 Review
Review

Affiliation:
1Department of Internal Medicine, Bronx Care Health System, Bronx, NY 10457, USA
2Department of Gastroenterology, Ahalia Hospital, Abu Dhabi, UAE
Email: george.lenx@yahoo.com
ORCID: https://orcid.org/0000-0001-5705-3275
Affiliation:
3Department of Anesthesiology, Ahalia Hospital, Abu Dhabi, UAE
ORCID: https://orcid.org/0009-0001-4217-2199
Explor Dig Dis. 2025;4:100573 DOI: https://doi.org/10.37349/edd.2025.100573
Received: January 31, 2025 Accepted: April 23, 2025 Published: May 08, 2025
Academic Editor: Jean Francois D. Cadranel, GHPSO, France
Hepatitis D virus (HDV), a satellite virus requiring hepatitis B surface antigen (HBsAg) for propagation, is a hepatotropic virus implicated in acute and chronic viral hepatitis, with an accentuated risk of cirrhosis and hepatocellular carcinoma. The epidemiology of HDV infection is underestimated owing to underdiagnosis and low screening rates. Being inherently defective, HDV depends on HBsAg, the envelope protein of the hepatitis B virus (HBV), for hepatocyte entry and exit. However, viral replication is then HBV-independent but dependent on the host cell RNA polymerases. Infection can either be a coinfection with HBV or superinfection in individuals with pre-existing HBV, with the latter exhibiting a higher propensity for progression to chronicity. Clinical manifestations could range from acute hepatitis to acute flares in chronic hepatitis to rapidly progressive chronic liver disease. For decades, the treatment of HDV infection relied heavily on conventional and pegylated interferons (PEG-IFNs), which, despite limited efficacy and high relapse rates, continue to be a therapeutic option in patients with compensated liver disease. The past decade witnessed an advanced understanding of HDV virology and pathogenesis, which led to the development of multiple specific and targeted therapeutic agents, most notably the HDV viral entry inhibitor, bulevirtide, and the prenylation inhibitor, lonafarnib. In 2020, bulevirtide became the first drug approved in the European Union to treat chronic HDV with compensated liver disease. The emergence of lambda interferons, nucleic acid polymers, RNA silencers, and immune modulators further expands the therapeutic landscape. Combination regimens leveraging complementary mechanisms are promising but require further validation to optimize dosing and treatment durations. While novel therapies provide hope, significant unmet needs remain, especially for patients with decompensated cirrhosis. Future research must prioritize comprehensive strategies to enhance treatment efficacy and accessibility, offering a brighter prognosis for those affected by this devastating virus.
The hepatitis D virus (HDV) is a negative-sense, single-stranded RNA virus belonging to the delta virus genus [1]. It is an incomplete virus, lacking envelope proteins and nucleic acid polymerases. Instead, it utilizes the surface antigen of hepatitis B virus (HBV) to envelope itself and the host RNA polymerase to aid replication. HDV can infect an individual only in the presence of HBV infection to provide hepatitis B surface antigen (HBsAg), making it a satellite virus to HBV. HDV infections may be coinfections, simultaneously acquired HBV-HDV duos, or superinfections, where HDV infects a person with a background HBV infection. It is estimated that about 5% of those chronically infected with HBV do have HDV infection as well [2]. The HDV prevalence rates are underestimated, thanks to the abysmally low testing rates. Recent studies reveal HDV screening rates ranging from 6.7% to 19.7% [3–5].
