 Review
Review

Affiliation:
1Posgrado en Biología Experimental, DCBS, Universidad Autónoma Metrolitana-Iztapalapa, Mexico City 09340, Mexico
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
ORCID: https://orcid.org/0000-0002-8978-3122
Affiliation:
1Posgrado en Biología Experimental, DCBS, Universidad Autónoma Metrolitana-Iztapalapa, Mexico City 09340, Mexico
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
ORCID: https://orcid.org/0000-0002-6678-0877
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
3Department of Biological Regulation, Weizmann Institute of Science, Rehovot 7610001, Israel
ORCID: https://orcid.org/0000-0002-1353-260X
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
4Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
ORCID: https://orcid.org/0000-0002-3923-6278
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
4Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
ORCID: https://orcid.org/0000-0001-8249-7257
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
4Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
ORCID: https://orcid.org/0000-0002-5704-5985
Affiliation:
2Área de Medicina Experimental y Traslacional, Departamento de Ciencias de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City 09340, Mexico
4Laboratorio de Medicina Experimental, Unidad de Medicina Traslacional IIB/UNAM, Instituto Nacional de Cardiología Ignacio Chávez, Mexico City 14080, Mexico
Email: mcgr@xanum.uam.mx
ORCID: https://orcid.org/0000-0003-0501-7226
Explor Dig Dis. 2022;1:21–39 DOI: https://doi.org/10.37349/edd.2022.00003
Received: May 14, 2022 Accepted: June 13, 2022 Published: July 29, 2022
Academic Editor: Matias A. Avila, University of Navarra, Spain
Hepatocellular carcinoma (HCC) is considered one of the most aggressive tumors worldwide. The consumption of lipid-enriched diets, mainly high cholesterol, induces oxidative stress and chronic inflammation, leading to HCC progression. Moreover, fatty acids and cholesterol could display differential responses on immune cells inside the tumor immune microenvironment (TIME). Tumor-associated macrophages (TAMs) represent one of the most critical leukocytes in the tumor microenvironment (TME) displaying pro-tumoral responses and one of the mainly cholesterol donors to cancer cells. Immunotherapy or cholesterol regulators, alone or combined, would represent an effective strategy for HCC treatment. Nonetheless, steatotic etiology from non-alcoholic fatty liver disease (NAFLD)-HCC tumors has been unexpectedly resulting in highly aggressive behavior.
Liver diseases represent one of the main challenges for health systems worldwide. Changes in human lifestyle, particularly those related to alcohol and unbalanced diet consumption, tend to increase the incidence and prevalence of liver diseases. The natural history of the liver disease starts with simple steatosis and then non-alcoholic fatty liver disease (NAFLD), which eventually progresses to steatohepatitis [non-alcoholic steatohepatitis (NASH)], fibrosis, cirrhosis, and ultimately, cancer.
Liver cancer is the sixth most diagnosed cancer and the third leading cause of death worldwide in 2020. It is estimated that 906,000 new cases and 83,000 deaths are related to this malignancy, particularly hepatocellular carcinoma (HCC) [1]. The highest incidence and mortality had been observed in Eastern Asia and Africa, but it has recently increased in Europe and North America [2, 3].
Among the main risk factors for HCC, it is the infection by hepatotropic viruses, such as hepatitis B virus (HBV) and hepatitis C virus (HCV), chronic ethanol consumption, aflatoxins exposure [4], and the one with the highest increase rate in the recent years, the consumption of hypercaloric diets, particularly with high in cholesterol content, which promotes NASH and HCC as we recently reported [5].
HCC, like any other cancer, is a proliferative disorder. Cancer cells exhibit a high capacity for survival, adaptation, and autonomy to escape any control that the system may exert [6]. Aberrant metabolism is also a key determinant in this malignancy [7]. We know this depends not only on the phenotypic changes in the tumoral cell but also on the tumor microenvironment (TME).
The tumor is not only composed of cancerous cells. There are a heterogeneous collection of resident cells and infiltrating ones, such as those of the immune system, which are one of the primary sources of growth factors, cytokines, and extracellular matrix components that favor the tumor progression, and, eventually, the metastasis [8].
The recruitment of inflammatory cells and fibroblast is a natural event in the wound-healing process. The mechanism is simple: cell arrival at the injured tissue, activation, repairment, and finally, resolution of those active cells. The resolution could be conducted by apoptosis or the reverse of the phenotype [9, 10].
In cancer, tumoral cells manipulate the resident and infiltrate members of the TME to gain support for growth and invasion. The mechanism of this manipulation remains partially characterized.
The natural history of liver disease, as previously stated, implies the progressive evolution of liver damage, ranging from a not-dangerous lipid accumulation and steatosis to cirrhosis and HCC. The advanced stage of steatosis is marked by the infiltration of immune cells such as macrophages and neutrophils, among others [11]. This inflammatory phase confers tumor aggressivity and poor prognosis [5].
Which could be the signal induced by the lipid overload that promotes inflammatory cell infiltration? The answer to this question is still elusive.
In the present work, we review the current knowledge of the relationship between lipids, specifically cholesterol, and the tumor immune microenvironment (TIME), in the progression to HCC, discussing some promising strategies to control this cancer.
Perhaps one of the emergent hallmarks of HCC is a metabolic adaptation, particularly for the lipid overload in the tumor. Lipids are fundamental as energy supply, building blocks, and lipid-based protein modification of remarkable oncogenic proteins such as Ras [12]. A dietary supply of lipid is an excellent source but is not limited to it.
Aberrant lipogenesis represents an extraordinary adaptation to provide free fatty acids (FFA) and cholesterol for tumor progression, aggressiveness, and metastasis, leading to poor prognosis [7, 13].
Cholesterol is positioning itself as one of the lipids cleverly used by the tumor. This sterol displays particular properties in the transformed cells, even at the early stages of differentiation [14]. The cholesterol overload in the hepatocytes induces functional changes favoring the transformation, such as oxidative stress, apoptosis resistance, and changes in mitochondrial dynamics, as observed in experimental models of mice fed with a high cholesterol diet (HCD) for 30 days [15].
