, inflammatory signaling, and extracellular matrix (ECM) production. This self-serving response, much like free-riding in societal systems, exacerbates dysfunction, reinforcing a cycle of fibrosis and organ failure. Moreover, the failure in MASLD extends beyond the liver itself. The liver’s cooperative role is integral to its participation in inter-organ axes, including those with the cardiovascular, gut, brain, and kidney systems. While the analogy has limitations—cells do not possess intent as humans do—the fundamental principle of cooperation breakdown leading to systemic instability holds across disciplines. An interdisciplinary approach integrating biological and sociological insights offers novel perspectives for therapeutic innovation. Sociological frameworks provide concepts such as incentive structures and collective action, which can be applied to cellular behavior. By restoring cooperative cellular networks, therapies like extracellular vesicle (EV) treatment, ECM remodeling, and receptor (ant)agonists mimic interventions in social systems that rebuild trust and sustainability. This review explores how biological and sociological models of cooperation breakdown align and how regenerative medicine can leverage these insights to develop strategies that restore cellular equilibrium and halt disease progression.
Sustainable cooperation, a key concept in sociology, refers to the persistence of mutually beneficial interactions among individuals or groups over time [1–4]. It is often analyzed through frameworks such as game theory, which models strategic decision-making among agents, mutualism, which describes cooperative relationships where all parties gain benefits, and collective action, which explores how individuals coordinate efforts to achieve shared goals despite potential conflicts of interest [3, 5, 6]. These principles, commonly used to understand human societies and ecosystems, can also provide insight into cellular interactions within complex biological systems [2–4, 6–8].
The liver, a highly organized and dynamic organ, relies on intricate communication networks among its resident cells to maintain homeostasis [8–10]. Hepatocytes, liver sinusoidal endothelial cells (LSECs), Kupffer cells (KCs), hepatic stellate cells (HSCs), cholangiocytes, and other innate immune cells engage in continuous cross-talk through signaling molecules, extracellular vesicles (EVs), and direct cell-cell interactions [10, 11]. Much like cooperative networks in human societies, these cells dynamically regulate each other’s functions, balancing regeneration, immune responses, and metabolic activities [10, 12]. However, when this cooperation breaks down—due to persistent stress, inflammation, or metabolic dysfunction—it can lead to disease progression, such as in metabolic dysfunction-associated steatotic liver disease (MASLD) [9, 10, 13, 14].
The idea that health depends on physiological harmony among body parts or organs has a long history, dating back to classical thinkers like Galen. Our aim is not to restate this holistic principle but to offer an updated, cross-disciplinary framework that connects modern sociological theories, such as the Tragedy of the Commons, institutional failure, and self-repair models, to specific disruptions in hepatic cell-cell communication seen in MASLD. This approach extends classical concepts by integrating contemporary systems biology with regenerative medicine and social theory.
In doing so, we go beyond metaphors by mapping concrete therapeutic strategies [e.g., EV-mediated immune recalibration, extracellular matrix (ECM)-based structural normalization, receptor (ant)agonist-driven metabolic reprogramming] onto sociological mechanisms such as conflict mediation, trust restoration, and collective-action repair. These analogies not only provide a conceptual lens for understanding intercellular dysfunction but also provide information on targeted interventions aimed at restoring hepatic cooperation.
This review, therefore, seeks to synthesize sociological and biomedical insights into a cohesive model of systemic breakdown and repair. By drawing parallels between sociological cooperation models and hepatic cellular networks, we aim to provide a novel perspective on the mechanisms governing liver homeostasis and disease development. Ultimately, we propose that viewing MASLD through this interdisciplinary lens may yield new therapeutic avenues by addressing the fundamental breakdown of cooperation within and outside the liver.
The liver functions as a highly coordinated microsystem, where multiple cell types interact dynamically to maintain homeostasis (Figure 1) [14, 15]. This balance is achieved through a continuous exchange of signals, nutrients, and metabolic byproducts among hepatocytes, LSECs, HSCs, KCs, and other immune cells [10, 15]. Each of these cell types plays a distinct yet interdependent role, mirroring the principles of mutualism in sociological theory, where different actors contribute to the stability of a shared system [9, 10, 16].