HDV infection is transmitted mainly through percutaneous exposure: contaminated needles or other sharps, followed by sexual transmission from infected partners [2]. Mother-to-child transmission is infrequent compared to HBV. Rare cases have been reported following mucosal exposure and breastfeeding [6]. Inside the host, being an incomplete virus that lacks an envelope and RNA polymerase, it is ineffective by itself in infecting or replicating. The HDV-ribonucleoprotein (HDV-RNP) depends on the hepatitis B antigens to facilitate hepatocyte infection. The HDV envelope is derived from the HBsAg, small, medium, and large. This HBsAg-enveloped HDV interacts with the hepatocyte receptors, heparan sulfate proteoglycan (HSPG), and sodium taurocholate co-transporting polypeptide (NTCP). Following the HBsAg-HSPG/NTCP interaction, the HDV-RNP comprising HDV-RNA and hepatitis D antigen (HDAg) is released into the hepatocyte. This complex migrates into the hepatocyte nucleus and utilizes the host RNA polymerase to replicate. HDV-RNA is transcribed into mRNA by host RNA polymerase and subsequently translated to generate both small and large HDAg (S-HDAg and L-HDAg). This HDAg integrates with HDV-RNA, forming nascent HDV-RNP. The HDAg undergoes post-translational modifications, most importantly, prenylation, which allows it to interact with HBsAg [7]. Subsequently, the HBsAg envelops this HDV-RNP core in the presence of concomitant HBV infection. Only these HBsAg-enveloped HDV-RNP can egress from the hepatocyte. In summary, HDV utilizes HBV-generated HBsAg to form an enveloped virus that enters and exits hepatocytes while replicating using host RNA polymerases independent of HBV [1, 2, 8].
The mean incubation period of HDV infection is 2–8 weeks; it tends to be longer in cases of coinfection but shorter in those of superinfection [9, 10]. The disease may manifest as acute hepatitis, or acute flare in chronic hepatitis, or as chronic hepatitis. Acute hepatitis typically results in the setting of HDV-HBV coinfection, while flare or chronic infection is more common with HDV superinfection. The possible outcomes of HDV-HBV infection are depicted in Figure 1.
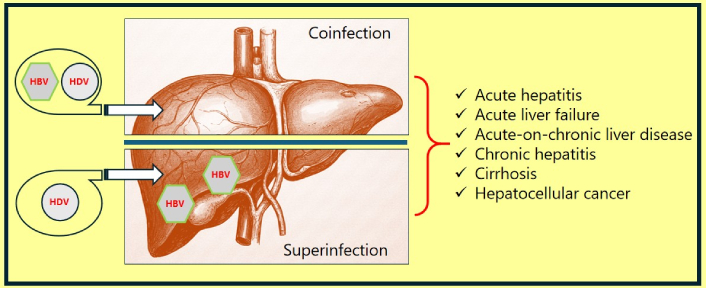
Clinical outcomes of HDV-HBV coinfection or superinfection. HBV: hepatitis B virus; HDV: hepatitis D virus
Coinfection results when an individual acquires hepatitis B and D simultaneously. The typical manifestation is acute or fulminant hepatitis. The manifestations include a prodromal phase characterized by fever, myalgia, and fatigue, followed by the development of jaundice. The infections resolve in most cases but may sometimes progress to acute liver failure. In an immunocompetent adult, coinfection rarely progresses to chronic illness; the estimated incidence of chronic HDV in coinfection is about 2% [11].
Superinfection results when an individual with pre-existing hepatitis B acquires an add-on HDV infection. The clinical manifestations can be acute hepatitis, or flare, or chronic hepatitis. The incidence of chronic hepatitis D, in the setting of HDV superinfection, is as high as 90% [11].
Chronic HDV infections are the most rapidly progressive form of chronic viral hepatitis, often culminating in cirrhosis in 5–10 years in 70% of cases [12]. HDV-HBV infection has a three times higher risk of cirrhosis compared to isolated HBV infection [13]. Though controversial, few studies even report a higher risk of hepatocellular carcinoma in HDV-related cirrhosis of the liver compared to isolated HBV cirrhosis [13, 14]. Chronic HDV infection is also associated with numerous autoimmune diseases; notably, approximately 15% of individuals are positive for anti-liver-kidney-microsomal antibodies (anti-LKM-3) [11].