In the presence of a well-known hepatic carcinogen, the diethylnitrosamine (DEN), this HCD increases DNA oxidative damage and inactivation of key DNA repair enzymes, eventually leading to hyper-vascularized liver tumors and lung metastasis at eight months under treatment [13]. Cholesterol displays pro-tumoral properties in animal studies and patients [7]; although it clearly favors HCC progression, dietary cholesterol supply is not necessarily determinant.
The main enzymes for cholesterol synthesis are in the mevalonate pathway, 3-hydroxy-3-methylglutaryl-coenzyme A (CoA) reductase (HMGCR); mevalonate kinase (MVK); squalene synthase (SQS), and those related to the FFA synthesis such as fatty acid synthase (FASN); acetyl-CoA carboxylase (ACAC); ATP citrate lyase (ACLY) is overexpressed in HCC regardless of the etiology, in order to ensure the constant production of cholesterol and FFA for proper tumor growth [7].
As mentioned above, cholesterol routes are involved in HCC cells, but interestingly, these enzymes are involved in cellular immunity. It has been described that sterol response element-binding proteins (SREBP), specifically SREBP2, regulate cholesterol by a constitutive expression; contrarily to SREBP1, which regulates fatty acids synthesis. SREBP2 induces immune activation of leukocytes, including T, natural killer (NK) cells macrophages with a subsequent inflammatory state [16]. Conversely, mevalonate pathway enzymes induce a trained immunity in monocytes mediated by the insulin-like growth factor 1 receptor (IGF1-R) [17].
These molecules have different regulators, including tumor necrosis factor-alpha (TNF-α), which induces late-phase activation of SREBP2 and cholesterol gene expressions in myelocytes, such as macrophages, which induce macrophage stimulation [16], which could be relevant in cancer. It is important to mention that mevalonate pathway activation is not only involved in cholesterol synthesis; for example, the metabolite mevalonate is the mediator of IGF1-R and mammalian target of rapamycin (mTOR) with a subsequent inflammatory pathway due to epigenetic program. Training is very relevant to the activation of the cholesterol synthesis pathway for the myeloid cells, but not the synthesis of cholesterol itself [17], although in the context of cancer could be different. As well as mevalonate, many other products of the same could modulate the immune system, but it is unknown in a large field how they could modulate by themselves or in cross-linking with other pathways in liver cancer.
We recently reported that lipid-enriched diets, particularly those with high cholesterol content, induce HCC tumors with high aggressivity and poor prognosis, judged by increased angiogenesis determined by CD34 marker and remarkable macrophage infiltration (F4/80), particularly pro-tumoral macrophages (M2, CD206+) [5].
Considering these findings, dietary and de novo synthesized cholesterol also affected the TME, particularly in the TIME.
The intricate relationship between tumor cells and the immune system is a multiphase process determined by spatial-temporal decisions. It is well-known that chronic inflammation is one of the leading causes of cancer initiation and progression due to the persistent local production of cytokines and growth factors and the increment of the reactive oxygen species (ROS) that eventually leads to DNA damage and the onset of aberrant cell proliferation. HCC mainly follows this mechanism of carcinogenesis [13].
The immune system usually eliminates initiated cells before becoming clinically detectable [18]; this represents one of the leading surveillance control mechanisms against tumor development. The next phase is determined by adapting tumor cells, leading to fewer immunogenic variants that eventually escape the immune surveillance [19, 20]. Finally, the tumor cell manipulates the immune system playing pro-tumoral roles allowing the progression and the metastasis by creating a particular microenvironment or TIME. Among the tumor-infiltrating immune cells, the tumor-associated macrophages (TAMs), the regulatory lymphocytes, the neutrophils, and the myeloid-derived suppressor cells (MDSC) (Figure 1A) stand out because they are associated with aggressivity and poor prognosis [5, 21].
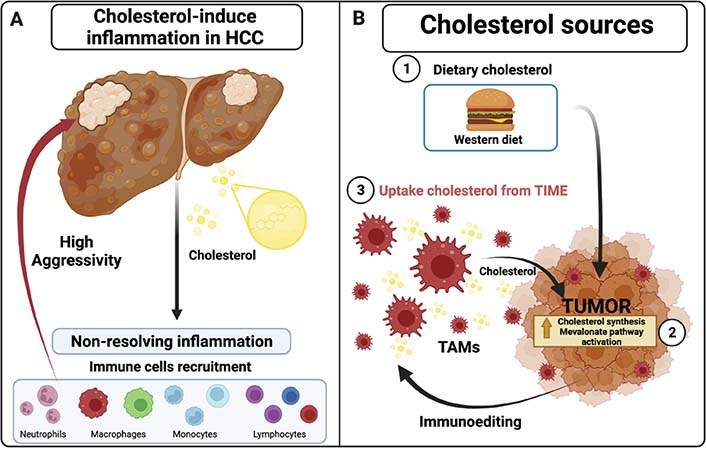
Inflammatory state by cholesterol in tumors. A. Cholesterol promotes and increases chronic inflammatory state (non-resolving inflammation) in HCC tumors, recruiting different immune cells that increase aggressive behavior; B. different cholesterol sources in solid tumors: 1) dietary cholesterol such as western diet (WD), 2) cholesterol synthesis and derivates molecules from exacerbating mevalonate pathway activation through HMGCR enzyme, 3) uptake cholesterol from immunoediting leukocytes such as TAMs and their cholesterol efflux in TIME. Created with BioRender.com
Dietary cholesterol promotes highly aggressive liver cancers, including HCC, with larger and highly vascularized tumors in mice models [5, 13, 22]. Interestingly, regardless of the diet, tumor cells increase cholesterol synthesis through the aberrant activation of the mevalonate pathway [22, 23], observing a similar phenotypic presentation as those raised in mouse models under HCD. Definitively, cholesterol plays a role in tumor promotion. Hypercholesterolemia induces the accumulation of cholesterol in macrophages and other immune cells, as observed in atherosclerosis and obesity [24]; this leads to the activation of these cells, increasing the synthesis and release of pro-inflammatory cytokines and growth factors. Similarly, the liver resident macrophages, also known as Kupffer cells (KC), are activated by the cholesterol overloading, contributing to the inflammatory state in NASH. Acquisition of cholesterol by KC is facilitated by receptor-mediated endocytosis of low-density lipoproteins (LDL) via LDL receptor (LDLR). The resulting endosome is merged with lysosomes, resulting in free cholesterol, managed in the cytosol by Niemann-Pick type C proteins [25]. This process is regulated by a feedback mechanism, inducing the downregulation of the expression of the gene encoding the Ldlr, preventing further cholesterol capture and foamy macrophage formation [26].
