 Review
Review

Affiliation:
1Laboratory of Translational Cellular and Molecular Biomedicine, National Research Tomsk State University, 634050 Tomsk, Russia
2Department of General and Molecular Pathology, Cancer Research Institute, Tomsk National Research Medical Center, Russian Academy of Sciences, 634009 Tomsk, Russia
ORCID: https://orcid.org/0009-0001-3835-5259
Affiliation:
1Laboratory of Translational Cellular and Molecular Biomedicine, National Research Tomsk State University, 634050 Tomsk, Russia
3Laboratory of Molecular Oncology and Immunology, Cancer Research Institute, Tomsk National Research Medical Center, Russian Academy of Sciences, 634009 Tomsk, Russia
ORCID: https://orcid.org/0009-0000-6159-5173
Affiliation:
1Laboratory of Translational Cellular and Molecular Biomedicine, National Research Tomsk State University, 634050 Tomsk, Russia
4Laboratory of Molecular Therapy of Cancer, Cancer Research Institute, Tomsk National Research Medical Center, Russian Academy of Sciences, 634009 Tomsk, Russia
ORCID: https://orcid.org/0000-0001-6637-4417
Affiliation:
1Laboratory of Translational Cellular and Molecular Biomedicine, National Research Tomsk State University, 634050 Tomsk, Russia
5Center for Systems Bioinformatics, Tomsk National Research Medical Center, Russian Academy of Sciences, 634009 Tomsk, Russia
ORCID: https://orcid.org/0000-0002-0646-6093
Affiliation:
6Abdominal Department, Cancer Research Institute, Tomsk National Research Medical Center, Russian Academy of Sciences, 634009 Tomsk, Russia
ORCID: https://orcid.org/0000-0002-2748-0644
Affiliation:
1Laboratory of Translational Cellular and Molecular Biomedicine, National Research Tomsk State University, 634050 Tomsk, Russia
4Laboratory of Molecular Therapy of Cancer, Cancer Research Institute, Tomsk National Research Medical Center, Russian Academy of Sciences, 634009 Tomsk, Russia
ORCID: https://orcid.org/0000-0001-5758-7330
Affiliation:
1Laboratory of Translational Cellular and Molecular Biomedicine, National Research Tomsk State University, 634050 Tomsk, Russia
7Institute of Transfusion Medicine and Immunology, Institute for Innate Immunoscience (MI3), Medical Faculty Mannheim, University of Heidelberg, 68167 Mannheim, Germany
8Bashkir State Medical University of the Ministry of Health of Russia, 450000 Ufa, Russia
Email: julia.kzhyshkowska@medma.uni-heidelberg.de
ORCID: https://orcid.org/0000-0003-0898-3075
Explor Dig Dis. 2026;5:1005124 DOI: https://doi.org/10.37349/edd.2026.1005124
Received: February 16, 2026 Accepted: April 14, 2026 Published: May 27, 2026
Academic Editor: Jose C. Fernandez-Checa, Institute of Biomedical Research of Barcelona (IIBB), CSIC, Spain
The article belongs to the special issue Immunotherapy for Cancer of Digestive System
Immunotherapy is a promising treatment strategy for treating colorectal cancer (CRC). Despite significant advances in this field, resistance and low efficacy of immunotherapy remain a principal problem. One of the most important factors affecting the response to immunotherapy is the tumor microenvironment (TME). Among the components of the TME, tumor-associated macrophages (TAMs) are key immune cells involved in cancer progression by stimulating tumor cell proliferation, angiogenesis, epithelial-mesenchymal transition, metastasis, and tumor immune evasion. This review presents currently investigated combination therapy based on the immune checkpoint inhibitors and inhibitors of diverse components of the TME, including TAMs, that can potentially increase the effectiveness of CRC treatment. Therapeutic efficacy, together with the functional activity of TAMs, is estimated in multiple preclinical data obtained with diverse in vitro and in vivo models. Ongoing clinical trials demonstrated the association of treatment effectiveness with TAM phenotypes and functions.
Immunotherapy is one of the promising methods for the effective treatment of many types of cancer. There are several strategies for cancer immunotherapy: oncolytic viral therapy, adoptive cell therapy, therapy with immune checkpoint inhibitors (ICIs), and therapy based on key cytokines and chemokines of the tumor microenvironment (TME) [1]. For decades, immunotherapy was a concept primarily based on the enhancement of cytotoxic immunity, where T cells and NK cells are considered the major anti-cancer immune cell subsets [2–7]. However, a number of promising and efficient approaches in animal studies failed in clinical trials. Only a few therapeutic strategies that came to clinical trials demonstrate effectiveness in a particular group of patients. Currently, the most challenging issue is to identify criteria discriminating responders from non-responders in order to efficiently apply expensive immunotherapy only to those patients who will benefit with high probability.
