 Original Article
Original Article

Affiliation:
1Posgrado en Biología Experimental, DCBS, Universidad Autónoma Metrolitana-Iztapalapa, Mexico City 09340, Mexico
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
ORCID: https://orcid.org/0000-0003-1348-0034
Affiliation:
3Departamento de Biología de la Reproducción, Universidad Autónoma Metropolitana, Mexico City 09340, Mexico
ORCID: https://orcid.org/0000-0003-3537-1576
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
ORCID: https://orcid.org/0000-0001-7773-9848
Affiliation:
1Posgrado en Biología Experimental, DCBS, Universidad Autónoma Metrolitana-Iztapalapa, Mexico City 09340, Mexico
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
ORCID: https://orcid.org/0000-0002-8978-3122
Affiliation:
1Posgrado en Biología Experimental, DCBS, Universidad Autónoma Metrolitana-Iztapalapa, Mexico City 09340, Mexico
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
ORCID: https://orcid.org/0000-0002-6678-0877
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
ORCID: https://orcid.org/0000-0002-3897-018X
Affiliation:
4Traslational Research Unit, Medica Sur Clinic & Foundation, Mexico City 14050, Mexico
ORCID: https://orcid.org/0000-0001-7616-2661
Affiliation:
5Laboratorio de Medicina Traslacional, Unidad de Medicina Traslacional, IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
ORCID: https://orcid.org/0000-0001-8671-3367
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
6Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
ORCID: https://orcid.org/0000-0002-3923-6278
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
6Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
ORCID: https://orcid.org/0000-0001-8249-7257
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
6Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
ORCID: https://orcid.org/0000-0003-0501-7226
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
6Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
ORCID: https://orcid.org/0000-0002-5704-5985
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
6Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
Email: lebo@xanum.uam.mx
ORCID: https://orcid.org/0000-0002-6963-2642
Explor Dig Dis. 2022;1:40–50 DOI: https://doi.org/10.37349/edd.2022.00004
Received: April 30, 2022 Accepted: June 23, 2022 Published: August 11, 2022
Academic Editor: Jose J. G. Marin, University of Salamanca, Spain
Aim: Cholestasis remains a partially characterized disease. Evidence has been gained that it is a systemic disease that begins in the liver but significantly impacts other organs and systems such as the kidney, heart, and intestine, among others. One of the primary damage mechanisms is the generation of reactive oxygen species (ROS), which eventually leads to oxidative stress, impacting canalicular morphology and actin cytoskeleton changes that could worsen the problem. These characteristics are also observed in the kidney and intestine. The work focused on addressing the intestine effects of intrahepatic cholestasis induced by α-naphthyl isothiocyanate (ANIT) and the protective response of the hepatocyte growth factor (HGF).
Methods: The 10- to 12-week-old CD1 male mice were treated with ANIT and then treated or not with HGF; intestine damage was addressed by histology, immunohistochemistry (IHC) of specific markers, oxidative stress, and apoptosis.
Results: Results show changes in the intestine histology, particularly the colon and ileum, induced by the cholestasis. HGF treatment restored the histology presentation and reverted the oxidative damage, clearly indicating a healing response. This observation was supported by an increment in anti-inflammatory macrophages (CD163+) in the HGF treatment.
Conclusions: The data prove that HGF induces a protective and repairing response in the intestine under cholestatic challenges.
Cholestasis is characterized by decreased bile flow that reaches the duodenum; an impairment could drive this in hepatocellular or cholangiocellular biliary secretion or the mechanical impediment of the passage of bile through intra- or extrahepatic bile ducts; this leads to accumulation of potentially toxic biliary constituents within the liver and in the bloodstream [1, 2].
The interplay of the liver and other organs in cholestasis was emerging in the last years as a relevant issue in the pathogenesis of this systemic disorder triggered by liver dysfunction. Cholestasis remains a poorly addressed liver disease compared with others. The increment in the risk factors associated with its initiation or complications, such as obesity, non-alcoholic fatty liver disease (NAFLD), and alcohol consumption, should also require an increment in the research in the field. It is well known that cholestasis impacts not only the liver but other organs such as the kidneys [1, 3], the cardiovascular system [4], the pancreas [5], the intestine [6], and even it could be associated with cognitive problems and locomotor activity [7].