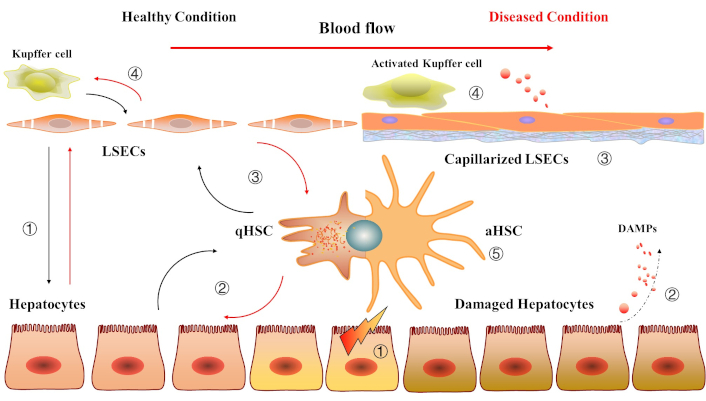
The intercellular communication and cooperation between liver cells under healthy and diseased conditions. Healthy condition: ① The dynamic crosstalk between hepatocytes and liver sinusoidal endothelial cells (LSECs) maintains liver metabolism, regeneration, and immune tolerance, ensuring a stable and functional hepatic microenvironment; ② The hepatocytes and quiescent hepatic stellate cells (qHSCs) engage in mutual regulation through paracrine signaling and extracellular matrix (ECM) maintenance, ensuring liver structure, metabolic balance, and protection from injury; ③ Functional LSECs maintain HSC quiescence through paracrine signaling, nitric oxide production, and vitamin A transfer, while qHSCs support the structural and functional integrity of LSECs; ④ The crosstalk between functional LSECs and Kupffer cells (KCs) in a healthy liver involves a balance of immunoregulation, anti-inflammatory signaling, and clearance of debris and endotoxins. LSECs maintain the immunotolerant environment by suppressing excessive KC activation via nitric oxide, anti-inflammatory cytokines, and antigen presentation. KCs, in turn, support LSEC function by clearing toxins and secreting growth factors, ensuring the stability of the liver sinusoidal microenvironment. Diseased condition: ① Chronic parenchymal cell damage; ② It leads to the release of danger-associated molecular patterns (DAMPs) and pro-inflammatory mediators by KCs; ③ LSECs capillarization; ④ KCs are activated and continuously secrete inflammatory mediators; ⑤ qHSCs are stimulated by DAMPs to transform into activated HSCs (aHSCs). They start to exhibit a state of rapid proliferation and secreting high amounts of ECM, triggering inflammation and fibrosis
Hepatocytes, the primary parenchymal cells of the liver, regulate metabolic functions, detoxification, and protein synthesis [17]. They rely on LSECs for nutrient and oxygen exchange, as well as on KCs for immune surveillance [9, 18]. HSCs contribute to ECM remodeling and respond to injury by supporting tissue repair, while cholangiocytes manage bile production and transport [19, 20]. In a healthy liver, these cells engage in reciprocal regulation, adjusting their behavior in response to environmental and physiological changes [9, 10, 21]. This cooperative equilibrium prevents excessive inflammation, fibrosis, or metabolic dysfunction [11, 21].
Sociologist Elinor Ostrom’s work on common-pool resource management provides an insightful parallel to liver cell cooperation [1, 22, 23]. Ostrom argued that sustainable resource management relies on collective governance, where individuals follow self-regulated rules to maintain long-term benefits for the group [24]. Similarly, liver cells maintain a self-organized regulatory network, where no single cell type dominates, but instead, each contributes to the stability of the system [9]. For instance, hepatocytes produce metabolic substrates that KCs and LSECs utilize, while LSECs secrete angiocrine factors that regulate HSC quiescence, preventing excessive fibrosis [9, 10]. When this delicate balance is disrupted—for example, due to chronic lipid overload, inflammation, or oxidative stress—cellular cooperation deteriorates, leading to pathological changes in MASLD, including hepatocyte dysfunction, LSEC capillarization, and uncontrolled HSC activation [10, 13].
By viewing the liver as a cooperative system governed by mutualistic interactions, we can better understand the mechanisms that sustain hepatic function under normal conditions and how their failure contributes to disease progression. This perspective also suggests that restoring cooperative dynamics, rather than targeting individual cell types in isolation, may be a more effective therapeutic approach for MASLD and other chronic liver diseases.
The liver operates as a highly coordinated system, where cellular interactions ensure homeostasis through specialized roles, signaling networks, and regulatory mechanisms (Figure 2) [9–11]. These cooperative behaviors resemble social systems, where resource sharing, communication, and conflict resolution sustain collective stability [1, 4].
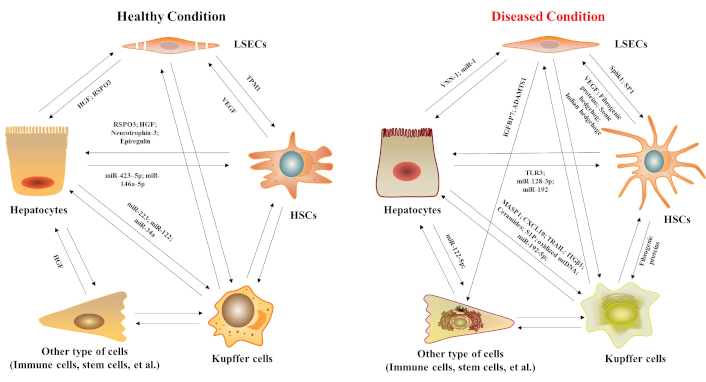
Regulatory mediators of intercellular communication between liver cells under physiological and pathological conditions. HGF: hepatocyte growth factor; RSPO3: R-spondin 3; MASP1: mannan-binding lectin serine protease 1; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; ITGβ1: integrin β1; IGFBP7: insulin-like growth factor-binding protein-7; ADAMTS1: ADAM metallopeptidase with thrombospondin type 1 motif 1; S1P: sphingosine 1-phosphate; TPM1: tropomyosin-1; SphK1: sphingosine kinase 1; TLR3: toll-like receptor-3; VNN-1: pantetheinase; VEGF: vascular endothelial growth factor; CXCL10: C-X-C motif chemokine 10; LSECs: liver sinusoidal endothelial cells; HSCs: hepatic stellate cells
In a functional liver, different cell types specialize in distinct yet interdependent roles, much like the division of labor in social economies [25]. LSECs play a crucial role in regulating nutrient and oxygen exchange between the bloodstream and hepatocytes, acting as metabolic gatekeepers [26]. Hepatocytes, in turn, process these nutrients and synthesize proteins, while HSCs contribute to ECM homeostasis and tissue repair [17, 19]. KCs, cholangiocytes, and other immune cells also fulfill essential roles in immune surveillance and bile production, respectively [18, 27].
