 Review
Review

Affiliation:
Department of Nutritional Sciences, Molecular Nutritional Science, University of Vienna, A-1090 Vienna, Austria
ORCID: https://orcid.org/0000-0002-4191-2570
Affiliation:
Department of Nutritional Sciences, Molecular Nutritional Science, University of Vienna, A-1090 Vienna, Austria
ORCID: https://orcid.org/0000-0002-3934-747X
Affiliation:
Department of Nutritional Sciences, Molecular Nutritional Science, University of Vienna, A-1090 Vienna, Austria
Email: ina.bergheim@univie.ac.at
ORCID: https://orcid.org/0000-0002-3356-4115
Explor Dig Dis. 2022;1:51–71 DOI: https://doi.org/10.37349/edd.2022.00005
Received: April 27, 2022 Accepted: July 14, 2022 Published: August 29, 2022
Academic Editor: Oren Tirosh, The Hebrew University of Jerusalem, Israel
Worldwide the number of individuals being overweight or obese has dramatically increased during the last decades, which is also associated with a similar dramatic increase of individuals afflicted with metabolic disorders like dyslipidemia, hypertension, and non-alcoholic fatty liver disease (NAFLD). Genetic predisposition may account for some of the increases in body weight and the development of metabolic disorders; however, much is probably also related to the changes in physical activity and dietary pattern. Indeed, results of epidemiological studies suggest that a ‘western-type dietary pattern’ composed of highly processed foods, sweetened foods, and beverages, all adding to a low fiber but high sugar and saturated fat intake, may increase the odd of developing overweight and metabolic disorders. Consumption of sugar, and especially, fructose has repeatedly been discussed to be a key contributor to the development of health disturbances including hypertension, dyslipidemia, insulin resistance as well as NAFLD. However, despite intense research effort, the question if and how (high) dietary fructose intake interferes with human health has not yet been fully answered also as findings are sometimes contradictory. In the present narrative review, results of recent studies assessing the effect of fructose consumption on the development of metabolic disorders including hypertension, dyslipidemia, cardiovascular diseases (CVDs), hyperinsulinemia, and NAFLD as well as underlying molecular mechanisms are reviewed, thereby, aiming to further address the question if (high) fructose intake is a trigger of metabolic diseases.
Along with the increases in prevalence of overweight and obesity, the prevalence of several metabolic disorders such as insulin resistance, dyslipidemia, hypertension, and even non-alcoholic fatty liver disease (NAFLD), has also increased (for an overview see [1]). The interactions between overweight and the development of metabolic disorders are complex and involve many factors like overall caloric intake, dietary pattern, physical activity, and genetic predisposition. While a lot of efforts have been admitted to unraveling mechanisms involved in the effects of general overnutrition and excessive consumption of certain dietary components such as saturated fatty acids, cholesterol, and sugars in the development of metabolic diseases, the understanding of the impact of single nutritional factors is still limited.
Results from some older epidemiological [2, 3] and intervention studies [4–7], as well as animal experiments (for an overview see [8]), suggest that the consumption of certain foods like sugary beverages may impact both the development of overweight and metabolic disorders. Furthermore, results of some more recent human and animal studies suggest that in settings of isocaloric nutrition, and even in the absence of marked weight gain intake of fructose-enriched diets might be causative in the development of metabolic disorders [7, 9]. Also, results of controlled intervention trials further suggest that dietary glucose and fructose intake may differ in their effects on glucose and insulin release and on satiety and appetite as well as the release of related gastrointestinal hormones [10–15]. However, there are also results suggesting that the effects of fructose and glucose on food intake and appetite as well as the development of metabolic diseases are much alike and that even a fructose-rich diet may have limited to no effects on the development of metabolic diseases in healthy individuals [16–19]. Still, the understanding of the effects of dietary fructose on the development of metabolic diseases is limited. Starting from this background, the aim of the present narrative review is to summarize contradictory data and recent findings of clinical and animal-based studies regarding the role of fructose in the development of selected metabolic disorders such as dyslipidemia, insulin resistance, hypertension, and NAFLD. Also, some of the underlying molecular mechanisms are discussed, thereby, also aiming to answer the question of how fructose is a trigger in the development of metabolic diseases.
Natural fructose is found in fruits, vegetables (e.g., in pepper or carrots), or honey. Results of the Dutch National Food Consumption Survey 2007–2010 suggest that fructose naturally found in fruits, juices, and vegetables accounts for ~26%/34% (male/female, ~12 g/14 g fructose per day) of the mean daily fructose intake [~48 g/42 g per day (male/female)] in adults [20]. In addition, added sugars, like sucrose or high-fructose corn syrup (HFCS) also markedly contribute to the dietary intake of fructose (see Table 1). In many countries, HFCS is used as a replacement for sucrose in soft drinks, sweets, bakery goods, and dairy products nowadays [21]. In the US, total daily HFCS intake is ~28 g/day and that of sucrose comes up to ~50 g/day (data from US Department of Agriculture [22]). These data suggest that despite a decrease in sugar intake during the last decades [23], total sugar intake in the US is still above the recommendations of the World Health Organization (WHO) [24] (energy derived from added sugar intake < 10% of total energy intake accounting to ~50 g of sugar in a 2,000 kcal diet [25]). These data also suggest that fructose intake is still rather high. Surveys conducted in Europe also report that total sugar intake in adults accounts for ~15–21% of total energy intake [26]; however, data on fructose intake in Europe are limited and are from before the coronavirus disease 2019 (COVID-19) pandemic. For instance, results of our own studies and those of others suggest that the average fructose intake in healthy middle-aged individuals in Germany ranges from ~40 g to ~48 g fructose/day [7, 27–29].