The scenario gets complicated when the oxidative stress observed in NASH induces protein oxidation of critical regulators, including the LDL. The system of the scavenger receptors internalizes the oxidized LDL. This system lacks feedback inhibition, leading to the accumulation of cholesterol in the macrophage and establishing foamy macrophages [25, 27]. This is also associated with lysosomal impairment and cholesterol accumulation in this organelle, contributing to the inflammatory state and NASH progression.
Conversely, in the cancer context, transformed cells can manipulate the TIME to obtain cholesterol from other sources such as myeloid cells. Macrophages can carry out cholesterol secretion to maintain the aggressive behavior of cancer cells observed in ovarian cancer models [28], and we have hypothesized that HCC macrophages perform a similar mechanism.
In summary, the HCC cell can obtain cholesterol from three sources: 1) from the diet, 2) de novo synthesis, and 3) delivery from microenvironment cells (Figure 1).
Chronic inflammation, also called non-resolving inflammation, is a process driven by key transcription factors such as nuclear factor kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3) in both cancer cells and immune cells, which promote the progression of HCC [29]. Inflammation and lipids work hand to hand, triggering higher aggressiveness. The relationship between inflammation and cholesterol in cancer is not entirely understood in gastrointestinal cancers, especially HCC. A non-resolving inflammatory state promotes carcinogenesis and progression of hepatic tumors. The activation of NF-κB induces transcription of Srebp2, Hmgcr, and Ldlr and, therefore, cholesterol synthesis and cholesterol uptake in HCC cell lines [30].
The relevance of cholesterol in HCC has been observed in a mouse model of c-Fos overexpression [31], which promotes hepatocyte transformation to a malignant phenotype and contributes to liver carcinogenesis, elevating alanine aminotransferase (ALT) activity, and the expression of glutamine synthetase (GS), cytokeratin 19 (CK19), SRY-box transcription factor 9 (Sox9), alpha-fetoprotein (AFP), and β-catenin, correlating with an increment on inflammation judged by CD45, an overall leukocyte marker, and myeloperoxidase (MPO), and the reduction of NK and B220 cells.
Interestingly, pro-inflammatory, pro-oncogenic, and genotoxic stress pathways are activated in this model, and an increment of STAT3, Jun N-terminal kinase 1/2 (JNK1/2), and AKT signaling were observed as tumor promoters compared with models using HFHCD [5]. On the other hand, they showed a cholesterol accumulation in hepatocytes due to the downregulation of Abcg5/Abcg8 transporters in a liver X receptor (LXR)-α dependent manner. This work observed a positive relationship with a possible immune effect. In addition, other noxious cholesterol metabolites such as oxysterols and bile acids also contribute to HCC development [31].
It was determined the influence of the cholesterol on immunity responses, and it has been studied in diabetic C57Bl/6 mice model induced with streptozotocin (STZ) and fed with high fat and HCD (HFHCD, 1.5%) or high fat and high sugar diet (HFHSD). Animals showed body and liver weight gain, liver damage, an increment in triglyceride content, oxidative stress, fibrosis, glutamine synthesis, and immune cell infiltration. HFHCD induced CD45+ leukocytes recruitment, macrophages, neutrophils, and various T cells, including regulatory T lymphocytes (Treg). Treg lymphocytes’ activation-induced immunosuppressive activities are related to an aggressive phenotype in HCC [32].
In the same way, the role of cholesterol in NASH status is evidently studied in mice under HFHCD, contrary to absent cholesterol diets with only steatosis development. Mice fed with HFHCD present an increment in the expression of genes related to inflammation [macrophage markers, cytokine-receptor interaction, such as transforming growth factor-β (TGF-β), interleukin-3 (IL-3), IL-4, IL-5 signaling], aberrant metabolism, and oncogenic pathways (Wnt/β-catenin, Myc, Rac2, etc.). Moreover, HFHCD dysregulates calcium, insulin, cell adhesion, and axon guidance. NASH-HCC tumors are more aggressive than steatosis-HCC [33].
Further studies show that using a high-fat diet (HFD) consumption in C57BL/6 mice and treated with DEN (25 mg/kg) promotes obesity, liver inflammation, and tumorigenesis in IL-6- and TNF-α-dependent manner; triggering the accumulation of myeloid cells such as macrophages and neutrophils; contrarily, observations were found using IL-6−/− and TNFR1−/− mice models [34].
The available information shows a positive correlation between cholesterol and immune system activation; cholesterol in the diet exacerbates aggressive phenotype by controlling the microenvironment and those effects observed in the transformed cell to harm the individual.