This specialization enhances efficiency, much like Adam Smith’s concept of division of labor, where task specialization increases productivity [28]. In economics, decentralized specialization allows societies to optimize resource use—similarly, in the liver, cellular division of labor prevents metabolic overload and ensures rapid adaptation to changing physiological demands [28, 29]. However, in conditions such as MASLD, hepatocyte lipid accumulation and metabolic dysfunction disrupt this balance, leading to excessive stress on other liver cells, much like economic systems failing due to inefficient resource distribution [30–32].
Effective cooperation requires continuous communication among liver cells, primarily mediated through EVs, cytokines, and direct cell-cell interactions [9, 11]. EVs, which carry bioactive molecules such as microRNAs and proteins, act as informational messengers, enabling cross-talk among liver cell types [11]. This resembles knowledge-sharing networks in social systems, where decentralized communication fosters stability and adaptability [33].
From a network theory perspective, the liver’s intercellular signaling resembles distributed information processing, where no single node (cell type) controls the entire system [34, 35]. Instead, multiple pathways ensure redundancy and adaptability [35]. For instance, LSECs secrete factors that keep HSCs in a quiescent state, while KCs modulate immune responses through cytokine signaling [27, 36]. When this informational balance is disrupted, such as in chronic inflammation, EV-mediated signals may shift towards pro-fibrotic or inflammatory cues, reinforcing pathological changes in MASLD [37]. This parallels failures in decentralized networks, where misinformation or communication breakdowns can destabilize entire systems [33, 34].
While cooperation is essential, conflict naturally arises in both social and biological systems. KCs and other immune cells act as regulatory agents, preventing excessive damage by clearing pathogens and apoptotic cells [18, 27, 38]. However, in conditions like MASLD, chronic stress, and lipid accumulation can lead to the overactivation of KCs, resulting in excessive inflammation and hepatocyte injury [27, 39].
This process mirrors challenges in social cooperation models, where systems must prevent exploitation and free-riding (as described in public goods theory) [1, 24]. In well-functioning societies, regulatory mechanisms such as legal frameworks and social norms prevent individuals from exploiting collective resources [1, 40]. Similarly, in the liver, negative feedback loops normally restrain immune activation, preventing excessive tissue damage [41]. However, in disease states, this self-regulation fails, leading to chronic inflammation, fibrosis, and loss of liver function [42, 43].
In a healthy liver, intercellular cooperation ensures homeostasis through resource sharing, communication, and self-regulation [9, 10]. However, in MASLD, prolonged metabolic stress disrupts this balance, leading to loss of LSEC integrity, HSC overactivation, chronic inflammation, and even hepatocyte death (Figures 1 and 2) [20, 30, 32, 39]. These pathological shifts resemble sociological collapse models, where the breakdown of cooperation destabilizes entire systems [44, 45].
LSECs play a central role in maintaining metabolic homeostasis by regulating nutrient and oxygen exchange, as well as preventing excessive immune activation [26, 46–48]. In early MASLD, chronic lipid accumulation and oxidative stress induce capillarization of LSECs, where they lose their fenestrae and adopt a more rigid, vascular-like phenotype [47]. This impairs nutrient exchange, leading to hepatocyte stress and metabolic dysregulation [47, 49]. Additionally, dysfunctional LSECs secrete pro-inflammatory signals, amplifying KC activation and perpetuating a cycle of inflammation [50].
This failure parallels Hardin’s “Tragedy of the Commons” (1968), a sociological model describing how unregulated resource consumption leads to collective system collapse [44, 51]. In MASLD, excessive lipid accumulation overwhelms hepatocytes, forcing LSECs to adapt maladaptively, much like how overexploitation of natural resources leads to environmental degradation [52–55]. Without intervention, this process escalates, further destabilizing the hepatic microenvironment [55].