The earlier HDV treatment efforts largely paralleled those of hepatitis B infection. Since HDV depends on HBsAg for its infectivity, it was previously believed that treating HBV infection would also eliminate its satellite infection. In 1994, Farci et al. [15] evaluated interferon alfa (IFNα) for the treatment of HDV infection, subsequently paving the way for pegylated IFN therapy. The early 21st century saw extensive trials of nucleic acid analogues in the treatment of HDV. The subsequent two decades witness no groundbreaking evolution in the treatment of the delta virus. However, since 2020, multiple new molecules have been evaluated, and many are still in different stages of clinical trials for managing HDV infection. Figure 2 illustrates the timeline of HDV treatment evolution.
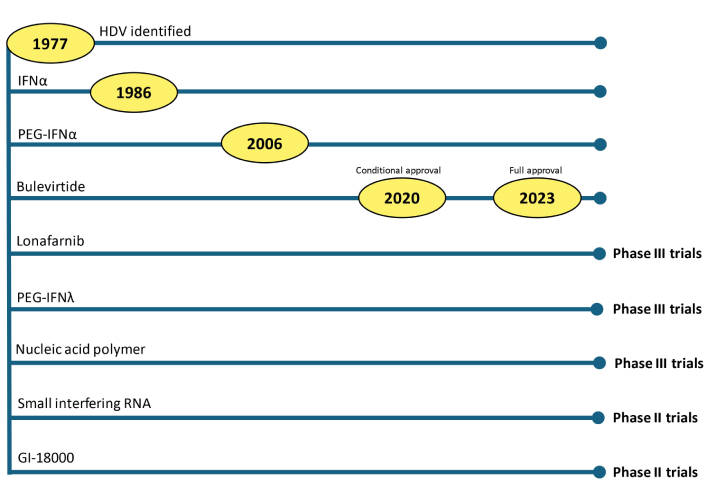
The timeline depicts the currently utilized and potential candidate medications for treating HDV infection. HDV: hepatitis D virus; IFN: interferon; PEG-IFNα: pegylated-IFNα
All patients with chronic HDV infection should be considered for antiviral therapy. Currently, available medications are validated for use in compensated liver disease. None of the medicines are licensed for use in decompensated cirrhosis, and hence, this group of patients should be evaluated for liver transplantation. The monitoring of patients includes virological, biochemical, and histological responses. The virological response is estimated using HDV-RNA and quantified every six months. Biochemical response involves monitoring of liver chemistries, typically 3–6 monthly. An ideal assessment of histological response requires a liver biopsy with an estimation of the histological activity index. Liver biopsy is an invasive procedure potentially associated with sequelae; hence, less invasive alternatives like hepatic Fibroscan® or Fibrotest® might suffice. A ≥ 2-log reduction in HDV-RNA, undetectable HDV-RNA at the end of treatment and 6 months post-treatment, and normalization of alanine aminotransferase (ALT) are utilized as markers in clinical trials. The ideal endpoint in HDV treatment is the loss of HBsAg, which is rarely achievable with currently available therapies [8].
IFNs are cytokines and a regular component of the human immune system with intrinsic antiviral properties. The IFNs interact with the cell surface receptors of virus-infected cells, upregulating the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway. This pathway downstream activates multiple IFN-stimulated genes, the products of which have numerous antiviral effects, including, but not limited to, inhibition of viral replication [16]. Pegylation of IFN results in slower clearance, thereby increasing its half-life [17]. IFN is the only class of medication recommended by the American Association for the Study of Liver Diseases (AASLD) HBV treatment guidelines, 2016 [18].
IFNα was the first molecule ever evaluated in the treatment of HDV. Administered subcutaneously at a dose of 9 million units (MU) three times weekly, it achieved approximately a 50% virological and biochemical response at 48 weeks of treatment. However, most of the patients relapsed weeks after cessation of therapy [15]. The unacceptable adverse effect profile, thrice weekly injection schedule, and high relapse rates post-treatment were the significant limitations. Long-term follow-up of IFNα-treated patients has reported better survival rates, lower HDV viral replication, and lower fibrosis rates years after treatment [19].