Fructose (and glucose) content of different foods
| Food | Free fructose (g)/100 g | Total fructose (g)/100 g | Free glucose (g)/100 g | Total glucose (g)/100 g |
|---|---|---|---|---|
| Apple juice | 6.8 | 8.3 | 2.4 | 3.9 |
| Orange juice | 2.5 | 4.2 | 2.6 | 4.3 |
| Banana | 3.4 | 8.6 | 3.5 | 8.7 |
| Kaki | 8.0 | 8.5 | 7.0 | 7.5 |
| Pear | 6.7 | 7.6 | 1.7 | 2.6 |
| Apple | 5.7 | 7.0 | 2.0 | 3.3 |
| Watermelon | 3.9 | 5.1 | 2.0 | 3.2 |
| Orange | 2.6 | 4.3 | 2.3 | 4.0 |
| Grapefruit | 2.1 | 3.6 | 2.4 | 3.9 |
| Strawberry | 2.2 | 2.7 | 2.2 | 2.7 |
| Beetroot | 0.3 | 4.3 | 0.3 | 4.3 |
| Sweet pepper (red) | 3.7 | 3.8 | 2.3 | 2.4 |
| Carrot | 0.8 | 3.2 | 0.8 | 3.2 |
| Sweet pepper (green) | 1.2 | 1.3 | 1.5 | 1.6 |
| Cucumber | 0.9 | 1.0 | 0.9 | 1.0 |
| Potato | 0.2 | 0.4 | 0.2 | 0.4 |
| Spinach | 0.1 | 0.2 | 0.1 | 0.2 |
| Yoghurt (fruit) | 0.5 | 5.9 | 0.2 | 5.6 |
| Milk | 0 | 0 | 0 | 0 |
| Yoghurt | 0 | 0 | 0 | 0 |
| Honey | 38.8 | 40 | 33.9 | 35.1 |
Data from software EBISpro (version 8.0, 2007, Germany) based on Bundeslebensmittelschlüssel, Germany.
Total fructose/glucose derived from sucrose and free fructose/glucose
With the diet, fructose is either ingested as free fructose (monosaccharide), sucrose (disaccharide), or fructan. It is well described that intestinal fructose and glucose uptake differs. Indeed, as firstly described by Crane [30] in 1962, glucose is predominately actively taken up into the enterocytes through an energy-dependent transporter, the so-called sodium-dependent glucose transporter 1 (SGLT1, for an overview see [31, 32]). In the presence of high luminal glucose concentrations, glucose transporter 2 (GLUT2) also seems to facilitate some of the glucose uptake (for an overview see [33]). In contrast, fructose is taken up by the enterocyte via the energy-independent transporter GLUT5, being proposed to be specific for fructose (for an overview see [31, 32, 34]). Results of in vivo and ex vivo studies also suggest that under certain conditions, e.g., when high glucose concentrations are present in the intestinal lumen [35], luminal fructose uptake into enterocytes may also be facilitated through GLUT2 [35, 36]. However, when compared to glucose uptake facilitated by SGLT1, fructose uptake through GLUT5 is slower than that of glucose [37]. Indeed, it has been shown that the absorptive capacity for fructose in the small intestine is lower than that of glucose [38], subsequently, leading to a loss of notable amounts of fructose into the colon, where the sugar is fermented [39]. At the basolateral side of the enterocytes, export of both fructose and glucose into blood is mediated through GLUT2 (for an overview see [32] and also Figure 1). Even after an elevated intake of fructose, concentrations in peripheral blood are rather low [40]. Indeed, recently it was shown in a study of administrating isotope-labeled fructose orally to healthy volunteers, that only ~4 g of a 30 g fructose load reached systemic circulation without being metabolized in splanchnic organs [41]. However, the debate whether this is related to a very effective clearance of fructose by the liver or to metabolism of the monosaccharide in enterocytes is still ongoing. Indeed, Bode et al. [42] reported in almost 40 years ago that fructose compared to glucose or starch contributes to adaptive changes of enzymes involved in fructose metabolism in jejunal mucosa of rats, thereby, may be contributing to maintaining fructose levels in peripheral blood rather low. Also, in obese subjects, the consumption of a fructose-sweetened beverage with a meal resulted in higher postprandial triglycerides levels compared to the consumption of the same meal in combination with a glucose-sweetened beverage [43]. In line with these findings, Jang et al. [44] recently reported that a gavage of low doses of fructose [0.5 g fructose/kg body weight (BW), equivalent to ~0.5% of the daily caloric intake of a mouse or 3 g of fructose in a human] in mice was almost completely cleared by the enterocytes (~90%) via fructokinase. Only when mice were exposed to doses > 1 g fructose/kg BW, fructose reached the liver (~30%) and was also cleared by the colonic microbiome, respectively, in this study [44]. Studies in mice further demonstrated that in the absence of fructose intake portal and systemic fructose levels are < 0.1 mmol/L and increased up to 0.2–1 mmol/L upon intake of fructose-enriched diets (20–40% fructose) [40]. Furthermore, it has been shown in studies with hamsters that after dietary fructose intake, intestinal de novo lipogenesis (DNL) and apolipoprotein B (ApoB) 48 synthesis are both induced [45]. These data suggest that enterocytes may play a pivotal role in metabolizing fructose (for an overview see [34, 46]). However, further studies are needed to determine the proportional contributions of the intestine, the liver, and extra-splanchnic tissue to fructose metabolism in different settings and especially in humans.