HSCs are normally quiescent and serve as the primary storage site for vitamin A in the liver, storing it in lipid droplets and regulating retinoid metabolism [20, 56]. In their resting state, HSCs contribute to liver homeostasis not only by maintaining ECM balance and providing structural support [19, 57], but also by promoting hepatocyte metabolism and regeneration through R-spondin 3 (RSPO3), an HSC-enriched modulator of WNT signaling [58]. However, in MASLD, chronic LSEC dysfunction and inflammatory signals drive persistent HSC activation, leading to excessive ECM deposition (fibrosis) [48, 59]. Over time, this reduces liver plasticity, impairing its ability to regenerate and respond to metabolic demands [21].
This breakdown of regulation mirrors sociological models of institutional failure, where self-organized systems collapse due to unchecked exploitation and rigidity [60, 61]. As Ostrom (1990) noted, sustainable governance relies on adaptive mechanisms that prevent the overuse of shared resources [1, 62, 63]. In the liver, HSCs should remain responsive to changing conditions, balancing ECM production and degradation [20]. However, in MASLD, the feedback loops that normally restore equilibrium fail, leading to a pathological fibrotic state akin to a society trapped in irreversible economic or environmental decline [20, 44, 64].
KCs are the liver’s resident macrophages, responsible for immune surveillance, clearance of apoptotic cells, and tolerance to gut-derived microbial products [27, 65]. However, in MASLD, excessive lipid exposure and oxidative stress trigger KC reprogramming into a pro-inflammatory phenotype, characterized by excessive secretion of tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6) [18, 65–67].
Chronic KC activation propagates hepatocyte injury and exacerbates fibrosis by stimulating HSC activation [67–69]. This loss of immune tolerance resembles sociological models of governance failure, where institutions meant to mediate conflicts instead contribute to systemic instability [70, 71]. In this analogy, KCs function like regulatory bodies that, when overwhelmed by excessive stimuli, shift from maintaining order to perpetuating dysfunction [27].
Hepatocytes, as the liver’s primary metabolic units, regulate lipid metabolism, glucose homeostasis, and detoxification [17]. Under physiological conditions, they maintain energy balance by coordinating with LSECs, KCs, and HSCs [9, 10]. However, in MASLD, sustained lipid accumulation and oxidative stress impair mitochondrial function, reducing ATP production and increasing reactive oxygen species (ROS) generation [30, 32].
This metabolic overload forces hepatocytes into maladaptive responses, including excessive triglyceride storage, apoptosis, and lipid peroxidation [17]. Eventually, these changes trigger hepatocyte ballooning and cell death, further exacerbating inflammation and fibrosis [72, 73]. The hepatocyte dysfunction in MASLD resembles economic collapse models, where excessive short-term resource consumption leads to irreversible decline [44, 74]. Just as economies suffer when industries prioritize immediate gains over long-term stability, hepatocytes under metabolic stress prioritize lipid accumulation at the cost of overall liver function [75, 76].
The liver in MASLD can be viewed as a failing common, where cooperative cellular behaviors that once sustained function are disrupted by metabolic stress, inflammation, and fibrosis [14, 39]. Just as societies collapse when individuals act in short-term self-interest at the expense of long-term stability, hepatocytes, LSECs, KCs, and HSCs in MASLD shift from cooperation to self-preservation, leading to organ dysfunction [11, 77]. This sociological perspective not only explains why MASLD progresses but also highlights potential therapeutic strategies: restoring LSEC function, reducing HSC activation, and modulating KC-mediated inflammation could help reestablish cellular cooperation. By addressing the breakdown of intercellular coordination, rather than just targeting isolated pathological features, we may identify more effective interventions to prevent liver failure in MASLD.
While much attention in MASLD research has focused on hepatocellular dysfunction, the progression and prognosis of the disease are deeply influenced by inter-organ dynamics [78, 79]. The liver does not operate in isolation but is embedded in a broader physiological network involving the gut, cardiovascular system, and other organs, each with tightly regulated feedback loops [78–80]. Disruption in this inter-organ “society” can exacerbate systemic failure, mirroring how the collapse of one sector in a sociopolitical system often cascades into wider instability, resulting not only in hepatic dysfunction but also in widespread consequences such as cardiovascular disease, cognitive disturbance, and gut-derived metabolic stress.
Cardiovascular disease is the leading cause of mortality in patients with MASLD [81, 82]. This reality underscores the pathophysiological entanglement between hepatic dysfunction and cardiovascular risk [83]. Hepatic steatosis and inflammation promote atherogenesis through increased secretion of pro-inflammatory cytokines, altered lipid metabolism, and insulin resistance [84]. Conversely, systemic hypertension and endothelial dysfunction can amplify hepatic injury by impairing blood flow and oxygen delivery to liver tissue [81–85]. Rather than coincidental comorbidities, MASLD and cardiovascular disease are co-evolving and mutually reinforcing conditions [14]. Their bidirectional influence reflects a collapse in systemic cooperation, where dysfunction in one organ destabilizes another, much like a financial crisis rippling through interconnected economic institutions. This breakdown underscores the need for therapeutic strategies that address the broader network of inter-organ dynamics, not just isolated hepatic pathology.