The TGF-β signaling displays differential functions in normal and transformed cells. In early-cancer stages, TGF-β induces cell cycle arrest and cellular cytotoxicity; these tumor-suppressor functions are well characterized; however, in late-cancer stages, TGF-β displays tumor promotion properties, including invasion, metastasis, epithelial-mesenchymal transition (EMT), and drug resistance [35, 36]. As mentioned before, TGF-β is up-regulated under the HCDs and tumor stages; the combined effects of TGF-β and cholesterol in HCC aggressiveness have been reported [33]. TGF-β could display immunomodulatory and immunosuppressive effects in leukocytes. The growth factors are secreted by the tumor and TME cells to inactivate the cytotoxic response of NK, NK T cells, and cytotoxic lymphocytes (CTL) and later activation of MDSC, Treg cells, and M2-macrophages [37]. Also, it has been described that M2-macrophages are activated following suppressor of mothers against decapentaplegic 2/3 (Smad 2/3) signaling and the activations of the SNAIL transcription factor, which drives EMT in cancer cells [38].
Moreover, the immunosuppressive effect of TGF-β1 has been quantified, and the relationship with elevated cholesterol biosynthesis has been observed in other cells such as human keratinocytes through HMGCR expression [39]. It has been reported that dickkopf-1 (Dkk1) is overexpressed in HCC patients promoting TGF-β secretion, indicating an immune cell activation [40].
Single-cell RNA sequencing from five different sites, including liver tumor, adjacent tissue, hepatic lymph nodes, blood, and ascites, reported high variability of leukocyte subsets [41]. It was cleared up that different HCC etiologies (alcohol, alcohol and hepatitis, HBV, HCV, and NASH) and tumor stages (I–IV) result in a similar immune cells activation without any significant changes in the immune contexture [42]. It has been studied that tumors formed from NASH-HCC represent more aggressive tumors than those of another etiology, which we will discuss later [43]. As mentioned before, in HCC, there are alternatively activated TAMs (M2 macrophages), MDSC, and Treg cells with cancer-associated fibroblasts (CAFs) reducing the antitumoral response of classical macrophages (M1 macrophages), NK cells, T helper (Th) and cytotoxic T lymphocytes (T CD4+ and CD8+) [44].
These wide leukocyte varieties can realize specific responses favoring or opposing tumor cells, but this scenario changes in the context of cholesterol and liver cancer. This section discussed how leukocytes work in an individual or particular form. It has been reported immunomodulating effects induced by cholesterol in leukocytes from mice fed with HCD or high-fat and cholesterol diet (HFCD) [45]. HFCD increases NK and memory cytotoxic T cells (CD122+ CD8+) and phagocytic activity of KC, suggesting that high cholesterol in the diet (1.25%) increases the functionality of mononuclear cells (MNCs) in the liver [45]. We know that cholesterol in TME is entirely different, inducing a diversity of responses. The cholesterol effects in each leukocyte in the TIME and the possible secretory factors are summarized in Figure 2; in the same way, we show in Table 1 a summary of this lipid in each cell.
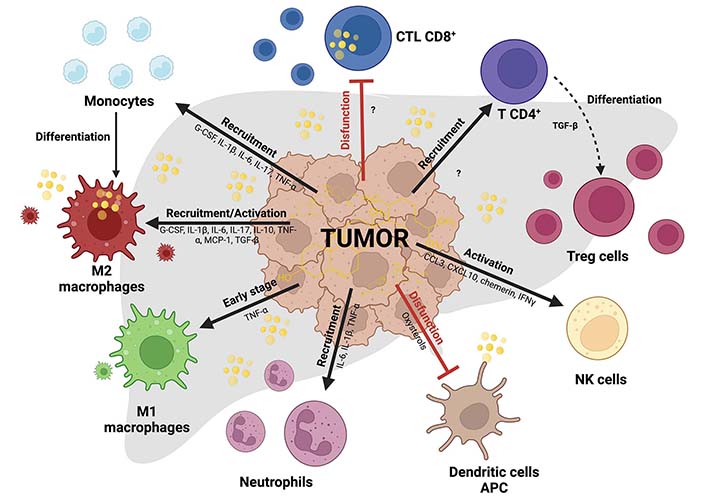
Immune cells in HCC tumors affecting cholesterol context. Differential effects of cholesterol in myeloid and lymphoid cells inside TIME in HCC. Black arrows indicate recruitment or activation, and flattened red lines indicate exhaustion or inactivity state leucocytes. M2 macrophages studied in ovarian cancer release cholesterol and provide aggressivity to the cancer cells. Cholesterol reduces cytotoxic activity in CD8+ T cells (CTL CD8+) through endoplasmic reticulum (ER) stress and affects antigen-presenting in dendritic cells (DC). In more cases, cholesterol promotes the recruitment and activation of different immune cells. Created with BioRender.com. CCL3: C-C motif chemokine ligand 3; G-CSF: granulocyte-colony stimulator factor; IFNγ: interferon gamma; MCP-1: monocyte chemoattractant protein-1
Role of cholesterol in TIME
| Cell type | Cholesterol role in cancer/HCC | References |
|---|---|---|
| Macrophages | Cholesterol induces macrophage recruitment and differentiation to M2 polarity in HCC.HCD induces M1 macrophages in early stages of HCC development, contrarily to M2 polarity in chronic phases.Cholesterol efflux is associated with aggressivity in cancer.HFHCD induce M2 macrophage polarization or CD206+ cells.The primary sources of oxysterols such as 27-HC are M2 macrophages. | [5, 28, 45, 47, 52, 59] |
| Neutrophils | HCD promotes neutrophil infiltration in HCC models.Oxidized cholesterol plays an essential role in neutrophils attracting in a CXCR2-dependent manner.Oxidized cholesterol correlates with tumor aggressivity.NASH-HCC tumors have high neutrophil infiltration. | [47, 58, 60] |
| NK | Cholesterol promotes NK cell activation and NKG2D, and NCR1 receptors are reflected in aggressivity reduction.Deletion of ApoE (ApoE−/−) provokes fewer and smaller liver tumors, augmenting effector functions of NK cells.The dysfunctional response of iNKT cells with high lipids and cholesterol biosynthesis in B16F10 is the opposite response in liver tumors. | [63, 64] |
| DC | Oxidized lipid forms inhibit antigen cross-presentation and peptide MHC class I complexes, reducing anti-tumor strategy.LXR affects DC in a CCR7-dependent mechanism, observing decrement in migration to lymphoid tissues. | [67, 68] |
| T lymphocytes | T lymphocytes have pleiotropic roles, activating a high variety of subsets of T cells in the NAFLD spectrum and HCC.HCD induces inflammation with T lymphocytes (CD3+ CD4+ CD8+) recruitment in the liver.Cholesterol accumulation in the microenvironment induces immune checkpoint inhibitors (PD-1, 2B4, TIM-3, and LAG-3) in CD8+ cells.Cholesterol overload is associated with an increment in Treg. | [72–75] |
27-HC: 27-hydroxy-cholesterol; ApoE: apolipoprotein E
Macrophages belong to innate immunity; they have high plasticity and phagocytic function for bacteria or other external pathogens. Macrophages respond according to different environmental conditions maintaining tissue homeostasis. These phagocytic cells display essential roles in diverse pathologies, including malignant neoplasm, where they exhibit changes to their function that allow them to be called TAMs [46]. Depending on the cancer stage and macrophage activation, macrophages can become allies or merciless enemies of the tumors [5, 47].