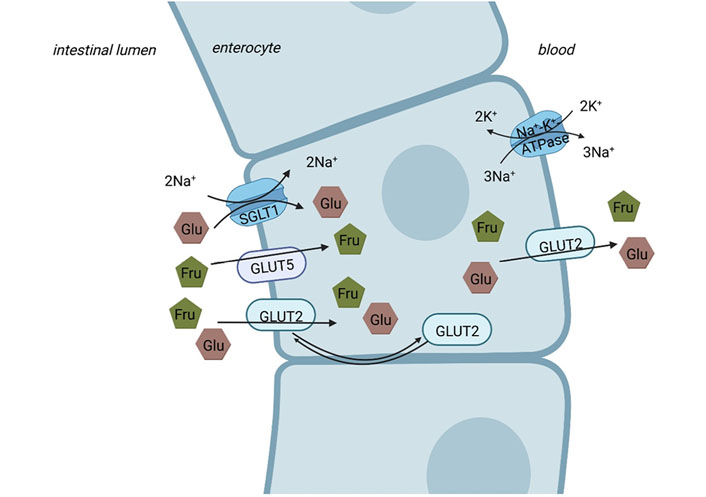
Intestinal sugar uptake. While glucose (Glu) is actively taken up via the SGLT1 from the intestinal lumen into enterocyte on the apical side (Na+ gradient maintained by Na+-K+-ATPase), fructose (Fru) is apically taken up into the enterocytes via GLUT5. At high concentrations of luminal saccharides, apical GLUT2 contributes to the uptake of fructose and glucose into the enterocytes. The basolateral transport from the enterocyte to the blood is facilitated via GLUT2 for both glucose and fructose. Figure was created based on [32, 47] with BioRender.com
Fructose and glucose metabolism also markedly differ within cells, which has been reviewed in great detail by others (for an overview see Figure 2 and [48–50]). In brief, fructose is phosphorylated to fructose-1-phosphate via fructokinase C. In contrast to the conversion of glucose to fructose-6-phosphate and via phosphofructokinase to fructose-1,6-bisphosphate, being tightly regulated by citrate and ATP, the conversion of fructose to fructose-1-phosphate via fructokinase is facilitated in the absence of any feedback control. Accordingly, intermediates of the metabolism of fructose like glyceraldehyde-3-phosphate and dihydroxyacetone-phosphate (DHAP) are built without regulation, bypassing phosphofructokinase, the major regulatory enzyme of glycolysis (for an overview see [48, 49]). Furthermore, metabolizing fructose to fructose-1-phosphate requires ATP as a co-substrate [51]. Indeed, in settings of high fructose intake, the depletion of ATP has been shown to result in an activation of adenosine monophosphate (AMP) deaminase, and subsequently, an induction of the purine nucleotide turnover. This may lead to an enhanced generation of uric acid [51, 52]. The monosaccharide is then metabolized to DHAP and glyceraldehyde through aldolase B. Glyceraldehyde is converted to glyceraldehyde-3-phosphate. Enzymes necessary for these metabolic steps are only known to be expressed in enterocytes, hepatocytes, and proximal tubular cells [48, 53]. However, extra-splanchnic tissue also has been shown to express fructose transporters and fructokinase A, the latter being an isoform of fructokinase (for an overview see [49]). From there on, fructose and glucose metabolism are alike (see also Figure 2) [48].
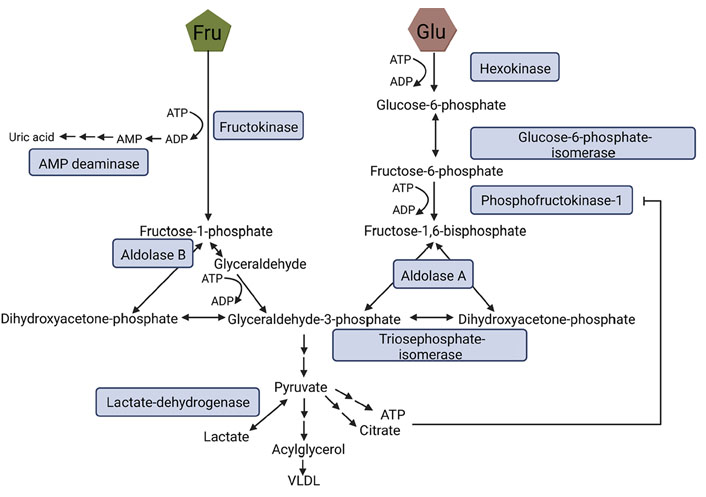
Fructose and glucose metabolism. During glycolysis Glu is metabolized to fructose-6-phosphate and, through phosphofructokinase-1 to fructose-1,6-bisphosphate being the rate-limiting step of this metabolic pathway which is strongly regulated by ATP and citrate. Fru bypasses this step and is metabolized to fructose-1-phosphate via fructokinase. Both fructose-1,6-bisphosphate and fructose-1-phosphate can be converted to DHAP and glyceraldehyde-3-phosphate, the latter being further processed to pyruvate ending up in the generation of lactate, very-low-density lipoprotein (VLDL), citrate, or ATP. Figure was created based on [54] with BioRender.com. ADP: adenosine diphosphate
Results of human intervention studies suggest that in healthy individuals, medium-term (> 7 days) intake of large amounts of fructose with fructose being exchanged isocalorically for other carbohydrates or when fructose is consumed in hypercaloric amounts increases fasting ApoB as well as triglyceride serum concentration levels [4, 55, 56]. As summarized in a meta-analysis, similar effects of fructose have also been reported in trials conducted in patients with type 2 diabetes [57]. However, a meta-analysis also suggests that fructose has only adverse effects on ApoB and triglycerides when being added to the diet at a high concentration, e.g., providing additional energy (e.g., + 21–35% energy) [58]. Recently, it has been reported that after the 10-week consumption of a fructose-rich diet but not after the intake of a glucose-rich diet, levels of ApoCIII were significantly increased, being also associated with a higher postprandial triglyceride content in low-density lipoprotein (LDL) [59]. Te Morenga et al. [60] provided evidence that a higher intake of sugars is associated with increased serum lipids. In contrast, LDL or high-density lipoprotein (HDL) cholesterol concentrations in blood were not reported to be affected by the monosaccharide [56]. When interpreting these results, it should be taken into consideration that some of the studies show no adverse effects of fructose intake on markers of lipid metabolism. Also, fructose provided by whole fruit was included in these studies and not only that derived through added sugar.