The gut microbiome plays a pivotal role in MASLD, functioning as a communication hub that links dietary inputs, metabolic regulation, and immune signaling [86]. In healthy states, the liver and gut maintain a cooperative relationship via the portal circulation, where microbial metabolites, such as short-chain fatty acids and bile acids, help modulate hepatic metabolism and immune tone [80, 87]. This bidirectional exchange preserves homeostasis and ensures that signals from the gut are appropriately interpreted by hepatic cells. However, gut dysbiosis disrupts this finely tuned relationship [80, 88]. Increased intestinal permeability (“leaky gut”) permits the translocation of bacterial endotoxins such as lipopolysaccharide (LPS), alongside microbial-associated molecular patterns (MAMPs), into the liver [89]. These stressors directly interfere with KC homeostasis and disrupt hepatocyte lipid metabolism, triggering chronic low-grade inflammation and metabolic dysfunction [90, 91]. Additionally, dysbiosis alters bile acid signaling, further impairing hepatic regulatory circuits [80, 92]. Collectively, these effects make gut dysbiosis a powerful upstream disruptor of intercellular communication in the liver, exacerbating MASLD progression [80]. From a sociological perspective, the gut microbiome can be likened to a decentralized network of informants and producers whose biochemical outputs influence central governance, represented here by the liver. When this network becomes dysregulated, distorted messages flood the system, mirroring the societal consequences of disinformation. As in political systems destabilized by misinformation, the liver’s capacity to coordinate and respond rationally is compromised, accelerating systemic breakdown [93]. Therapeutically, restoring liver–gut cooperation involves recalibrating this informational axis. Interventions targeting the microbiome, such as prebiotics, probiotics, postbiotics, or microbial-derived EVs, aim to reduce inflammatory inputs and restore signal fidelity [94]. By reestablishing a healthier microbial environment, these strategies seek to interrupt the pathological feedback loop and promote cooperative stability across the liver–gut axis.
The liver–brain axis is increasingly recognized as a bidirectional communication pathway involved in regulating appetite, cognition, and systemic homeostasis [95]. The liver influences central nervous system (CNS) function through cytokines, metabolic hormones [e.g., insulin, leptin, fibroblast growth factor 21 (FGF21)], and detoxification of neuroactive substances [96, 97]. In MASLD, disrupted hepatic signaling and chronic low-grade inflammation can alter vagal tone, impair neuroendocrine feedback, and contribute to neuroinflammation, cognitive impairment, and mood disorders [96, 98]. This systemic crosstalk breakdown parallels sociological dysfunction, where impaired “policy implementation” (liver signaling) undermines executive function (brain regulation). Thus, the liver–brain axis represents another site of inter-organ cooperation whose destabilization amplifies MASLD’s multisystem burden.
The liver and kidneys cooperate in key metabolic functions, including ammonia detoxification, glucose homeostasis, and xenobiotic clearance [99]. In MASLD, particularly in advanced stages with fibrosis or cirrhosis, this interdependence is strained [99, 100]. Systemic inflammation, disrupted bile acid metabolism, and impaired renal perfusion contribute to hepatorenal dysfunction and cardiorenal complications [99–101]. Analogous to a destabilized central bank burdening a dependent economy, hepatic failure transfers metabolic stress to the kidneys, accelerating their decline and reinforcing systemic collapse. The liver–kidney axis thus exemplifies how inter-organ cooperation, once disrupted, can amplify disease progression across organ systems.
Adipose tissue, particularly visceral fat, is a key regulator of hepatic function through the release of adipokines (e.g., adiponectin, leptin, resistin) and free fatty acids [102, 103]. In MASLD, dysfunctional adipose tissue acts as a metabolic saboteur, contributing to insulin resistance, lipotoxicity, and systemic inflammation [104]. This reflects a breakdown in metabolic diplomacy, where formerly cooperative energy reservoirs now release antagonistic signals that destabilize hepatic homeostasis. Sociologically, this resembles a once-productive trading partner devolving into a hostile neighbor, flooding markets (the liver) with toxic goods (lipids and cytokines) and undermining internal regulatory systems.
The liver and skeletal muscle maintain a dynamic partnership in regulating systemic metabolism, particularly in glucose and amino acid homeostasis [105, 106]. Skeletal muscle acts as a major reservoir for glucose disposal and protein storage, while the liver orchestrates gluconeogenesis and nutrient distribution [105, 107, 108]. In the context of MASLD, this inter-organ axis becomes strained. Chronic inflammation, insulin resistance, and altered amino acid metabolism contribute to sarcopenia—a progressive loss of muscle mass and function, which in turn reduces peripheral glucose uptake and increases metabolic burden on the liver [109, 110]. This feedback loop represents a failure in systemic resource allocation, akin to the breakdown of infrastructure-sharing in a complex federation. When skeletal muscle, normally a cooperative energy sink, begins to degrade, it no longer fulfills its role in buffering postprandial glucose or maintaining metabolic stability. The liver, already under lipotoxic and inflammatory pressure, is forced to compensate, worsening hepatocellular stress and accelerating disease progression.