Disturbances of cholestasis in the different segments of the intestine have been poorly addressed. However, there is a clear relationship between the liver and the gut (the gut-liver axis) in the well-known enterohepatic circulation of bile acids. This process is a great example of physiological homeostasis, which, when disrupted, leads to disorders such as gallstone formation [8].
One of the main mechanisms of cholestatic damage is the generation of reactive oxygen species (ROS), which induces apoptosis and the aggravation of the problem [9], but also induces the internalization of critical transporters close associated with ROS-responsive signaling pathways [10, 11].
We have extensively reported that the hepatocyte growth factor (HGF) and its receptor c-Met display remarkable antioxidant and repair response, particularly in epithelia, by a mechanism associated with nuclear factor kappa B (NF-κB) and nuclear factor erythroid 2-related factor 2 (Nrf2) activation and the expression of antioxidants enzymes and increment in glutathione synthesis [12].
HGF/c-Met proves its invaluable effect in cholestasis in obstructive cholestasis, in a bile duct ligation model, and the well-characterized intrahepatic cholestasis induced by α-naphthyl isothiocyanate (ANIT), not only in the liver but in the kidneys [1].
Our central hypothesis is that HGF/c-Met also displays protective effects in the intestine in cholestasis-induced oxidative damage.
In the present work, we focused on gaining more evidence of the crosstalk between cholestasis and the gut and addressing the protecting effect of HGF.
ANIT-induced cholestasis model was implemented as previously reported [13]. Briefly, forty 10- to 12-week-old CD1 male mice (ten animals per group) were maintained in pathogen-free conditions with controlled temperature and humidity on a 12 h light-dark cycle in the animal facility at the Universidad Autónoma Metropolitana Iztapalapa (UAM-I). The mice experimental protocol was approved by the academic ethic commission of the UAM-I (CECBS22-02). We followed the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Mice had free access to food and water.
Except where noted, all reagents were obtained from Sigma-Aldrich (Saint Louis, MO, USA).
To properly recover the stool of mice, one animal per group was placed in individual metabolic cages (Figure S1A). Animals were treated (or not) with ANIT (60 mg/kg, ig, Sigma-Aldrich, N4525) for 48 h. After 24 h of ANIT treatment, HGF (10 μg/kg, i.v., PeproTech, Cranbury, NJ, USA, #100-39) was administrated. We collected urine and stool over the last 12 h of the study. Animals were euthanized forty-eight after ANIT treatment (Figure S1B).
We used at least eight mice per group. Liver tissue was recovered to confirm the cholestasis model as previously published [13].
The intestine (small and large) was recovered and sectioned according to Ruehl-Fehlert et al. [14], in the duodenum (1 cm distal to the pyloric sphincter); jejunum (central section); ileum (1 cm proximal to the cecum), and colon (central section). Cecum, rectum, and anus were not considered.
Histology was performed by routine hematoxylin and eosin (H&E) staining as previously reported [15]. Villus height and crypt depth measures were made from images obtained from a microscope Axio Vert.A1 (Carl Zeiss, Jena, Germany) and processed with ZEN software (Carl Zeiss, Jena, Germany). Villus height (villus tip to crypt junction) and crypt depth (depth of the invagination between adjacent villi) were measured on 20 villi per sample (Figure S2).
As previously reported, immunohistochemistry (IHC) was performed [16]. Briefly, 5 μm tissue samples were fixed in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS), washed in PBS, and permeabilized with 0.1% Triton X-100 in PBS. Slides were blocked for 1 h with a blocking solution [1% bovine serum albumin (BSA) in PBS]. Slides were probed for 1 h with primary antibody (anti-CD163, Abcam, Cambridge, UK, #182422; and anti-cleaved caspase 3, cell signaling, Danvers, MA, USA, #9661) solution at room temperature, extensively washed with PBS, and incubated for 1 h with secondary antibody solution at room temperature. IHC was observed in a microscope Axio Vert.A1 (Carl Zeiss).
Samples were deparaffinized by the routine procedure. Once hydrated, slides were covered with 3% acetic acid for 3 min, and the tissue was immediately immersed in 1% alcian blue 8GX in 3% acetic acid solution at room temperature for 30 min. The tissue was washed with tap water for 5 min and incubated with hematoxylin. The sample was quickly immersed in absolute ethanol and allowed to dry prior to mounting and microscopic observation in a microscope Axio Vert.A1 (Carl Zeiss). Quantification was performed by using ImageJ software (NIH).