Results of studies in healthy individuals also suggest that an acute intake of fructose is associated with decreased free fatty acid levels [61]. In line with these findings, Abdel-Sayed et al. [62] also reported that after 7 days of consuming a high-fructose diet (3 g fructose/kg BW, on average 234 g fructose/day), lipolysis and lipid oxidation were markedly inhibited in healthy lean males. Molecular mechanisms underlying this effect of fructose remain to be determined. The release of insulin, shown to be the key suppressor of lipolysis in adipose tissue [63], has been reported to be limited after fructose ingestion (also see below) which might, at least in part, explain these findings.
However, despite the contradictory findings of human intervention studies and some knowledge gaps, the American Heart Association and other large health societies by now incorporated recommendations regarding a reduction of fructose or sugar in general in their guidelines (e.g., American Heart Association no more than 150 cal/day from added sugars for men and 100 cal/day for women) [64–67].
For several decades, fructose was recommended in the therapy of type 2 diabetes due to its insulin-independent metabolism but also as it does not stimulate pancreatic insulin secretion directly [68, 69]. However, results of animal studies have shown that a chronically elevated intake of fructose induces insulin resistance [70, 71]. These findings are also supported by human studies showing a relation between the intake of fructose (total fructose including fructose consumed as sucrose and HFCS, and as free fructose) and impaired peripheral and hepatic insulin sensitivity, which may be independent of body weight gain [29, 72, 73]. Results of our own studies in animals suggest that fructose even in the absence of a marked body weight gain may lead to the development of metabolic alterations [74, 75]. Contrasting these findings, results of human intervention studies summarized in a meta-analysis of Cozma et al. [76] showed that an isocaloric exchange of fructose for other carbohydrates improves glycemic control as determined by hemoglobin A1c (HbA1c) levels in patients with type 1 and 2 diabetes, respectively, while not affecting fasting insulin or glucose levels. Furthermore, while in the prospective cohort studies of Della Corte et al. [16], no association between total intake of fructose-containing sugars was found, the meta-analysis of prospective cohort studies of Imamura et al. [77] focusing on sugar-sweetened beverages (SSB) reported an adverse association between intake of SSB and the incidence of diabetes. Similar relations were also reported for the intake of SSB and the metabolic syndrome [78, 79] but not for other foods like yogurt, fruit or 100% fruit juices, and mixed fruit juices at moderate doses [79]. Indeed, in the latter study, a U-shaped dose-dependent association between the intake of mixed fruit juices and 100% fruit juices was reported with a protective association against the metabolic syndrome with intakes of less than 200 mL of fruit juice [79].
Despite intense research efforts throughout the last decade, mechanisms underlying the effects of elevated fructose intake on insulin, and even more so, the development of hyperinsulinemia and insulin resistance are not yet fully understood. Results of some animal studies suggest that hepatic insulin resistance may be related to fructose-induced hepatic steatosis (for an overview see [80, 81] and below). Indeed, results of studies suggest that the intake of fructose adds through an increase in hepatic diacylglycerol accumulation, activation of protein kinase C and alterations of insulin-mediated Akt to the development of steatosis [82–84]. Furthermore, in recent years, fructose-induced insulin resistance has also been associated with changes of fecal microbiota composition (for an overview see [85, 86]). Recently, a study employing Wistar rats reported that a fructose-rich diet induced marked changes of fecal microbiota within days and increased protein glycation in intestine [87]. Employing in vitro studies, glycated proteins were shown to alter microbial growth in the same study. Results of our own group suggest that an activation of bacterial endotoxin-dependent signaling cascades in liver, e.g., the activation of toll-like receptor 4 (TLR4) and the subsequent induction of inducible nitric oxide synthase (iNOS) and tumor necrosis factor alpha (TNFα) may also be critical in the development of hepatic insulin resistance in rodents (also see section Fructose: a trigger in the development of NAFLD?) [71, 88, 89]. Indeed, we have shown that the alterations of insulin receptor expression and related genes in whole liver tissue are found after both short-term and long-term intake of diets rich in fructose and can be attenuated when animals are concomitantly treated with non-resorbable antibiotics [90] or through genetic deletion of genes involved in endotoxin-dependent signaling cascades like in lipopolysaccharide (LPS) binding protein, iNOS or TLR4 knockout or mutant mice [71, 88, 91]. Results of Kim et al. [92] further suggest that through carbohydrate responsive element-binding protein-dependent mechanisms, glucose-6-phosphatase is induced in livers of fructose-fed animals, which in turn may add to an enhanced glucose production. However, if mechanisms alike are also involved in hyperinsulinemia and the development of insulin resistance in humans consuming large amounts of fructose remains to be determined.