Although less frequently discussed, the liver and lungs are deeply interconnected, especially in the context of systemic inflammation and hypoxia [111]. In MASLD, circulating pro-inflammatory cytokines and altered coagulation profiles can affect pulmonary microvasculature, increasing the risk for conditions such as pulmonary hypertension and obstructive sleep apnea [112, 113]. Conversely, chronic hypoxia from lung disease can exacerbate hepatic steatosis and fibrosis through oxidative stress [114, 115]. This reflects a feedback loop where two interdependent systems collapse under shared inflammatory pressure, much like adjacent urban sectors in crisis amplifying each other’s vulnerabilities.
MASLD is not merely a condition of individual cellular malfunction but a broader failure of intercellular cooperation within the liver microenvironment [9, 10]. Effective treatment, therefore, must extend beyond correcting isolated pathways to restoring the systemic interactions that underlie hepatic function. Drawing inspiration from sociological models of intervention, such as policy reform, economic restructuring, and conflict mediation, we can conceptualize therapeutic strategies like EVs, ECM, and receptor (ant)agonists as tools for rebuilding cooperative networks among liver cells.
EVs mediate intercellular communication by transferring proteins, microRNAs, and lipids, helping maintain homeostasis under physiological conditions [11, 116]. In MASLD, the loss of functional EV-mediated signaling contributes to inflammation, fibrosis, and metabolic dysfunction [11]. Studies suggest that stem cell-derived EVs or engineered EVs could [117, 118]:
Reprogram KCs toward a pro-resolving phenotype, reducing excessive inflammation [27, 65, 119, 120].
Suppress HSC activation, preventing excessive ECM deposition [119, 121].
Enhance LSEC function, restoring endothelial integrity and nutrient exchange [50, 122, 123].
Promoting hepatocyte function recovery and regeneration [124–126].
This strategy mirrors sociological policy interventions, where external mediation helps restore communication between conflicting groups, reducing systemic instability [127]. Just as diplomatic negotiations or economic aid can rebuild cooperative structures in failing societies [127, 128], EV therapy aims to reestablish intercellular dialogue, preventing disease progression [129].
A key challenge in MASLD is the loss of liver plasticity due to fibrosis, which limits tissue regeneration [130]. ECM hydrogels offer a biomimetic environment, supporting hepatocyte function, reducing HSC activation, and improving overall tissue remodeling [131–133]. By providing a structural and biochemical niche, ECM hydrogels may:
Promote hepatocyte regeneration, counteracting metabolic dysfunction [131, 134, 135].
Suppress fibrogenic signaling, preventing irreversible ECM accumulation [57, 136].
Support LSEC stability, facilitating vascular homeostasis [57, 131, 137, 138].
This approach is analogous to institutional interventions in sociology, where rebuilding public infrastructure (e.g., education systems, healthcare networks) restores long-term societal function [74, 139]. Just as stable institutions enable communities to recover from economic or environmental crises, ECM hydrogels provide a microenvironment that allows hepatocytes and other liver cells to regain functionality [140, 141].
The liver’s ability to maintain homeostasis and respond to injury is governed by complex signaling among diverse cell types. Receptor (ant)agonists, which activate/inhibit specific signaling pathways, can be viewed as molecular analogues to public policy—strategically deployed to incentivize constructive cellular behavior and restore communication. These agents help orchestrate a return to physiological balance, much like systemic reforms aimed at stabilizing failing social institutions.
In the context of MASLD, receptor (ant)agonists that modulate pathways related to metabolism, inflammation, and fibrosis show particular promise. Agonists of the peroxisome proliferator-activated receptors (PPARs), for instance, not only enhance lipid metabolism but also exert anti-inflammatory effects within hepatic cells [142–144]. Likewise, glucagon-like peptide-1 (GLP-1) receptor agonists improve hepatocyte survival and reduce hepatic steatosis, contributing to a more resilient and functionally coordinated liver environment [145, 146]. By recalibrating intercellular signaling networks, these agents foster conditions that support collective regeneration and reduce fibrotic progression. Many drugs under investigation have demonstrated promising therapeutic effects by targeting various key pathways in the pathogenesis of MASH (Table 1) [147]. Among these, resmetirom, a thyroid hormone receptor β (THRβ) agonist developed by Madrigal Pharmaceuticals, has become the first and only officially FDA-approved drug for treating MASH [148]. Other candidates, such as PPARs agonists, GLP-1 analogs, and FGF21 analogs, are awaiting approval [147, 149]. Despite these advances, developing pharmacotherapeutics for MASH remains a significant challenge due to the complexity of its pathogenesis, heterogeneity, and the side effects associated with existing treatments [39, 147, 149–151]. Metabolic modulators improve insulin sensitivity and reduce hepatic steatosis but are associated with weight gain, while GLP-1 receptor agonists (e.g., liraglutide, semaglutide) and sodium-glucose transport protein 2 (SGLT2) inhibitors enhance glycemic control, reduce liver fat, and demonstrate cardiovascular benefits [147, 152]. Anti-fibrotic agents like obeticholic acid, a FXR agonist, and PPAR agonists like lanifibranor target lipid metabolism and fibrosis but may cause adverse effects like pruritus [147, 149, 153, 154]. Resmetirom and other lipid metabolism modulators have shown efficacy in reducing steatosis and fibrosis during clinical trials, and antioxidants like vitamin E alleviate oxidative stress in some patients. However, their long-term safety and limited clinical efficacy require further evaluation [39, 147, 149].