The macrophage marker F4/80 was determined by immunofluorescence (IF), as previously reported [17]. Briefly, tissue was fixed in 4% formaldehyde and preserved in the Tissue-Tek® optimal cutting temperature (OCT) compound (Sakura Finetek, Torrance, CA). We obtained 5 μm cryosections. Subsequently, these were washed with PBS-tween for 5 min. The F4/80 antibody (Abcam, Cambridge, UK, #60343, dilution 1:1000) was prepared in PBS with 0.1% horse serum and incubated overnight at 4°C in a humid chamber. The tissues were rewashed with PBS and incubated with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min. Finally, slides were examined using the Carl Zeiss LSM 780 NLO confocal microscope (Carl Zeiss).
After mice euthanasia, fresh intestine tissue was rapidly obtained and sectioned, frozen in liquid nitrogen, and embedded in the Tissue-Tek® OCT compound. Immediately, 8-μm frozen sections were obtained in a cryostat (Leica CM-3050S, Wetzlar, Germany) at −20°C, and the slides were incubated for 15 min in the dark at room temperature with dihydroethidium (DHE, 5 μmol/L) for determination of superoxide radical detecting ethidium fluorescence. Samples were observed using a confocal microscope (Carl Zeiss 780 LSM-NLO) at excitation and emission wavelengths of 485 nm and 570 nm, respectively, as we previously reported. Arbitrary fluorescence units were analyzed by using ImageJ software.
Lipid peroxidation was assayed by determining thiobarbituric acid reactive substances (TBARS) using spectrophotometry as previously reported [18].
Data are presented as mean ± standard error of the mean (SEM) for at least four different animals; each experiment was conducted in triplicate. An analysis of variance (ANOVA) was used to compare the means of different groups, followed by multiple comparisons by the Tukey test. The GraphPad Prism version 8 software for macOS was used. Differences were considered statistically significant when P ≤ 0.05.
We previously reported that the ANIT treatment in mice induces cholestasis resembling the human clinical presentation, even in the systemic compromise presented in these diseases, affecting, in addition to the liver, the kidneys in a clear presentation of cholemic nephropathy [1]. We observed in this model some changes in other organs, such as the intestines; in fact, animals under the ANIT treatment were observed fecal retention judged by a reduction in the content of stool and presented a lighter color (Figure 1A). As observed in our previous reports, HGF induced a remarkable reversion of the cholestatic damage in the liver [13] and kidneys [1]. These protective effects were also observed in feces production, significantly restoring the content.
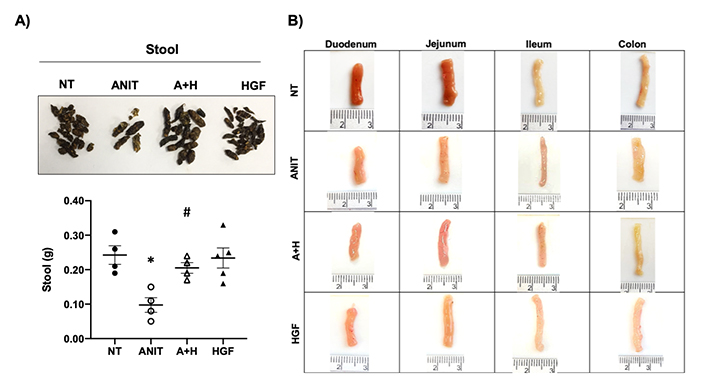
Cholestasis induces stool retention and macroscopic changes in the intestine in mice treated with ANIT. A). Aspect and quantification of stool from animals under different treatments; B). macroscopic inspection of the different segments of the intestine from mice under different treatments. Images are representative of at least four mice. *: P < 0.01 vs. not treated (NT); #: P < 0.01 vs. ANIT; A+H: ANIT + HGF treatment
We carefully sectioned the different segments of the intestine according to the recommendations of the organ trimming reported by Ruehl-Fehlert and collaborators [14]. The gross inspection of all segments of the intestine revealed some differences, mainly in the colon, observing an increase in thickness in animals treated with ANIT (Figure 1B) regarding the NT group. We also observed a slight thinning of the ileum, but we did not find any visible difference in the jejunum and duodenum. These data suggest that ANIT-induced cholestasis particularly impacts the colon. Interestingly, colon changes in the ANIT group were restored by the HGF treatment (A+H).