Macrophages could present, according to the stimulus, different phenotypes or polarities. M1 or activated macrophages display bactericidal and antitumoral responses. Unlike M1 polarity, M2 macrophages possess pro-tumoral activity. They are considered M2 polarity or M2-TAMs macrophages under anti-inflammatory stimuli or cytokines such as IL-4 and IL-13 or TGF-β secreted by tumoral cells [38]. It is suggested that mitochondria also play an essential role in this macrophage activation and aggressivity behavior by the preference for the use of the β-oxidation of fatty acids [48].
Some reports sustain the participation of mitochondria in macrophage polarity; Bao and coworkers [49] showed that changes in polarity are associated with mitochondrial fission and mitochondrial DNA delivery by a mechanism dependent on the toll-like receptor 9 (TLR9)/NF-κB signaling pathway activation; this induced recruitment of pro-tumoral macrophages with CCL2 secretion in HCC in vitro and in vivo models, demonstrating the remarkable participation of the mitochondrial fission as a mechanism for macrophage infiltration in HCC [49]. Moreover, HCC has a vast heterogeneity of macrophages, including resident or KC and blood-recruited monocyte-derived macrophages observed in the study of CCL2, CCL5, MCF, and CD11b, a specific infiltrating macrophage marker in carbon tetrachloride (CCl4) mice models [50].
As mentioned above, monocyte-derived macrophages can also be implicated in NASH, and inhibition of this monocyte recruitment could reduce NASH-related complications and late HCC onset [51]. In normal conditions, cholesterol-enriched diets increase the content of secretory (CD11b+) and decrease (CD68+) phagocytic macrophages subsets; strikingly, CD68+ cells increase their phagocytic activity [45]. Non-healthy diet combinations such as the high fat-high cholesterol-high sugar diet (HF-HC-HSD) or WD induce NASH development demonstrated in C57BL/6 mice models with DEN (75–100 mg/kg). It is particularly relevant to activating the hypoxia-inducible factor 1α (HIF-1α) and IL-10 secretion from hepatocytes to macrophages. Autocrine and paracrine communication of IL-10 in macrophages induced M2 phenotype and provided cell aggressivity [52].
As mentioned above, it has been demonstrated that HCC-generated mice at eight months under HC and WD diets (cholesterol 1%) and treated with DEN induce secretion of inflammatory factors such as G-CSF, IL-1β, IL-6, IL-17, and TNF-α, and the exacerbated macrophage infiltration (F4/80+) with specific M2 polarity (CD206+). Also, high vascularity (CD34) and IL-6/Janus kinase (JAK)/STAT3 upregulated signaling pathway, relevant in liver tumors and inflammation, were associated with the increment in the expression of Slc41a3, Fabp5, Igdcc4, and Mthfd1l, genes strongly associated to metabolic adaptation that favors an aggressive subtype of HCC with poor prognosis in human HCC; interestingly this study showed a positive correlation between the expression of these genes and the content of cholesterol in human HCC cell lines, and the signature correlated with a poor outcome in HCC patients. This signature could be considered cholesterol specific [5]. This clarifies the long-term effect of cholesterol in the context of liver cancer. It has been demonstrated that M1 or TNF-α positive macrophages are involved in short early responses in hepatocarcinogenesis in an HCD study in zebrafish [47]. These two works demonstrate opposite macrophage polarities and effects.
The fascinating immune dissection in ovarian macrophages has shown us that ovarian cancer cells scavenge membrane cholesterol from TAMs, inducing immune-suppressive polarity and aggressive phenotype in cancer cells. Cholesterol efflux via ATP binding cassette subfamily A member 1 (ABCA1)/G1 transporters in macrophages and anti-inflammatory stimulus with IL-4/13 increase specific oncogenic signaling pathways such as AKT and STAT6 to turn it into pro-tumoral macrophages phenotype [28]. The straightforward questions: are the liver’s TAMs governed by the exact mechanism or an alternative system? The evidence indicates that TAMs represent more than 50% of the tumor mass, while macrophages are 90% of all of them in the human body. Does it have any disadvantages?
Evidently, cholesterol is significantly collaborating with tumor aggressivity partly through pro-tumoral macrophages. The best way to reduce this aggressiveness promoted by TAMs could be the specific elimination of some critical molecular machinery used by the macrophages, for example, the components of the nicotinamide adenine dinu-cleotide phosphate (NADPH) oxidases (NOX), a pro-oxidant system that provides ROS for signaling purposes and as a determinant factor in phagocytic activity as showed by some authors in the Nox1−/− system [53], or the complete depletion of TAMs for greater effectiveness on available therapies against metastasis such as sorafenib [54]. As mentioned before, cholesterol synthesis in macrophages is driven by SREBP2 transcription factor, and the inhibition of the TNF-α/SREBP axis induces M2 macrophage activation and wound healing [16]. However, it is possible that the contrary effect could induce different responses with beneficial effects.