List of ongoing pharmacotherapeutic agents in phase II–IV trials for MASH/MASLD
| Target | Agent | Latest phase | NCT | Sponsor | |
|---|---|---|---|---|---|
| Nuclear receptor agonists | THRβ agonist | Resmetirom | FDA-approved | NCT04951219NCT04197479NCT05500222NCT03900429 | Madrigal Pharmaceuticals |
| VK2809 | Phase II | NCT04173065NCT02927184 | Viking Therapeutics | ||
| ASC41 | NCT05462353 | FirstWord Pharma | |||
| TERN-501 | NCT05415722 | Terns Pharmaceuticals | |||
| PPARα/δ/γ agonist | Pioglitazone | Phase IV | NCT00994682 | University of Florida | |
| IVA337 (Lanifibranor) | Phase III | NCT04849728NCT05232071NCT03459079 | Inventiva Pharma | ||
| Elafibranor | NCT02704403NCT03883607NCT01694849 | Genfit | |||
| Saroglitazar | Phase II | NCT03061721NCT03863574 | Zydus Therapeutics | ||
| Pemafibrate | NCT05327127NCT03350165 | Kowa Research Institute, Inc. | |||
| FXR agonist | Obeticholic acid | Phase III | NCT02548351NCT01265498 | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) | |
| Cilofexor | Phase II | NCT02854605 | Gilead Sciences | ||
| HPG1860 | NCT05338034 | Hepagene (Shanghai) Co., Ltd. | |||
| Tropifexor | NCT02855164NCT04147195NCT03517540 | Novartis Pharmaceuticals | |||
| TERN-101 | NCT04328077 | Terns, Inc. | |||
| MET409 | NCT04702490 | Metacrine, Inc. | |||
| CS0159 | NCT05591079 | Cascade Pharmaceuticals, Inc | |||
| GLP-1R agonist | Semaglutide | NCT04822181NCT02970942 | Novo Nordisk A/S | ||
| Liraglutide | NCT01237119NCT02654665 | University of Birmingham | |||
| Dulaglutide | Phase IV | NCT03648554 | Central Hospital, Nancy, France; Eli Lilly and Company | ||
| A3 adenosine receptor agonist | Namodenoson | Phase III | NCT02927314 | Can-Fite BioPharma | |
| FGF mimetics | FGF21 analogue | Efruxifermin | Phase III | NCT06215716NCT06161571NCT03976401NCT04767529 | Akero Therapeutics |
| Pegozafermin | Phase II | NCT03486912NCT03486899NCT03400163NCT02413372 | 89bio | ||
| Inhibitors | SCD-1 inhibitor | Aramchol | Phase III | NCT04104321 | Galmed Research and Development |
| CCR2/CCR5 inhibitor | Cenicriviroc | NCT03028740 | Tobira Therapeutics, Inc. | ||
| Galactin-3 inhibitor | Belapectin (GR MD-02) | NCT06035874 | Galectin Therapeutics | ||
| ASK1 inhibitor | Selonsertib | CTR20230344 | The First Hospital of Jilin University | ||
| ACC inhibitor | GS-0976 | NCT02856555 | Gilead Sciences | ||
| MK-4074 | NCT01431521 | Merck Sharp & Dohme LLC | |||
| FASN inhibitor | Denifanstat | NCT04906421 | Sagimet Biosciences Inc. | ||
| MPC inhibitor | MSDC-0602K | NCT02784444 | Cirius Therapeutics, Inc. | ||
| ACLY inhibitor | Bempedoic acid | NCT06035874 | Medanta - The Medicity | ||
| BGT-002 | CTR20230344 | The First Hospital of Jilin University | |||
| Combination therapy | ACC inhibitorDGAT2 inhibitor | PF-05221304PF-06865571 | Phase III | NCT04321031NCT03248882NCT03776175 | Pfizer |
| ASK1 inhibitorACC inhibitorFXR agonist | SelonsertibFirsocostatCilofexor | NCT02781584 | Gilead Sciences | ||
| FXR agonistHMGCR inhibitor | Obeticholic acidAtorvastatin | NCT02633956 | Intercept Pharmaceuticals | ||
| FXR agonistGLP-1R agonist | CilofexorSemaglutide | NCT03987074 | Gilead Sciences | ||
MASLD: metabolic dysfunction-associated steatotic liver disease; THRβ: thyroid hormone receptor β; PPARα/δ/γ: peroxisome proliferator-activated receptor α/δ/γ; FXR: farnesoid X receptor; GLP-1R: glucagon-like peptide-1 receptor; FGF: fibroblast growth factor; SCD-1: stearoyl-CoA desaturase-1; CCR2/CCR5: C-C chemokine receptor type 2/5; ASK1: apoptosis signal-regulating kinase 1; ACC: acetyl-CoA carboxylase; FASN: fatty acid synthase; MPC: mitochondrial pyruvate carrier; ACLY: ATP-citrate lyase; DGAT2: diacylglycerol O-acyltransferase 2; HMGCR: 3-hydroxy-3-methylglutaryl-CoA reductase
The potential of receptor (ant)agonists is further amplified when integrated with EV therapy and ECM hydrogels. EVs—natural carriers of proteins, lipids, and RNAs—facilitate targeted communication between cells and have been shown to modulate fibrosis and support tissue repair. When embedded in ECM hydrogels, which provide mechanical support and biochemical cues, EVs can more effectively propagate regenerative signals across damaged hepatic regions. This integrated approach creates a supportive niche analogous to a social safety net, enabling liver cells to resume cooperative functions and self-organize toward recovery.