ANIT treatment-induced mild colitis (Figure 2A) is characterized by histological changes, including inflammatory infiltrate, disrupted mucosa, and crypt architecture changes judged by the decrement in depth (Figure 2B). We observed the correction of crypt depth, even over the NT tissue in animals treated with ANIT and HGF. Similar findings we observed in the ileum (Figure S3). Interestingly, we found an increment in the inflammatory infiltrate in the A+H group (Figure 2A).
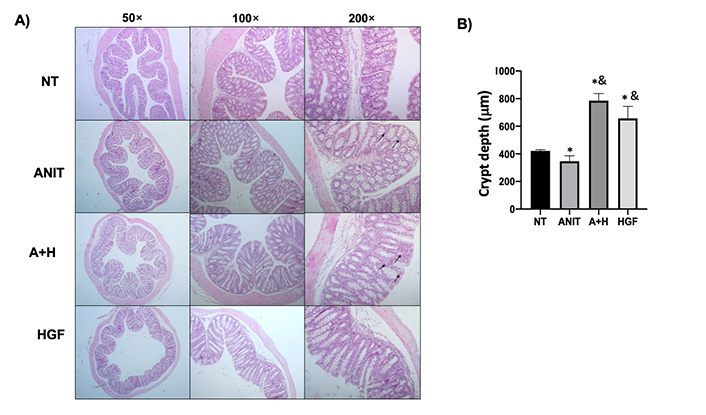
Histomorphological characteristics of colon tissue from mice under cholestasis and HGF treatment. A). Histology appearance by H&E routine staining; black arrows show inflammatory cell infiltration; B). crypt depth measurement. The images are representative of at least four mice. Each column represents the average ± SEM in at least three mice. *: P < 0.05 vs. NT; &: P < 0.05 vs. ANIT
To confirm the inflammatory phenotype, and because macrophages are the primary gatekeeper of the intestinal innate immune system, we performed IF of the overall macrophage marker F4/80; we confirmed the observation finding an increased presence of macrophages in A+H (Figure 3A, B) comparing with NT and ANIT groups; this was intriguing. We decided to address the macrophage phenotype by IHC of CD163, a specific marker of anti-inflammatory macrophages (M2 polarity), which could fit with the protective effect elicited by HGF. The result depicted in Figure 3C, D shows the increment of M2 macrophages; these cells are associated with repairing processes [19], strongly suggesting that HGF is driving a wound-healing response. Interestingly, conversely to the colon result, the ileum observed the F4/80 marker elevated in mice samples from the ANIT group, and HGF decreased the macrophage infiltration (Figure S4).
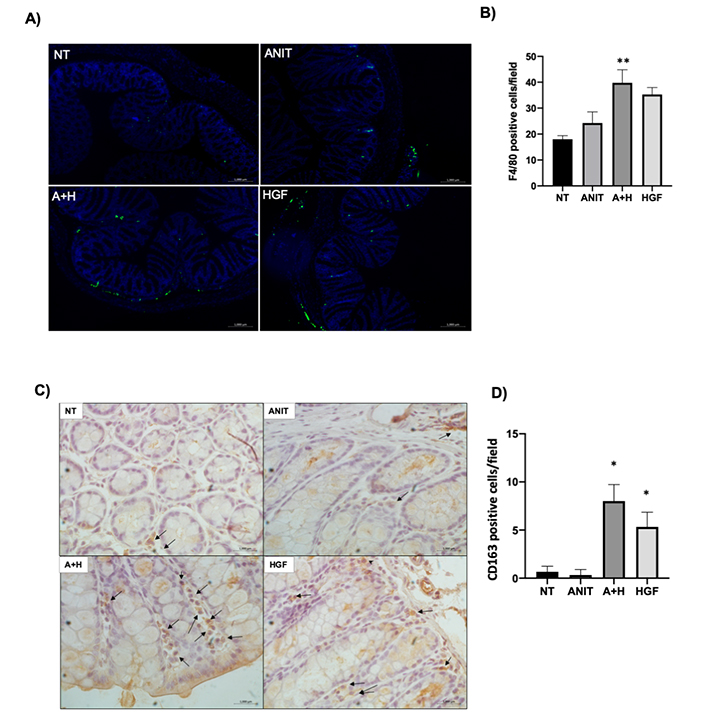
Macrophages infiltration. A). Representative image of IF of marker F4/80 (green) in colon tissue; B). quantification of the F4/80 positive cells per field; C). IHC of CD163, a marker of anti-inflammatory macrophages (M2 polarity). Black arrows indicate positive cells. Original magnification 200×; D). percentage of CD163 positive cells. Each bar represents the average ± SEM in at least three mice. *: P < 0.05 vs. NT; **: P < 0.01 vs. NT
Next, we focused on knowing if the tissue was functional in each treatment. The colon is a section of the intestine that relies heavily on its secretory function to prevent the translocation of bacteria that lodge within the lumen. This led us to observe mucin secretion as a protective factor in the colon and determine whether HGF also affected this process. What was observed was a slight but significant increase in mucin secretion in the colon as a repair effect of HGF after ANIT damage (Figure 4).