Somewhat contrasting the above reported findings, it has repeatedly been suggested that when ingested in small amounts (≤ 10 g/meal), fructose and its epimers including allulose, tagatose, and sorbose may dampen glycemic response to meals with high glycemic index. Indeed, results of acute glucose challenge tests have shown that small doses of ≤ 10 g fructose/meal may decrease postprandial glycemic response to glucose tolerance tests up to 30% in patients with type 2 diabetes and healthy individuals [93–95]. Furthermore, it has been suggested by the results of randomized controlled human intervention trials that a so-called ‘catalytic’ dose of fructose defined as an intake of ≤ 36 g/day separated in three meals with ≤ 10 g/meal and two snacks with ≤ 3 g/snack of fructose in exchange for other carbohydrates like starch may decrease HbA1c levels by ~0.4% (for an overview see [93, 96]). However, underlying mechanisms have not yet been fully understood.
While results of several animal studies suggest that a diet rich in fructose may lead to the development of hypertension [97–99], findings regarding the association of fructose and cardiovascular disease (CVD) and blood pressure are also still controversial [100]. For instance, results of some cohort studies suggest that total dietary intake of fructose including fructose derived from fruits and vegetables as well as from added fructose (including fructose contained in sucrose) may increase the odds of CVD in those consuming large amounts (e.g., > 7.4% of total energy intake) [101]. However, it was also suggested that effects of ‘natural’ fructose, e.g., fructose consumed as fruits and vegetables, may differ from that of added fructose [101]. As already pointed out in other sections of this review, data regarding effects of ingesting large amounts of fructose on risk factors of CVD including blood lipid levels, LDL, and HDL cholesterol are contradictory (see section Effect of fructose on lipid metabolism and the development of dyslipidemia). Still, in intervention studies exposing normal and overweight individuals to large amounts of fructose (200 g/day for 2 weeks) or sucrose (120 g/day), blood pressure was shown to significantly increase [5, 102]. The meta-analysis of Ha et al. [103] suggests that even excessive amounts of fructose may have no adverse effects on blood pressure, while in a study published by the same group in 2019, a positive association was reported for the consumption of SSB and the prevalence of hypertension [104]. Also, in the latter study, for the consumption of moderate amounts of 100% fruit juice, a protective association was reported. Furthermore, Te Morenga et al. [60] reported in their meta-analysis that dietary sugars may be independent of body weight and influence blood pressure. Somewhat in line with these findings, Gugliucci et al. [105] reported that in overweight children with metabolic disorders, replacing dietary fructose (11.8% to 3.8% of total daily energy intake) was associated with a reduction of risk factors of CVD (e.g., a reduction of ApoB, increases in large HDL, improved insulin sensitivity). Also, it has been shown that decreasing dietary fructose intake results in lower blood pressure [106, 107], further suggesting that fructose may be a target in the treatment and also in the prevention of hypertension. Indeed, dietary patterns are recommended to prevent and treat CVD and also include an avoidance of a daily intake of free sugar of > 10 g/day [108].
During the last years, possible mechanisms regarding the interaction of fructose and blood pressure have repeatedly been reviewed (for an overview see [50, 109, 110]). Therefore, in the present review, we will only briefly highlight some of the possible mechanisms involved in the fructose-induced elevation of blood pressure found in some human studies. Disturbances of sodium homeostasis are thought to be among the key factors in the development of hypertension (for an overview see [111]). The WHO [112] strongly recommends a reduction of dietary salt intake to < 5 g/day. Results of animal studies have shown that fructose feeding stimulates both sodium and chloride uptake in small intestine and kidney [113–115]. A genetic deletion of GLUT5 and putative anion transporter 1 in mice was associated with an attenuation of fructose-induced hypertension [113, 114]. Furthermore, it was reported that in animals fed a 60% fructose diet, renin expression in kidney was reduced [113], and that fructose feeding in rodents reduced sodium excretion by the kidney [116]. In support, Catena et al. [116] showed that in rats fed a 66% fructose-containing diet in combination with either low (0.07%), normal (0.03%), or high (7.5%) NaCl content, only those fed the normal or high-salt diet developed hypertension being associated with a lack of reduction in density of insulin receptor and messenger RNA (mRNA) in kidney. The insulin-induced sodium reabsorption in a feedback manner normally limits sodium reabsorption when high levels of dietary salt are consumed (for an overview also see [109]). Recently, it was shown that chronic intake of fructose (20% in drinking water) also increases sensitivity of proximal tubules to angiotensin II (AngII) in rats fed a high-salt diet, being also associated with increased markers of oxidative stress, e.g., AngII-dependent induction of O2– production and diminished glutathione [117, 118]. Taken together, these data further suggest that high fructose diets, at least in rodents, may alter kidney function and sodium homeostasis thereby adding to the development of hypertension.