Limitations associated with the lack of efficiency of immunotherapy include components of the TME, the microbiome, intratumor heterogeneity, and tumor mutational load [8]. TAMs are the most abundant immune cell population in the TME and represent a promising therapeutic target for improving tumor resistance to immunotherapy [9]. The role of tumor-associated macrophages (TAMs) at each stage of solid tumor progression was recognized and demonstrated in multiple animal models. In cohorts of cancer patients, the correlation of TAM phenotypes with poor prognosis and poor responses to conventional anti-cancer therapies was found [10]. Surprisingly, in contrast to other cancer types, the total number of TAMs in colorectal cancer (CRC) correlates with a favourable prognosis. However, M2-like macrophages were indicative of poor prognosis in multiple CRC cohorts [11]. Low-grade glycolysis was found to provide the metabolic conditions for TAMs to support tumor progression [12, 13].
Macrophages are conventionally classified into two states of polarization: M1-like macrophages and M2-like macrophages [14]. M1-like macrophages, or classically activated macrophages, possess antimicrobial, pro-inflammatory, and antigen-presenting properties, while M2-like macrophages, also known as alternatively activated macrophages, exhibit anti-inflammatory, pro-tumor, and antimicrobial properties [15]. Macrophages can be repolarized under diverse factors secreted by immune cells, tumor cells, microorganisms, and other components of the tissue microenvironment [15].
Macrophages in the TME are primarily recruited from circulating bone marrow-derived monocytes and differentiate into TAMs [16, 17]. For a long time it was believed that TAMs have an M2-like phenotype expressing major pro-tumor molecules, e.g., mannose receptors (CD206), scavenger receptors (CD163), vascular endothelial growth factor (VEGF), interleukin (IL)-10, C-C motif chemokine receptor 2 (CCR2), CD204, and C-C motif chemokine ligand 22 (CCL22) [18, 19] However, recent single cell and spatial transcriptomic data allowed identifying that pro-tumoral TAMs have a mixed M1/M2-like signature, and the origin of TAMs is critical for their pro-tumoral activation [10]. Functional activity of TAMs is formed by transcriptional, epigenetic, and metabolic reprogramming [20]. The activation state of TAMs becomes an effective predictor for the immunotherapy used. Moreover, TAMs are an attractive target for the development of new immunotherapy. However, multiple clinical trials with agents targeting viability, differentiation, recruitment, or activation state of TAMs failed [10]. Despite this, more and more TAM-based approaches are under development to overcome resistance to conventional anti-cancer therapy.
In our review, we combined recent data on the state-of-the-art in the progress and challenges in immunotherapy of CRC, highlighting novel TAM-based approaches and potential clinical significance of TAMs for the efficiency of existing immunotherapy with ICIs. For this purpose, we focused on the relevant articles containing major research terms including “colorectal cancer”, “tumor-associated macrophages”, “immunotherapy”, “immune checkpoint inhibitors”, and “targeted therapy”. The review elucidates the mechanisms of the synergistic effects of combined therapeutic schemas where TAMs are used to enhance the effects of ICIs.
CRC is the third most common cancer and the second most deadly cancer among all cancer types [21]. The number of new cases of CRC is projected to increase from 1.93 million cases per year to 3.2 million by 2040 [22]. CRC usually begins as a benign tumor, which develops into a malignant one within 10–20 years [23].
CRC is classified according to the CpG island methylation phenotype (CIMP) and microsatellite instability (MSI) status, and is divided into four molecular subtypes: CIMP+/MSI+, CIMP+/MSI–, CIMP–/MSI+, and CIMP–/MSI– [24]. It was shown that the number of intraepithelial TAMs detected by the pan-macrophage marker CD68, as well as the number of the intraepithelial marker of M2-like macrophages detected by CD163, was elevated in patients with high MSI (MSI-H) and high CIMP levels [25].
A classification of CRC based on genomic and transcriptional profiling data was proposed in 2016, and includes four consensus molecular subtypes (CMSs): CMS1 (MSI immune), CMS2 (canonical), CMS3 (metabolic), and CMS4 (mesenchymal) [26]. CMS1 tumors are hypermutated and characterized by MSI, high levels of CIMP cluster, the presence of BRAF mutations, and diffuse infiltration of immune cells (primarily T-helper 1 and cytotoxic T cells). The CMS2 tumors are characterized by the activation of downstream targets of the WNT and MYC pathways, as well as a loss of tumor suppressor genes and an increase in the number of oncogene copies. KRAS mutations and metabolic deregulation predominate in CMS3 tumors. CMS4 tumors have increased expression of proteins involved in various oncogenesis pathways: epithelial-mesenchymal transition (EMT), activation of the transforming growth factor β (TGF-β) signaling pathway, angiogenesis, matrix remodeling pathways, and the inflammatory complement system [26]. Among all CRC subtypes, CMS4 tumors are highly infiltrated with CD68+ macrophages, as well as myeloid cells, stem cells, and stromal clusters, enriched with the M2-like CD163+ subset of macrophages [27, 28]. Transcriptional profiling revealed that CMS4 CRC tumors were highly enriched in signatures associated with altered macrophage activation and downregulation of phagocytosis [28]. CRC metastases to the lung and liver are usually classified as CMS2. However, peritoneal metastases are characterized by CMS1, CMS2, and CMS4 signatures. CMS4 peritoneal metastases are enriched in macrophages (CD163+ and CD206+), cancer-associated fibroblasts (CAFs), and endothelial cells, as well as have increased expression of the immune checkpoint molecules (TIGIT and PD-L2) [29].