The macrophage “TAMpering” could represent other therapeutic options moving from M2 to M1 polarity playing cholesterol homeostasis.
Neutrophils are the most abundant circulating leukocytes in humans (50–70%); they are part of the innate immunity displaying microbicide and phagocytic effects. Tumor-manipulated neutrophils, also known as “tumor-associated neutrophils (TAN)” [55], display special functions in the TME, favoring tumor progression. The TAN could be activated into N1 or N2 phenotypes, like macrophages. The N1 neutrophils display cytotoxic and antitumoral activities, while the N2 exerts pro-tumoral responses, immunosuppression, tumor growth, angiogenesis, and metastasis [56].
A variety of mechanisms have been currently reported in human and mice models of HCC, which were observed an increment of neutrophil extracellular traps (NETs) that provide metastatic advantages for cancer cells by activating the TLR4/9/NF-κB/cyclooxygenase 2 (COX2) signaling pathway; this observation associates to a poor prognosis in patients [57].
It is observed that neutrophils are recruited in hepatic tumors in zebrafish fed with HCD [47]. Also, it was observed that MPO-positive cells in the c-fos overexpression mice model were previously described [31].
In Lewis lung carcinoma, oxidized cholesterol or oxysterols play an essential role in neutrophils attracting in a C-X-C chemokine receptor 2 (CXCR2)-dependent manner, triggering tumor growth and survival reduction [58]. The primary sources of these oxysterols, specifically 27-HC, are M2 macrophages with high cytochrome P450 family 27 subfamily A member 1 (Cyp27a1) and Cyp27b1 expression levels; interestingly, they secrete more 27-HC than breast cancer cell lines [59]. Similarly, a high macrophage and neutrophil recruitment process was found in zebrafish models, but with specific significance in neutrophil numbers with HCD, inducing NASH-HCC tumors [60].
Both phagocytic cells mentioned act as a double-edged sword [61] harming or benefiting the tumor’s aggressiveness. In the same way as TAMs, it will be a good strategy for neutrophil elimination or TAMpering.
NK cells represent 5–20% of human circulating lymphocytes and are classified as part of the innate immunity with cytotoxic activities. The most important differences with CTL cells are that NK lymphocytes do not require prior antigen exposure for their activation and anti-tumor functions [62]. It has been studied the relationship between cholesterol and NK cell activation. HCC mice models initiated by DEN treatment or Hepa1–6 cell line transplantation and fed with HCD (cholesterol 2% and sodium cholate 0.25%) revealed that NK cells uptake cholesterol from serum, promoting their accumulation in the tumor and enhancing receptor NK group 2, member D (NKG2D), and natural cytotoxicity triggering receptor 1 (NCR1) function owing to the lipid rafts formation in these cells decreasing tumor aggressivity. NK cells are activated with CCL3, CXCL10, chemerin, and IFNγ. Specific deletion ApoE−/− provoked the same behavior inducing the fewer and smaller developed of liver tumors and lung metastasis process and augmenting effector functions of NK lymphocytes [63].
Similar results were analyzed in the B16F10 melanoma model with the dysfunctional response of invariant NK T (iNKT) cells, reporting a high lipid and cholesterol biosynthesis in these cells and activating peroxisome proliferator-activated receptor gamma (PPARγ) and promyelocytic leukemia zinc finger (PLZF) to transcription of Srebf1 [64]. A similar process shows high cholesterol uptake for the activation of NK cells in normal conditions [45]. In the same way, studies of c-fos genetic overexpression reduce the activity of NK cells [31].
DC are innate cells responsible for capturing the antigen and subsequently presenting it to T cells through the major histocompatibility complex (MHC). DC have a heterogeneous response and bone marrow origin functions [65]. Subsets such as blood dendritic cell antigen 2 (BDCA2+) plasmacytoid DC promote poor prognosis in HCC patients with intra-tumoral location and are associated with Treg and Th17 cells [66].
DC have opposite effects with lipids, mainly oxidized lipid forms, inhibiting antigen cross-presentation and peptide MHC class I complexes, and reducing anti-tumor surveillance in different tumor models [67]. As we mentioned before, oxysterols inhibit antitumoral function in leukocytes and exacerbate pro-tumoral behavior in tumors. However, sterol metabolism and LXR also affect DC in a C-C chemokine receptor-7 (CCR7) dependent manner, affecting migration to lymphoid tissues in mice and humans [68].
It is necessary to investigate the relationships between the DC and cholesterol in HCC and clarify why antigen presentation is annulated. Vaccine designs with HCC tumor-associated antigens (TAA) coupled with cholesterol complexes could represent an excellent therapy to active this “immune gossip”, as it was observed using cholesterol-coupled “New York esophageal squamous cell carcinoma 1” (NY-ESO-1), promoting CD4+ and CD8+ cells cross-presentation [69]. These data show the contradictory effects of cholesterol on tumor aggressiveness and therapeutic strategy like some studies performed in NK cells.
T lymphocytes encompass adaptive immunity with a wide variety of responses and subsets. These cells have bone marrow origin progenitors and migrate to the thymus for maturation. Only selected cells are exported to secondary lymph organs. T cell activity depends on the contact microorganisms or pathology. Th1, Th2, and Th17 respond to intracellular microbes, parasites, and extracellular pathogens. They have diverse roles in the cancer context. For example, Th2 cytokines represented a poor prognosis in HCC patients concerning Th1 secretion in normal health. Th2 responses decrease after the treatment with radiofrequency ablation (RFA) [70]. Finally, Treg cells subsets represent an immunosuppressive response to these last cells [71]. T cells have differential roles and activities in liver diseases such as NAFLD, obesity, and NASH-HCC progression [72].