Besides effects on sodium homeostasis, results of experimental studies also suggest that an elevated fructose intake may affect endothelial function [109]. Through the synthesis and release of mediators like endothelin-1 (ET-1) and nitric oxide (NO/NOx), vascular endothelial cells play a crucial role in the regulation of vascular tone, thereby not only affecting blood flow but also blood pressure (for an overview see [119, 120]). Studies in rats suggest that chronic intake of a fructose-rich diet can lead to impairments of endothelial function after 18 days followed by the development of hypertension after 28 days [121]. Results of animal studies have further shown that the intake of a fructose-rich diet is associated with a lower formation of NO and lower activity and expression of endothelial nitric oxide synthase (eNOS) [122–124]. Recently, we showed that consuming a diet containing 25% of energy as fructose for only 3 days leads to a significant reduction of NOx levels in blood in healthy young adults, an alteration not found when subjects ate an isocaloric glucose-enriched diet [7]. Molecular mechanisms involved in this ‘loss’ of NO have not yet been clarified. Results obtained in rats suggest that fructose intake may not directly decrease NO production but rather may impair the NO-hydrogen sulfide (H2S) interaction [125]. Klein et al. [109] proposed in their overview of the mechanisms underlying fructose-induced hypertension that through insulin resistance and the associated hyperinsulinemia, expression of ET-1 is induced subsequently leading to (1) an increase in AngII, (2) an increase of cyclooxygenase-2 (COX2) expression and the production of thromboxane A2 (TXA2), and (3) a suppression of eNOS expression (see also Figure 3). Indeed, it has been shown in fructose-fed hypertensive rats that COX2 expression and levels of TXA2 are elevated and that blocking TXA2 formation can prevent the development of fructose-induced hypertension [126]. However, data on humans are still limited. Furthermore, Ehrlich et al. [127] showed that different angiotensin-converting enzyme inhibitors attenuated the development of fructose-induced hypertension in rats supporting the hypothesis that AngII may be critical in mediating the effects of fructose regarding blood pressure. It was also shown that bosentan almost completely blocked the increase of plasma AngII found in fructose-fed rats, further suggesting that insulin-induced expression of ET-1 may be critical in the fructose-induced up-regulation of AngII [128]. Taken together, these findings together with rodent studies, showing that ET-1 expression in mesenteric vascular tissue is higher in fructose-fed rats than in controls and that blocking of the ETA and ETB receptor attenuated fructose-induced hypertension in rats [128, 129], suggest that ET-1 indeed is critical in the development of fructose-induced hypertension. In contrast, in our own studies showing a marked drop in circulating NOx serum levels, ET-1 protein levels were not altered in healthy subjects after the 3-day consumption of the fructose-rich diet [7]. However, none of the study participants in our short-term study had yet developed signs of insulin resistance.
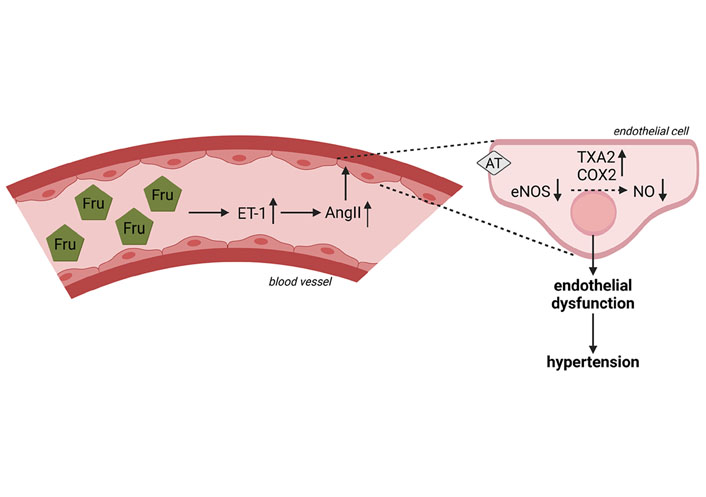
Effect of fructose on the formation of NOx and blood circulation and possible mechanism underlying fructose-induced hypertension. The intake of a Fru-rich diet leads to an increased ET-1 expression, a lower expression of eNOS, lower NOx levels in blood as well as an increase in AngII and production of COX2 and TXA2. Figure was created based on [109] with BioRender.com. AT: AngII receptor
NAFLD is by now the most prevalent liver disease in the world. Indeed, it is assumed that ~25% of the general global population is affected by NAFLD [130]. While known to be multifactorial, results of several animal studies suggest that dietary fructose may contribute to the development of NAFLD, too [9, 74]. Results of human studies are not quite clear. Indeed, results of some clinical and epidemiological studies reported a higher intake of fructose in both adults and pediatric patients with NAFLD [27, 62, 131–133], whereas others found no association [134, 135], especially, when fructose intake was normalized to overall calories consumed [136]. It has been discussed that patients with NAFLD are frequently found to be overweight, and accordingly, tend to have a higher total caloric intake; relative intake of sugar/fructose may also be higher, when compared with normal-weight healthy subjects. Furthermore, results of some human intervention studies suggest that contrary to the findings in animal experiments, even an extended and several-week-long intake of high amounts of fructose has no effect on liver fat content [19, 74, 134]. Indeed, recently, Smajis et al. [19] reported that in young healthy men, even an 8-week-long intake of 150 g fructose/day had no effect on liver lipid or glycogen content. However, Maersk et al. [6] assessing the effects of different sucrose-containing beverages—but not free fructose—showed an increase of ectopic fat in overweight subjects consuming regular cola while neither isocaloric semi-skim milk nor the intake of aspartame-sweetened diet cola or water had any adverse effects on the liver. Furthermore, there are also some data suggesting that even within 9 days of consuming a fructose-rich diet (25% of energy derived from fructose), DNL in liver and liver fat levels in healthy men may significantly increase [137]. In this study, it was not determined if fructose directly went into DNL in the liver or if DNL was altered through indirect measures like increased lipids derived through intestinal metabolism of fructose or lactate or other metabolites being built from fructose. Somewhat in line with these findings, we recently reported that increasing dietary fructose intake for only 3 days is associated with a slight but significant increase in serum alanine aminotransferase activities (~+ 10%) in healthy young adults. Similar effects were not found when the same subjects consumed glucose at concentrations alike [7]. Also, in the rare studies comparing NAFLD patients with weight-matched overweight but otherwise healthy controls or controls displaying other metabolic disorders but not NAFLD, the overall intake of sugar-rich beverages was higher in NAFLD patients despite similar caloric intake [138]. In line with these findings in adults, we recently reported that in overweight children with and without early signs of NAFLD, those with NAFLD had a slightly higher body mass index (BMI) and higher intake of fructose, mainly stemming from beverage consumption [133]. In support of the hypothesis that fructose and/or sugar intake may be critical in the development of NAFLD, results of dietary intervention studies in which adult and pediatric NAFLD patients were instructed to reduce their daily fructose or sugar intake, showed a marked improvement in disease-related parameters like liver fat content or transaminase activity in serum [139–142]. However, in the latter studies, reduction of fructose and/or sugar intake was frequently associated with a reduction of overall caloric intake, and subsequently, body weight, both being critical factors in the development of NAFLD.