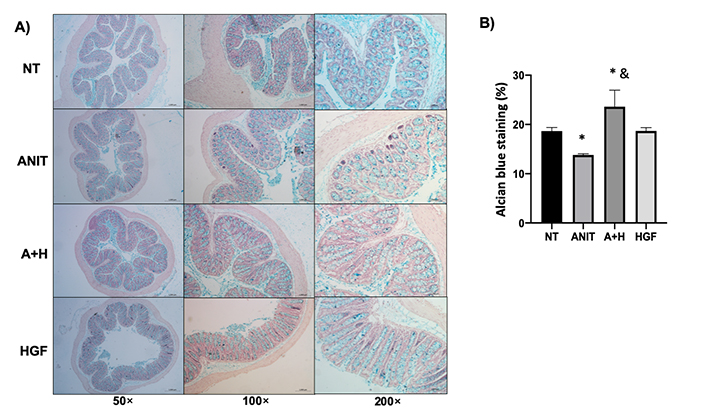
HGF restores mucin production altered by cholestasis. A). Mucin content determined by alcian blue staining (blue); B). percentage of alcian blue staining. The images are representative of at least three mice. Each column represents the average ± SEM in at least three mice. *: P < 0.05 vs. NT; &: P < 0.05 vs. ANIT
To further characterize, we determined apoptosis by IHC of cleaved caspase 3; ANIT treatment increased caspase 3 activity (4-fold change regarding the NT), the effect diminished to control levels by the HGF treatment (Figure 5). This was associated with an increment of ROS production judged by DHE fluorescence in ANIT-treated mice. Lipid peroxidation confirmed the oxidative damage related to ROS increment (Figure 5E). HGF treatment restored the ANIT-induced ROS production and oxidative lipid damage. ROS content in the ileum exhibited the same effect; ANIT induced oxidative stress in this tissue, and HGF treatment significantly decreased the ROS production (Figure S5).