In experimental animals fed with WDs, the progression from NAFLD to NASH involves cirrhosis and tumor promotion. Diet components such as cholesterol induce ballooning hepatocytes, fibrosis, and inflammation with specific T cells (CD3+, CD4+, and CD8+) recruitment to the liver [73]. Cholesterol accumulation in the TME in other melanoma or pulmonary tumor models induces immune checkpoint inhibitors in CD8+ T cells such as programmed death 1 (PD-1), 2B4 (CD244), T cell immunoglobulin 3 (TIM-3), and lymphocyte-activation gene 2 (LAG-3) on another form of cholesterol accumulation disrupts lipid metabolism and causes their exhaustion through XBP-1 increasing ER stress in lymphocytes. Dysfunctional cells are not capable of delivering TNF-α, granzyme B precursor (GzmB), or IFNγ to eliminate cancer cells [74].
Other reports show that cholesterol synthesis increases Treg cell activity. Liver kinase B1 (LKB1) regulates the mevalonate pathway in Treg cells inducing cholesterol and geranylgeranyl pyrophosphate (GGPP). Deficient LKB1 produces Th1 and Th17 secretion [75]. Even though LKB1 protein is considered a tumor suppressor [76], different reports show that upregulation of LKB1 has been demonstrated in HCC tumors, indicating oncogenic functions such as larger tumor size and tumor foci and poorer tumor differentiation [77, 78].
The design of new therapies against tumors led researchers James P. Allison and Tasuku Honjo to be awarded the Nobel prize in physiology or medicine in 2018. The innovative approach consists of specific monoclonal antibodies against immune checkpoint inhibitors. Immunotherapies could represent the most effective strategy to treat different tumors, including HCC. Nevertheless, these molecules are limited to early-stage development. These therapies currently trigger specific T-cell responses against tumors, inhibiting PD-1 and cytotoxic T-lymphocyte antigen 4 (CTL4) receptors achieve 15–30% efficacy in HCC patients [43].
The second-line drugs combination of monoclonal antibodies atezolizumab (1,200 mg/kg) plus bevacizumab (15 mg/kg), an anti-programmed death-ligand 1 (PD-L1) and anti-vascular endothelial growth factor A (VEGFA) respectively, intravenously every three weeks, is promising molecules to treat HCC tumors, improving results and reflected in overall and progression-free survival outcomes than other multikinase inhibitors such as sorafenib (400 mg/kg) orally twice daily. These results have yielded an overall survival at six months of 84.8% for antibodies combination vs. 72.2% for only sorafenib; and progression-free survival at six months of 54.5% vs. 37.2% in the same manner [79].
As mentioned before, HCC etiology and tumor stage are not linked or associated with significant changes in the immune contexture, observing the presence of cytotoxic and T cells or macrophage and Th2 cells absence positively correlated with patient survival [42]. The above gives us light for the design of new strategies for the HCC elimination, but regarding immunotherapies, it has been reported only in NASH etiology limiting anti-tumor surveillance; thus, NAFLD/NASH-HCC tumors have been classified as the less responsive to treat this pathology and maybe the most aggressive HCC-phenotype. In mice, a choline-deficient HFD (CD-HFD) results in NASH, with high macrophages and T cells infiltration. Contrary to what is observed in the literature, cytotoxicity by CD8+ lymphocytes decreased the patient survival in NASH-HCC tumors or increased tumor lesion incidence only in NASH using anti-PD-1 monoclonal antibodies. Anti-PD-1 raises ALT activity, steatosis, fibrosis, NAFLD activity score (NAS), and specific PD-1+ TNF+ CD8+ T cell subsets with pro-inflammatory molecules such as TNF-α. The authors used anti-TNF-α and anti-CD8 antibodies to reduce tumor formation, with excellent results observed in aggressivity reduction and tumor lesion incidence.
It was studied the role of lipids and cholesterol on T CD8+ lymphocytes. Cholesterol induces an exhausted state in CD8+ T cells and reduces TNF-α or GzmB, contrary to what is expected using CD-HFD mentioned above in this review [74].
The effect of lipids in NAFLD-HCC treated with monoclonal antibodies compared with other HCC etiologies is summarized in Figure 3.
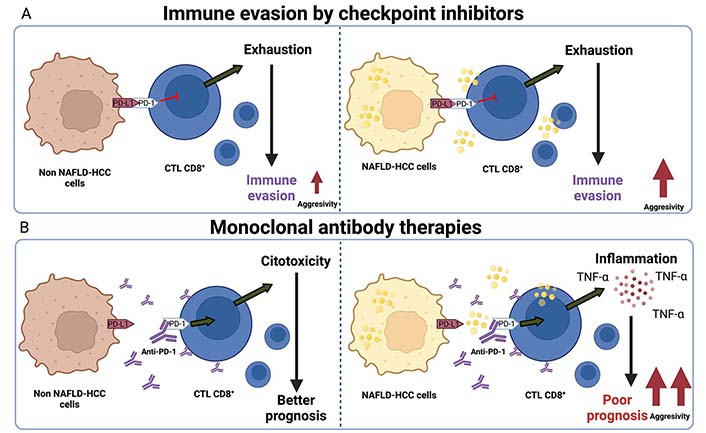
Immune checkpoint inhibitors therapies impair aggressivity in NAFLD-HCC. A. Non-NAFLD-HCC cells induce T cell exhaustion in CD8+ lymphocytes (CTL CD8+) and promote immune evasion and increment of tumor growth. For another hand, NAFLD-HCC cells performed the same effect, but an increment of aggressivity; B. monoclonal antibodies against PD-1 that inhibits the junction of PD-L1 present in cancer cells and macrophages; this allows CTL CD8+ activation and cytotoxicity effect reflected in a better prognosis in patients. Conversely, this effect is not observed in NAFLD-HCC with immune-therapies, analyzed by high inflammation reflected in TNF-alpha secretion, poor prognosis, and high aggressivity in HCC patients. Created with BioRender.com
The other novel strategy is using cholesterol synthesis inhibitors, for example, statins. It has been reported that statins reduce cardiovascular morbidity and cancer diagnosis mortality risk for HCC patients [80], or in combination with other drugs, increase their efficacy, which is reflected in the non-small cell lung carcinoma (NSCLC) patients’ survival and decreased mortality [81].