PEG-IFNα, the advent of which allowed a shift from thrice weekly to once-a-week injection therapy. Trials with PEG-IFNα-2a (180 µg/week) or PEG-IFNα-2b (1.5 µg/kg/week) for 48 weeks to 96 weeks reported HDV-RNA negativity rates of 17% to 46% at 6 months to 12 months post-treatment follow-up [20]. A meta-analysis comprising 13 studies involving 475 patients treated with PEG-IFNα revealed a pooled virological response of 29% and a biochemical response of 33% at 24 weeks post-treatment [21]. The European Association for the Study of the Liver (EASL), 2023 practice guidelines recommend considering PEG-IFN, preferably PEG-IFNα, for 48 weeks to treat chronic HDV infection with compensated liver disease [8]. The treatment limitations include an overall suboptimal response, an adverse effect profile, and the need for parenteral administration of the medication. IFN therapy is not recommended in patients with decompensated liver disease. Also, IFN treatment has been linked with the onset of autoimmune hepatitis [8].
PEG-IFNλ has demonstrated antiviral effects with a more favorable adverse effect profile compared to PEG-IFNα. The IFNλ receptor is preferentially expressed in the liver, lung, and gut, compared to the ubiquitous expression of the IFNα receptor, which explains its better adverse effect profile [22]. Human liver chimeric mouse model studies have reported reduced intrahepatic HDAg and HDV RNA levels with PEG-IFNλ, which are comparable to those observed with PEG-IFNα [23]. The LIMT-1, phase II trial, with PEG-IFNλ, reported a 24-week post-treatment virological response of 36% and 16%, respectively, with subcutaneous injections of 180 µg/week and 120 µg/week, for 48 weeks. Hepatobiliary adverse effects requiring drug interruption was encountered with both dosages in this trial [24]. The LIMT-2 phase III trial, which evaluated PEG-IFNλ at 180 µg/week in compensated liver disease, was discontinued in 2023 due to concerns about hepatobiliary complications, which led to hepatic decompensation [25]. Considering the overall mediocre response rates, a narrow therapeutic index, the need for parenteral therapy, and the emergence of safer, novel medications, PEG-IFNλ may be pushed into oblivion in the HDV therapeutic landscape. However, future research may reveal that its immunological effects are beneficial when combined with direct antiviral agents, possibly at a lower dose and in specific patient populations.
Several trials have evaluated the role of nucleos(t)ide analogues (NA) in treating HDV-HBV infection. Lamivudine, famciclovir, entecavir, and tenofovir have been evaluated as monotherapy, with no clinically meaningful response in HDV infection [26–29]. The EASL, 2023 recommends using NA in patients with HBV-HDV infection, guided by the indications for HBV and not targeting HDV [8].
Bulevirtide interacts with the hepatocyte NTCP and blocks the entry of the HDV/HBV into the hepatocyte [30]. Hepatocyte entry blockade results in diminished immune-mediated hepatocyte injury, subsequent viral replication, and a reduction in viral load, leading to a biochemical and virological response. Multiple MYR trials since 2015 have demonstrated the safety and efficacy of bulevirtide [31–33]. The MYR 202 dose-finding trial estimated that subcutaneously administered doses of 2 mg and 10 mg per day were effective in HDV infection [32]. In a phase III trial, Wedemeyer et al. [34] reported that at least a 2 log10 IU/mL reduction in HDV-DNA levels and normalization of ALT were achieved in nearly half of the subjects at 48 weeks. Studies have demonstrated a good safety profile, with non-specific symptoms, such as leukopenia and thrombocytopenia, reported in 2–8% of the subjects. Dose-dependent elevation of bile acid levels has been reported consistently across studies, but asymptomatic and reversible upon drug discontinuation [35]. The fact that NTCP is a natural bile acid transporter explains the elevation of bile acids with bulevirtide therapy [36]. Due to its efficacy and safety profile, bulevirtide received conditional approval for treating chronic HDV infection in Europe in 2020. The EASL Practice Guidelines, 2023, suggest considering bulevirtide in all patients with chronic HDV and compensated liver disease; however, it notes that the duration and dose have yet to be defined [8]. Beyond the trials, real-world studies from France, Italy, and Austria reported excellent virological response rates and safety profiles with bulevirtide administered at 2 mg and 10 mg daily [37–39]. Bulevirtide monotherapy in cirrhosis reportedly reduces the progression to decompensation and mortality [40]. The off-label use of bulevirtide in patients with decompensated cirrhosis was associated with similar biochemical and virological responses; however, further data from randomized trials would be required to confirm these benefits [41].