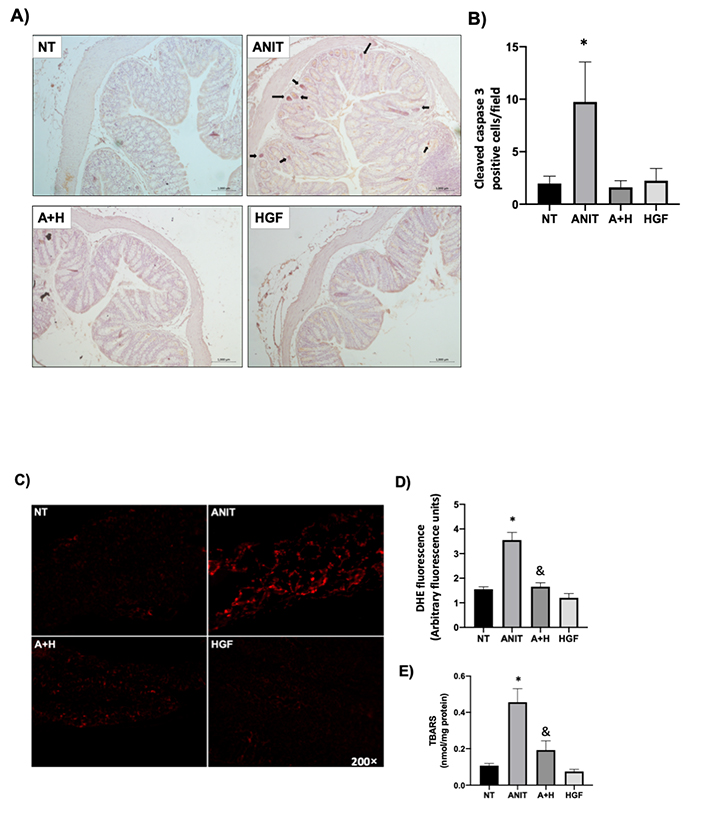
HGF decreases colon apoptosis and oxidative stress induced by ANIT treatment. A). IHC of cleaved (active) caspase 3 in colon samples from mice under different treatments. Black arrows indicate positive cells. Original magnification 200×; B). percentage of caspase 3 positive cells; C). ROS, determined by DHE fluorescence in fresh colon tissue from all groups; D). quantification of DHE fluorescence in arbitrary fluorescence units determined by ZEN software (Carl Zeiss). Images are representative of at least three experimental animals. Original magnification 100×; E). lipid peroxidation was determined in colon tissue as TBARS. Each column represents the average ± SEM in at least three mice. *: P < 0.05 vs. NT; &: P < 0.05 vs. ANIT
We have extensively reported the remarkable capacity of tissue-protective and repair effects induced by HGF/c-Met in many epithelial systems such as the liver, including fibrosis [16], alcoholic liver disease [20], steatosis [21]; but also in the lugs [15], and the pancreas [22]. The primary HGF repair process is mediated by canonical signaling pathways such as Akt, signal transducer and activator of transcription 3 (Stat3), and extracellular signal-regulated kinase 1/2 (Erk1/2) by using a mechanism dependent on cellular redox regulation [12, 23].
In cholestasis, we have demonstrated the protective effect mediated by HGF. The growth factor induced a significant decrement of necrotic areas, restored the bile flow impeded by the ANIT-induced cholestasis, and improved values of alanine aminotransferase, aspartate transferase, alkaline phosphatase, ɣ-glutamyl transpeptidase activities, total and direct bilirubin concentration, and serum and tissue levels of bile salts [13]. These findings were closely associated with a decrement in ROS production and an improvement in oxidative tissue damage, preserving the canalicular structure.
Cholestatic damage extends to the kidneys. We observed a decrement in renal function judged by a decrement in urinary flow and creatinine clearance and a significant increment in urine bile salts; all these parameters were restored to normal levels by HGF treatment [1].
On the other hand, we also observed in c-MetKO mice a clear cholestatic phenotype in animals fed with a high cholesterol diet [21], a feature that we did not find in WT animals under the same diet, confirming the anti-cholestatic effect elicited by HGF signaling. Our previous works clearly indicate that HGF induces protective and repair responses in cholestasis damage.
Cholestasis exhibits complex pathophysiology. It displays deleterious effects in other organs, as stated. In the present work, we were focused on figuring out the impact of cholestasis on the intestine, particularly in the colon. We were directed to confirm whether HGF can also mediate a healing response in this organ.
Our data show no significant effects observed macroscopically, only some mild changes we observed in the colon and ileum. Interestingly, these changes were opposite of each other; the colon tended to increase the thickness, and the ileum was thinner than the control animals.
The microscopic inspection addressed by H&E routine staining revealed architecture and inflammatory infiltrate changes. Although, in both cases, macrophages were detected, it was surprising in the increment in the treatment with HGF, even in the case of the growth factor alone in the colon.