In general, as already discussed above, elevated fructose intake may be due to its insulin-independent metabolism and through bypassing the regulated step of glycolysis, increase DNL in liver leading to lipid accumulation (for an overview see [143] and Figure 4). In addition, it is also discussed that during progression of NAFLD, plasminogen activator inhibitor-1 (PAI-1) dependent signaling cascades contribute to modulation of hepatic VLDL export thereby promoting the development of hepatic steatosis (for an overview see [144]). Indeed, it has been shown by us that in mice chronically fed a 30% fructose solution, PAI-1 through its inhibitory effects of urokinase-type plasminogen activator may attenuate the activation of pro-hepatocyte growth factor subsequently, leading to an inadequate upregulation of the activity of the microsomal triglyceride transfer protein in hepatocytes and export of triglycerides [145]. These data suggest that chronically elevated intake of fructose may (1) lead to an increased formation of triglycerides through an induction of DNL and (2) impair the export of fat from the liver.
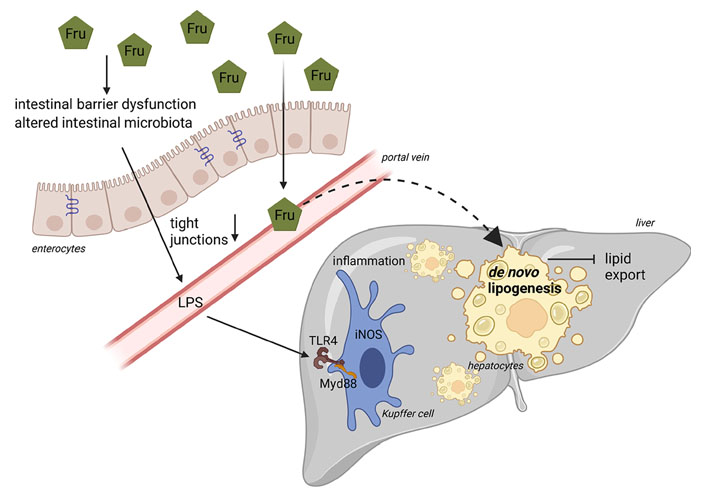
Schematic drawing on the role of fructose in the pathogenesis of NAFLD. Diets rich in Fru may alter intestinal microbiota composition and intestinal barrier function associated with a loss of tight junction proteins and subsequent increase in portal endotoxin (LPS). Through TLR4-dependent signaling cascades in the liver, endotoxin leads to an induction of iNOS, formation of reactive oxygen species, and release of proinflammatory cytokines. Due to its unregulated metabolism, Fru may also stimulate hepatic DNL. Figure was created based on [146] with BioRender.com. Myd88: myeloid differentiation primary response 88
Interestingly, already more than 10 years ago, results of our own group using an ad libitum feeding mouse model employing different mono- and disaccharides, showed that the development of fructose-induced NAFLD is associated with increased portal endotoxin levels [74]. We further showed that a concomitant treatment of fructose-fed and fructose- and fat-fed mice with non-resorbable antibiotics abolished the development of fructose-induced NAFLD [74, 90]. In line with these findings, Kavanagh et al. [9] in some years later also reported that in monkeys consuming a high-fructose diet, the development of liver damage was associated with increased microbial translocation and endotoxemia. These data suggest that other factors besides those discussed above may contribute to the development of fructose-induced NAFLD. Using animal models, it was shown by us and others that the intake of fructose-enriched diets within days to weeks leads to a loss of tight junction proteins in the upper parts of the small intestine being associated with increased levels of bacterial endotoxin in portal vein, an induction of TLR4 but also other TLRs in the liver as well as of iNOS and TNFα [88, 147–150]. Using knockout mice, e.g., LPS binding protein, TLR4, iNOS, and TNFR1, it was further shown by us that a disruption of this signaling cascade markedly attenuated the development of fructose-induced NAFLD [71, 88, 89, 91]. Furthermore, a treatment with metformin or antibiotics as well as certain amino acids like citrulline and arginine not only prevented the loss of tight junction proteins in small intestinal tissue but also attenuated the increase in bacterial endotoxin levels and the development of NAFLD found in animals fed a fructose-enriched diet [90, 147, 149–151]. Recently, we showed that fructose at concentrations that can be reached through dietary intake, e.g., 5 mmol/L, alters intestinal permeability within 40–50 min in an ex vivo everted sac model of small intestinal tissue and that this can be prevented when tissue is concomitantly treated with the amino acids citrulline or arginine [149, 150]. Jegatheesan et al. [152] and Zhang et al. [153] showed that the intake of fructose-rich diets in rats was associated with a decrease of Bifidobacterium and Lactobacillus in feces. These findings were associated with decreased expression of claudin-1 and increased expression of TNFα and TLR4 suggesting that diets enriched with fructose may alter intestinal bacterial colonization [154]. Supporting the hypothesis that dietary fructose through mechanisms depending upon changes of intestinal barrier function and an increased translocation of bacterial endotoxin may add to the development of NAFLD, Jin et al. [155] showed in adolescent NAFLD patients enrolled in a randomized cross-over trial that a challenge with fructose-containing beverages (33% fructose of estimated calorie intake) for 24 h and 2 weeks, respectively, results in elevated bacterial endotoxin levels while similar changes were not found in healthy subjects. Results of studies of our own group suggest that in dietetically standardized young healthy adults, consumption of a fructose-rich diet (25% of total energy intake) for only 3 days results in significantly increased