Ezetimibe is a lipid-lowering drug that acts by inhibiting the Niemann-Pick C1-Like 1 protein receptors on enterocytes, which are essential for the absorption of luminal cholesterol. Additionally, it exhibits inhibitory effects on NTCP, the hepatocyte entry receptor for HBV and HDV. Abbas et al. [42] reported a ≥ 1 log reduction in HDV-RNA with a 12-week course of ezetimibe in a phase II trial. Further evaluation, with a larger number of subjects, is required to prove the efficacy of ezetimibe in HDV infection.
Lonafarnib, an orally administered farnesyltransferase inhibitor, is being tested for the treatment of HDV infection. Prenylation refers to a post-translational modification that involves the covalent addition of lipid groups to the polypeptide chains, mainly at the C-terminus. Farnesylation is a type of prenylation in which a farnesyl group is added and catalyzed by the enzyme farnesyltransferase [43]. Prenylation of HDAg is a critical post-translational modification that facilitates its interaction with HBsAg and the subsequent release of virions from the hepatocyte. Blockade of prenylation prevents the release of nascent HDV virions from hepatocytes. Hence, prenylation inhibitors result in a lower circulating viral load and a better virological response [7]. Phase II trials have yielded a statistically significant reduction in HDV-RNA, with no major adverse effects; however, the ALT trends did not differ from those of the placebo [44]. The phase III D-LIVR trial evaluated ritonavir-boosted lonafarnib, PEG-IFNα monotherapy, the combination of ritonavir-boosted lonafarnib with PEG-IFNα, and placebo in patients with compensated liver disease. The composite endpoint, ALT normalization, and at least a 2-log drop in HDV-RNA at 48 weeks was achieved in nearly 10% and 20% of the ritonavir-boosted lonafarnib and ritonavir-boosted lonafarnib with PEG-IFNα arms, respectively [45]. Higher doses of lonafarnib reportedly have higher gastrointestinal disturbances. Ritonavir boosting allows for lower lonafarnib dosage and, thereby, better tolerability [46].
Nucleic acid polymers (NAPs) are amphipathic oligonucleotides with a polyanionic backbone that interact with the viral envelope proteins, including the HBsAg. After interacting with NAPs, the envelope proteins fail to bind to host receptors, thereby preventing the virus from entering the cell. Again, the NAPs migrate into hepatocytes and interact with the nascent HBsAg, preventing the exit of newly formed virions [43]. Furthermore, limited data suggests engagement of HSPG/NTCP by NAPs, further impeding viral entry [47]. REP 2139 is a candidate NAP currently under evaluation for HBV and HBV-HDV coinfections. Reports of phase II trials, with REP 2139 chelates combined with PEG-IFN, are promising in HBV-HDV coinfection [48, 49]. However, the combination therapy of REP 2139 and PEG-IFN was associated with significant ALT elevations, requiring further evaluations for its safety [50].
Viral RNA silencers utilize small interfering RNAs to silence the viral RNA. ALN-HDV belongs to this class of medications and is currently in preclinical trials for the treatment of HDV [51].