The critical step for the resolution of inflammation is the efficient removal of dead and dying cells [24, 25]; this process, called efferocytosis, is driven by alternatively activated macrophages [26]. It is possible an apoptotic cell uptake by CD163 positive macrophages due to an efficient tissue clearance. This evidence was observed in Figure 5, and data suggest cooperation between HGF and indirectly activated macrophages reduces cleaved-caspase 3 positive cells. These data show the immunomodulatory effect in phagocytic cells to speed up tissue homeostasis in the intestine. In normal conditions, M2 macrophage encompassed the majority gut macrophage population [27]. Strikingly, the repair activities have been studied, all macrophage phenotypes triggered epithelial repair in vitro, but M2 phagocytes show the most substantial repair activity. M2 cells in dextran sulfate sodium (DSS)-treated mice models accelerated the restoration of colon ulcers regarding M1 macrophages (pro-inflammatory) without any beneficial effects [28].
It was remarkable that HGF induced macrophage activation (CD163+). It is well-known that the M2 macrophages induce a repair response, particularly in acute injury in epithelia [19], where they can promote a wound milieu with a pro-resolution molecular profile. As we clearly show, HGF significantly increased the CD163+ macrophage infiltration in the colon.
We confirmed this repair process by restoring the typical histological presentation, mainly in the crypt depth and mucin production. It has been reported that HGF induces mucin synthesis in the lungs [29], indicating a regulatory function of HGF on respiratory mucin metabolism. In fact, we previously observed similar findings in the repair process and lung physiology in a model of progressive pulmonary tuberculosis under the HGF treatment [15]. The exciting finding of the increment of mucin content in the damaged colon supports this poorly explored effect of HGF; in fact, some evidence is reported regarding the protective role of this growth factor in the colon, the HGF displayed its repair capacity in the DSS model [30]. Here we report the protective effect on the colitis induced by chemical-induced intrahepatic cholestasis.
The crosstalk between the cholestasis and the intestine could also be the opposite; it has been observed that patients with ileal and colonic diseases are with differential levels of bile acids, while patients with ileal disease presented elevated levels of primary, secondary, and tertiary bile acids; those with colonic disease exhibited higher levels of conjugated bile acids [31].
In conclusion, we prove that ANIT-induced cholestasis is an excellent model for studying the systemic impact of this disease. Principally the colon was affected by cholestasis, followed by the ileum. Cholestasis induced mild colitis, particularly related to macrophage infiltration, ROS production, and apoptosis. HGF exogenous treatment restores the typical architecture of the colon, decreases oxidative stress, and induces a healing response mediated by CD163+ macrophages.
Cholestasis should be revisited and viewed as a systemic disease initiated in the liver that profoundly impacts the system. The main damage mechanisms are ROS production and apoptosis. HGF significantly influenced the oxidative stress and induced antiapoptotic and repair effects.
A+H: α-naphthyl isothiocyanate + hepatocyte growth factor treatment
ANIT: α-naphthyl isothiocyanate
DHE: dihydroethidium
H&E: hematoxylin and eosin
HGF: hepatocyte growth factor
IF: immunofluorescence
IHC: immunohistochemistry
NT: not treated
PBS: phosphate buffered saline
ROS: reactive oxygen species
SEM: standard error of the mean
TBARS: thiobarbituric acid reactive substances
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/10054_sup_1.pdf.
We thank the confocal microscopy core unit, UAM-I, and Maricruz García Barrera, for the support and assistance in animal handling.
JLR: investigation, methodology, and writing; RLL: tissue research and histology; MGR: immunohistochemistry; AEC: ROS experiments; LCR: statistics and writing review; SSS: animal management and methodology; NNL: investigation and methodology; FM: fluorometry experiments; VSA: animal care and tissue collecting; RUML: ROS studies; MCGR: writing review and editing; LEGQ: founding, conceptualization, methodology, and writing; LBO: founding, conceptualization, project administration, initial draft preparation, and paper submission.
The authors declare that they have no conflicts of interest.
The study “The hepatocyte growth factor induces an anti-inflammatory and repairing response in the cholestasis-induced colon damage” was approved by the Academic Ethic Commission of DCBS, Universidad Autonoma Metropolitana Iztapalapa (CECBS22-02).
Not applicable.
Not applicable.
The data that support the findings of this study are available from the corresponding author on reasonable request.
This work was partially funded by a grant from the Consejo Nacional de Ciencia y Tecnología (CONACYT): CB-A1-S-38154, CB-252942, Infra-2017 280788, “Estímulo Antonio Ariza Cañadilla 2017” from FUNDHEPA, Mexico, and Universidad Autónoma Metropolitana-Iztapalapa (UAM-I). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