Ultimately, systemic remodeling aims not merely to suppress individual pathological pathways but to reestablish a functional and collaborative cellular ecosystem. By treating the liver as a dynamic, self-regulating system—much like a society recovering from systemic breakdown—therapeutic strategies that rebuild intercellular cooperation hold significant potential in addressing the complex, multifactorial nature of MASLD.
Acknowledging these inter-organ axes demands a therapeutic strategy that transcends hepatocentric approaches. Receptor agonists, EV therapies, and ECM scaffolds must be evaluated not only for their intrahepatic effects but for how they modulate systemic physiology. Targeting the gut microbiome, restoring endothelial function, modulating neuroimmune signaling, and reducing pulmonary inflammation represent coordinated interventions—akin to inter-ministerial policies aimed at restoring national coherence. Viewing MASLD as a systemic disorder opens new pathways for organ-crossing therapies and diagnostic frameworks.
The concept of sustainable cooperation, widely studied in sociology, provides a compelling framework for understanding liver homeostasis and its breakdown in MASLD. Just as societies rely on resource sharing, communication networks, and conflict resolution to maintain stability, the liver depends on cooperative interactions between hepatocytes, LSECs, HSCs, KCs, and cholangiocytes to function effectively. When this cooperation fails, due to metabolic stress, inflammation, and fibrosis, MASLD progresses, resembling sociological models of systemic collapse.
Moreover, the failure in MASLD extends beyond hepatic disintegration. The liver’s cooperative function is embedded in a network of inter-organ axes, including the liver–cardiovascular, liver–gut, liver–brain, liver–lung, liver–kidney, and so on. These axes represent critical lines of metabolic, immune, and neuroendocrine communication. Disruption along these pathways mirrors the collapse of interdependent institutions within a society, where dysfunction in one domain precipitates cascading failures across the whole system. For instance, gut dysbiosis, cardiovascular inflammation, and neuroimmune signaling disruptions not only influence liver pathology but are also amplified by it, creating feedback loops of escalating dysfunction.
By applying sociological theories to liver biology, we gain new perspectives on disease progression and potential interventions. Viewing MASLD as a failure of cellular cooperation shifts the therapeutic focus from merely targeting isolated pathological features to restoring intercellular communication and metabolic balance. Strategies such as EV therapy, ECM hydrogels, and receptor (ant)agonist, which reestablish liver cell interactions, mirror policy interventions in sociology, where external support can stabilize failing systems.
This interdisciplinary perspective—linking systems biology, network theory, and cooperative game theory with sociological models of governance and collapse—opens new avenues for research. Future efforts should prioritize restoring cooperative equilibrium not only at the cellular level but also across the organ axes that define the body’s systemic integrity. In doing so, we may move closer to therapies that don’t just treat liver disease, but reestablish the inter-organ harmony essential to long-term health.
ECM: extracellular matrix
EVs: extracellular vesicles
FGF21: fibroblast growth factor 21
GLP-1: glucagon-like peptide-1
HSCs: hepatic stellate cells
KCs: Kupffer cells
LSECs: liver sinusoidal endothelial cells
MASLD: metabolic dysfunction-associated steatotic liver disease
PPARs: peroxisome proliferator-activated receptors
THRβ: thyroid hormone receptor β
During the preparation of this work, the authors used GPT-4o (San Francisco, USA) in order to improve language. After using this tool/service, all authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
JW and JL: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. MCH and HM: Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
Prof. Han Moshage is an Associate Editor of Exploration of Digestive Diseases. However, he was not involved in any aspect of the peer review or editorial decision-making process for this manuscript. The other authors declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This project is financially supported by the China Scholarship Council, File No. [202006250036] (J.W.) & No. [202106200024] (J.L.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