GI-18000, a candidate immune response stimulator, is undergoing preclinical trials for the treatment of various diseases, including HBD-HDV infections. The proposed mechanism involves the activation of specific host T-cells to target hepatocytes infected with HBV and HDV [52].
Combination therapy, utilizing the synergic and complementary effects of multiple HBV/HDV-targeted medications, should technically offer advantages over monotherapy. Following this principle, numerous drug combinations have been and are being evaluated in the management of HDV.
NA-IFN/PEG-IFN: Several studies evaluated this combination in HDV infection, with most reporting futility or clinically irrelevant results. Wedemeyer et al. [53], 2011, evaluated Adefovir and PEF-IFN as monotherapies and combination therapies in HBV-HDV infection. There was no significant difference in HDV-RNA negative status at 48 weeks between PEG-IFN and Adefovir-PEG-IFN subgroups [53]. Boyd et al. [29] failed to demonstrate a clinically relevant response with tenofovir-PEG-IFN therapy. The study by Wolters et al. [54] was unable to support the use of lamivudine-IFN in HBV-HDV infection. The meta-analysis by Rong et al. [55] reported no additional benefit in adding NA to IFN preparation.
Bulevirtide-PEG-IFN: Multiple recent trials have reported favorable outcomes when bulevirtide is combined with PEG-IFN to treat HDV infection. Asselah et al. [56] evaluated the benefits of combination therapy compared to bulevirtide or PEG-IFN monotherapy. They reported a significantly higher undetectable status for HDV-RNA at 24- and 48-week post-treatment follow-up. Both 2 mg and 10 mg of bulevirtide yielded superior results at 24 weeks, but only 10 mg dosage had benefits at 48 weeks [56]. A meta-analysis of multiple drug combinations in HDV infection reported a combination of bulevirtide and IFNs to be the most potential treatment option [55]. The results of the French Early Access Program, with bulevirtide at 2 mg/day, combined with PEG-IFNα at 180 μg/week, estimated HDV virological responses of 84% and 94%, respectively, at 24 weeks and 48 weeks of treatment [37].
Bulevirtide-NA: In case reports, the bulevirtide-tenofovir combination was associated with improved liver function and virological response [57]. The MYR202 evaluated different doses of bulevirtide in combination with tenofovir and tenofovir monotherapy. The trial reported significantly higher levels of HDV virological response at the end of therapy with all combinations; however, 24 weeks post-treatment, most patients experienced viral rebound [32]. Hence, the beneficial effects of this combination require further validation, including for the optimal duration of therapy.
Lonafarnib-IFN/PEG-IFN/NA: A phase II study evaluating the combination of lonafarnib and lonafarnib-PEG-IFNα reported virological response rates of 46% and 89%, respectively, at 24 weeks of therapy [58]. The LOWR HDV-1 study reported a better virological response with ritonavir- and lonafarnib and an even more substantial response with lonafarnib-PEG-IFNα [46]. The phase II LIFT-HDV study analyzed a combination of PEG-IFNλ, ritonavir, and lonafarnib and reported a 77% virological response at the end of 24 weeks of therapy, as well as a 23% response 24 weeks after the end of treatment [59].
In brief, combination therapies for HDV infection are likely more efficacious than monotherapy owing to the synergistic effects of medications. The pharmacotherapeutic options for patients with HDV infection are summarized in Table 1, while Figure 3 depicts the mechanism of action of these medications.