Statin synthesis derived from fungal fermentation performed competitive and reversible inhibition of the limiting rate enzyme of the mevalonate pathway, HMGCR, to form mevalonate or mevalonic acid. Statins can be classified depending on their chemical characteristics: lipophilic such as lovastatin, simvastatin, fluvastatin, atorvastatin, cerivastatin, and pitavastatin; and non-lipophilic such as pravastatin and rosuvastatin [82]. Regarding statin types, it was reported that lipophilic statins had been associated with a significant reduction in HCC incidence and mortality with chronic viral hepatitis, but not using hydrophilic statins [83].
In the same way, it has been demonstrated in meta-analysis studies that statins decreased HCC occurrence, showing beneficial chemopreventive effects. Specifically, dose-dependent and lipophilic statins are associated with HCC reduction incidence [84]. To demonstrate statin efficacy after HCC diagnosis, patients were treated using statin varieties finding better responses reflected in patient survival [80].
Regardless of the reduction in cholesterol and derivatives, atorvastatin reduces small GTPases activity such as Rac, and the phosphorylation of MYC by glycogen synthase kinase β (GSK3β); as a consequence, cell proliferation of HCC cell lines is decreased, and in vivo tumors [85]. It has been reported that simvastatin with sorafenib combination enhances apoptotic sensitivity and reduces proliferation in the LM3 HCC cell line, reducing HIF-1α/PPAR-y/PKM2-mediated glycolysis [86].
In the same way, it is surprising how the simple treatment using statin reduces mortality and incidence. It has been demonstrated the reduction of HCC in NASH patients with advanced liver fibrosis and cirrhosis. The relevance of this retrospective study was found to be an association statin and a lower risk of developing HCC (23.6%) [87]. As the most interesting data for this review, it is important to mention that statin use also decreased the risk of NAFLD and liver fibrosis [88]. Nevertheless, it is important to mention the dose-dependent use of statins due to possible liver toxicity and ALT detection [89].
On leukocytes, specifically in myeloid cells treated with statins decreased the HMGCR enzyme activity, clearly impacting macrophages’ infiltration and chemotaxis. Lovastatin treatment induces a genetic program inhibiting M2 macrophages phenotype and triggers T lymphocytes recruitment into mouse mammary tumors, as we expected in liver tumors [90]. Same data were observed in lung adenocarcinoma; statin decreases M2 macrophages (CD68+, CD163+) [91]. Similar results were observed in specific genetic methods (Hmgcrm-/m-). This deletion reduces hepatic steatosis and adipose tissue macrophages in HFD-fed mice [92]. The anti-inflammatory effects of pitavastatin have been observed to reduce the transcriptional activity of NF-κB in the nucleus and the production of IL-6 induced by TNF-α [93]. It was reported that atorvastatin diminished expression and content levels of PD-L1 with IFNγ and TNF-α cotreatment in HepG2 cells, indicating a possible reduction in immune exhaustion in cytotoxic cells [94]. These recent data show that removing inflammation and specific response is vital to treating hepatic pathologies, including HCC.
Statins are well accepted and present minimal adverse effects, but it is important to address the impact of these molecules in the context of cancer, particularly in HCC and their microenvironment.
Currently, immune-oncology research is needed in HCC and other liver cancers, but there is a modest relationship between cholesterol/lipids and inflammation in HCC progression compared with other tumor types. Each cell leukocyte triggers different responses, depending on the stage of this pathology, such as M1 or M2 macrophages in early and late stages, respectively. Leukocytes are responsible for aggressiveness because they represent the infiltrating population, and the stability of tumors depends on them and provides cholesterol to the microenvironment and cytokine secretion. The use of lipid and cholesterol inhibitors combined with other immunomodulators would effectively eliminate cholesterol biosynthesis and inflammation in liver tumors such as HCC.
27-HC: 27-hydroxy-cholesterol
ALT: alanine aminotransferase
ApoE: apolipoprotein E
CCL3: C-C motif chemokine ligand 3
CTL: cytotoxic lymphocytes
DC: dendritic cells
DEN: diethylnitrosamine
FFA: free fatty acids
G-CSF: granulocyte-colony stimulator factor
HCC: hepatocellular carcinoma
HCD: high cholesterol diet
HFD: high-fat diet
HFHCD: high fat and high cholesterol diet
HMGCR: 3-hydroxy-3-methylglutaryl-coenzyme A reductase
IFNγ: interferon gamma
IL-3: interleukin-3
KC: kupffer cells
LDL: low-density lipoproteins
LDLR: low-density lipoproteins receptor
LKB1: liver kinase B1
LXR: liver X receptor
MDSC: myeloid-derived suppressor cells
MHC: major histocompatibility complex
NAFLD: non-alcoholic fatty liver disease
NASH: non-alcoholic steatohepatitis
NF-κB: nuclear factor kappa B
NK: natural killer
PD-1: programmed death 1
PD-L1: programmed death-ligand 1
SREBP: sterol regulatory element-binding proteins
STAT3: signal transducer and activator of transcription 3
TAMs: tumor-associated macrophages
TGF-β: transforming growth factor-β
Th: T helper
TIME: tumor immune microenvironment
TME: tumor microenvironment
TNF-α: tumor necrosis factor-alpha
Treg: regulatory T lymphocytes
WD: western diet
AEC conceptualized the manuscript, wrote the draft, and revised the final manuscript. LCR and ASN wrote and revised the draft. VSA and RUML provided the input in writing the paper. LEGQ conceptualized the manuscript and revised the draft and the final manuscript. MCGR conceptualized the manuscript, designed the outline, and coordinated the writing and financial support.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was funded by “Universidad Autónoma Metropolitana” and “Conacyt-Fronteras de la Ciencia-1320”. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