plasma endotoxin concentrations while similar effects were not found when the same subjects consumed isocaloric amounts of glucose [7]. These findings are in contrast to those recently published by others, reporting that in a double-blind, cross-over design study with 10 obese subjects, the isocaloric substitution of complex carbohydrates with 75 g of either fructose or glucose for 2 weeks had no effect on fecal microbiota composition, gut permeability, or indices of endotoxemia [156]. Differences between our study [7] and that of Alemán et al. [156] might have resulted from the study design (standardized nutrition vs. exchange of parts of the complex carbohydrates), the dose of fructose (25E% vs. ~20E%) but also the parameters detected (bacterial endotoxin vs. CD14, intestinal fatty acid binding protein and LPS binding protein). However, the divagating results also underlie the need for further well-designed studies.
In summary, while results of epidemiological studies are somewhat contradictory and molecular mechanisms are not yet fully understood, recommendations to reduce dietary fructose intake are now found in several guidelines for the treatment of NAFLD. Indeed, Zhu et al. [157] summarizing recommendations for the treatment of NAFLD of different countries-stated that along with lifestyle interventions focusing on weight reduction and increased physical activity, patients with NAFLD should also be encouraged to generally reduce their free fructose intake.
In conclusion, while being intensely studied for several decades, understanding of the effects of dietary fructose on human health and disease development is still limited. Indeed, while some data suggest that an intake of large amounts of fructose can lead to the development of adverse effects like hypertension and NAFLD, there are also other data suggesting that even when consumed for several weeks the intake of large amounts of fructose has no effects on general health in healthy humans. Further studies are needed to determine long-term effects of fructose-rich diets and the comparison of fructose- vs. glucose-rich diets under isocaloric conditions. Indeed, as fructose is predominantly consumed either as sucrose or HFCS, both containing glucose in concentrations alike to fructose or ranging from 40–55%, interpretation of epidemiological studies assessing the effects on health and the development of metabolic diseases is difficult. Also, up to now, only limited data are available assessing what could be called the ‘matrix’ effects, e.g., if fructose consumed in isolated form in SSB or candy has a similar effect to those found after the consumption of natural foods like fruit and vegetables. Indeed, a recent systematic review by Semnani-Azad et al. [79] suggests that fructose and fructose-containing sugars when consumed in a ‘matrix’ of yogurt or fruit may have no adverse association with metabolic diseases. Also, Yan et al. [158] recently pointed out that maybe the WHO guidelines for sugar intake for adults and children recommending to reduce free sugar intake to less than 10% of total energy intake [25], resulting in a maximum of ~50 g of free sugar in 2,000 kcal/day diet, might need to be revised due to an overextrapolation of results from studies focusing on SSB. Recommendations regarding sugar and/or fructose intake are by now also found in some national nutritional recommendations, e.g., Austria: 50 g free sugar/2,000 kcal, Germany: 50 g free sugar/2,000 kcal, France: 100 g sugar/day for adults [65, 159] and in the guidelines for the treatment of several metabolic diseases [25, 157]. However, recommendations vary considerably, and results of studies are still contradictory, further suggesting that there is a great need for further studies to the better understanding of the effects of dietary fructose on humans.
AMP: adenosine monophosphate
AngII: angiotensin II
ApoB: apolipoprotein B
BW: body weight
COX2: cyclooxygenase-2
CVDs: cardiovascular diseases
DHAP: dihydroxyacetone-phosphate
DNL: de novo lipogenesis
eNOS: endothelial nitric oxide synthase
ET-1: endothelin-1
GLUT2: glucose transporter 2
HDL: high-density lipoprotein
HFCS: high-fructose corn syrup
iNOS: inducible nitric oxide synthase
LDL: low-density lipoprotein
LPS: lipopolysaccharide
NAFLD: non-alcoholic fatty liver disease
NO/NOx: nitric oxide
SGLT1: sodium-dependent glucose transporter 1
SSB: sugar-sweetened beverages
TLR4: toll-like receptor 4
TNFα: tumor necrosis factor alpha
TXA2: thromboxane A2
VLDL: very-low-density lipoprotein
WHO: World Health Organization
A Baumann: visualization, writing original draft preparation and review, and editing; A Brandt: visualization, writing original draft preparation and review, and editing; IB: conceptualization, supervision, writing original draft and review, and editing. All authors reviewed and approved to the final version of the manuscript.
The authors declare that they have no conflicts of interest related to the present work.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