Summary of current and potential future pharmacotherapies for HDV infection
| Drug | Class | Route | Status | Dose | Duration |
|---|---|---|---|---|---|
| PEG-IFNα [15–21] | Immunomodulator | Subcutaneous | Off-label# | PEG-IFNα-2a: 180 µg/weekPEG-IFNα-2b: 1.5 µg/kg/week | 48 weeks |
| PEG-IFNλ [22–25] | Immunomodulator | Subcutaneous | Phase III | 120 or 180 μg/weekly‡ | 48 weeks |
| Bulevirtide [30–40] | NTCP receptor blocker | Subcutaneous | Approved°° | 2 mg daily** | Unclear |
| Lonafarnib [43–46] | Prenylation inhibitor | Oral | Phase III | 50 mg to 200 mg, twice daily‡ | Unclear |
| REP 2139 [43, 47–50] | Nucleic acid polymers | Intravenous | Phase III | 500 mg weekly‡ | Unclear |
| ALN-HDV [51] | Viral RNA silencers | Subcutaneous | Phase II | Trials ongoing | |
| GI-18000 [52] | Immune response stimulators | Subcutaneous | Phase II | Trials ongoing | |
‡ Limited data from trials; °° approved by European Medicines Agency, not US-FDA approved; ** recommended by European Medicines Agency, higher doses of 5 mg and 10 mg have been evaluated in trials; # recommended off-label in the treatment of HDV, not formally approved. HDV: hepatitis D virus; IFN: interferon; NTCP: Sodium Taurocholate Co-transporting Polypeptide; PEG-IFNα: pegylated-IFNα
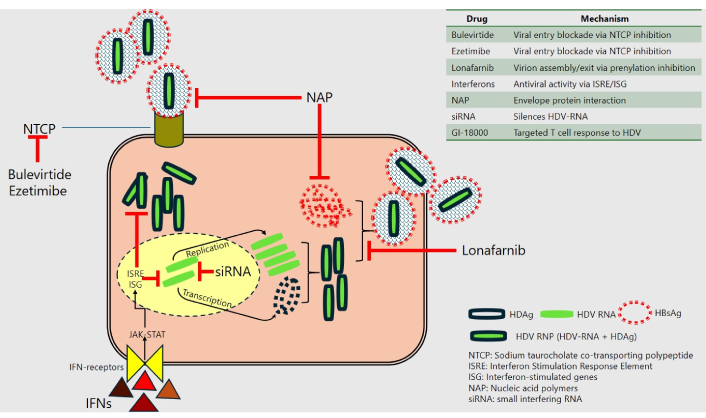
A summary of HDV virology and the current therapeutic targets. The HDV-ribonucleoprotein (HDV-RNP), enveloped by the HBsAg, interacts with the NTCP on hepatocytes to facilitate hepatocyte entry. Within the hepatocytes, the HDV utilizes the host enzymes to replicate and transcribe, forming nascent HDV-RNP. Farnesylation of the HDAg on the HDV-RNP allows it to interact with HBsAg and form nascent virions, which exit the hepatocytes. Bulevirtide and ezetimibe inhibit the NTCP, thereby preventing viral entry into hepatocytes. Lonafarnib, a farnesyltransferase inhibitor, prevents the prenylation of HDAg, the assembly of nascent HDV-RNP-HBsAg, and the subsequent release of virions from the cell. IFNs act through the JAK-STAT pathway, causing the activation of ISREs and ISGs, which results in numerous antiviral effects. The nucleic acid polymers link to and prevent the HBsAg from enveloping the HDV-RNP, as well as its interaction with NTCP. HBsAg: hepatitis B surface antigen; HDAg: hepatitis D antigen; HDV: hepatitis D virus; IFN: interferon; JAK-STAT: Janus kinase-signal transducer and activator of transcription
Human immunodeficiency virus (HIV): The management of HDV in patients with HIV is an area of interest as both infections, at least to some extent, share similar modes of transmission. IFN-based monotherapies have been utilized in the treatment of HIV-coinfected, non-decompensated liver diseases with modest response rates. Bulevirtide monotherapy in five patients infected by the HDV-HBV-HIV triad complicated with cirrhosis and clinically significant portal hypertension was associated with a progressive reduction in HDV RNA and transaminases, with good tolerance and no detectable drug-to-drug interactions [60]. de Lédinghen et al. [61] administered bulevirtide, 2 mg, with or without PEG-IFNα in HIV-coinfected patients. At 44 weeks, 52.6% and 71.4% of patients achieved virological response with monotherapy and combination therapy, respectively. The regimens were well-tolerated and had no deleterious effects on CD4 counts or HIV viral loads [61]. However, these studies were non-randomized and involved only a small number of patients; larger, better-designed studies are needed to substantiate the benefits. There is a lack of data to suggest a role for other novel molecules in the treatment of HDV in people living with HIV infection.
Decompensated liver diseases: Regardless of etiology, the optimal management of decompensated liver disease is hepatic transplantation. The use of IFN-based therapies is generally discouraged in such cases owing to the risk of worsening hepatic function [62]. Bulevirtide, though approved for treatment in compensated liver disease, has been used off-label in the treatment of decompensated diseases with similar virological and biochemical response rates [41]. Data are lacking regarding the role of other agents in this subset of patients. Hepatitis B immunoglobulin and NA is associated with a lower risk of HDV recurrence post-liver transplantation. The European Liver and Intestine Transplantation Association recommends lifelong combination therapy of immunoglobulin and NA following liver transplant in HDV infected [63].
Pediatric: Currently, there is a dearth of data regarding HDV management in children. Trials from Greece and Pakistan reported the safety of conventional IFN in children; however, the efficacy was not encouraging [64, 65]. PEG-IFNs were also employed in the treatment of HDV in the pediatric population; again, they were not supported by governing bodies or guidelines [66]. The European Medicines Agency permits the use of bulevirtide at a dose of 2 mg daily for children over three years of age and weighing more than ten kilograms. However, the safety and efficacy of bulevirtide remain unestablished in individuals under 18 years of age [67].
Pregnancy: Convention and PEG-IFN are contraindicated, while there is no published literature to support the use of novel anti-HDV agents during pregnancy. Universal precautions and optimal management of HBV infection are believed to prevent the other to child transmission of HBV infection [68].
Pharmacotherapy of hepatitis D has been a formidable challenge in hepatology due to its unique virology, significant disease burden, and limited therapeutic armamentarium. However, recent advances in molecular virology and immunology have catalyzed the development of targeted therapies, such as bulevirtide and lonafarnib, which promise to improve patient outcomes. Combination regimens, through synergistic mechanisms, are poised to become the cornerstone of future HDV management. Despite these advancements, unmet needs persist for patients with decompensated cirrhosis. Ongoing research and clinical collaboration will be crucial in achieving sustained virological control and enhancing long-term outcomes for individuals infected with HDV.
AASLD: American Association for the Study of Liver Diseases
ALT: alanine aminotransferase
Anti-LKM-3: anti-liver-kidney-microsomal antibodies
EASL: European Association for the Study of the Liver
HBsAg: hepatitis B surface antigen
HBV: hepatitis B virus
HDAg: hepatitis D antigen
HDV: hepatitis D virus
HDV-RNP: hepatitis D virus-ribonucleoprotein
HIV: human immunodeficiency virus
HSPG: heparan sulfate proteoglycan
IFNα: interferon alfa
JAK-STAT: Janus kinase-signal transducer and activator of transcription
NA: nucleos(t)ide analogues
NAPs: nucleic acid polymers
NTCP: Sodium Taurocholate Co-transporting Polypeptide
PEG-IFNs: pegylated interferons
During the preparation of this work, Figure 1 was created using OpenAI’s ChatGPT version April 2025. This tool was utilized to visually communicate a complex concept for which no suitable copyright-free image was available from conventional sources. After using the tool, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the content of the publication.
GSZ: Conceptualization, Investigation, Data curation, Writing—original draft. AJ: Investigation, Data curation, Writing—original draft, Writing—review & editing. Both authors read and approved the submitted version.
The authors have no conflicts of interest to declare.
Not applicable.
Not applicable.
Not applicable.
The data analyzed in the preparation of the manuscript is mostly available free of cost over the Internet. The corresponding author can be contacted for any concerns or clarifications regarding data availability.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