The term CRC encompasses two types: colon cancer (CC) and rectal cancer (RC). However, accumulated data suggest that they should be considered as two separate diseases [30, 31]. CC and RC differ in anatomy and topography, epidemiology, risk of carcinogenesis, histology, as well as symptoms, primary prevention, and treatment [30, 31]. Immune-associated differences were found for these two histological locations. Compared to RC, in CC, the neutrophil-to-lymphocyte ratio and platelet-to-lymphocyte ratio, as well as the systemic inflammatory response index, systemic immune inflammation index, and total systemic inflammation index, were significantly increased. At the same time, blood monocyte levels did not differ significantly between patients with CC and RC [32]. CD3+ and CD8+ T lymphocyte infiltration in the tumor nest and CD3+ lymphocytes in the stroma of CC, but not RC tumors, correlated with better overall survival [33]. High tumor infiltration with NK cells, macrophages, and CD4+ T cells correlated with a low probability of local recurrence in RC [34]. High numbers of both peritumor and intratumor macrophages and T cells (CD3+, CD8+) correlated with a low probability of distant recurrence in RC [34]. Our recent study demonstrated an increased number of peripheral blood monocytes and an accumulation of intratumor TAMs expressing glycolysis activator PFKFB3 in patients with CC compared with those with RC [12]. We demonstrated that PFKFB3 may induce changes in lipid and amino acid metabolism in macrophages, but it had a dual effect in the context of pro- and anti-tumor effects [13]. Differences in gene expression (such as S100P, Reg family, ACTN1, CAMK2G, ACAT1) and signaling pathways (metabolic pathway, cell cycle pathway, p53 pathway) between CC and RC have also been demonstrated [35, 36]. However, non-hypermutated CC and RC tumors cannot be distinguished at the genomic level [37]. Right-sided and left-sided СС are distinct in genomics. Right/ascending colon is more often hypermethylated and has elevated mutation rates [37]. Right-sided CC had MSI-H and gene mutations in BRAF, KRAS, SMAD4, TGF-β, PIK3CA, PTEN, and AKT1 compared to left-sided CC. No significant differences were observed in HRAS, NRAS, APC, TP53, and FBXW7 between the right-sided CC and left-sided RC groups [38]. Tumors with a high mutational load and overexpression of tumor neoantigens are more sensitive to immunotherapy. Approximately 80–85% of patients with CRC are unresponsive to ICIs because they have “cold” tumors with microsatellite stable (MSS) or low MSI (MSI-L) [39]. “Cold” tumors often harbor immunosuppressive cell populations, such as TAMs, regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs), which can potentially be targets for immunotherapy to induce tumor cold-to-hot transition [40]. Both deficient mismatch repair (dMMR) and MSI-H tumors can provide de novo tumor antigens caused by accumulating mutations, leading to tumor immunogenicity. In contrast, mismatch repair proficient (pMMR) CRC cells exhibit weak immunogenicity and are resistant to immunotherapy [41]. Tumors with MSI-H typically respond well to ICIs, including antibodies against programmed cell death-1 (PD-1), programmed cell death ligand-1 (PD-L1), and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4). However, in MSI tumors, immunotherapy may also be ineffective due to the reprogramming of key metabolic pathways in CRC. This results in the formation of by-products that affect the immune response, reducing the anti-tumor activity of immune cells and contributing to resistance to ICIs [42, 43].
The development of CRC is a multi-stage process. Focal changes in the intestinal mucosa lead to the formation of benign polyps, followed by the active proliferation of cells that can eventually acquire malignant properties [44]. Chronic inflammation is one of the causes of CRC. Chronic inflammation may be associated with changes in the levels of inflammatory markers in immune and tumor cells [45]. Furthermore, chronic inflammation facilitates DNA damage, resulting in the activation of pro-oncogenes and the inactivation of tumor suppressor genes [46].
The inflammatory process involves various immune cells, such as macrophages, lymphocytes, as well as proinflammatory factors—tumor necrosis factor (TNF)-α, IL-6, and IL-1β, and reactive oxygen species [47, 48]. Immune cells play a key role in facilitating the transition from inflammation to carcinogenesis [47]. In the early stages of CRC development, M1-like macrophages play a key role, creating an inflammatory environment that promotes mutational changes [49, 50]. The transcription factor (TF) NF-κB and the inflammatory cytokine TNF-α are key regulators of the inflammatory process in the intestinal mucosa. Specifically, activation of the NF-κB pathway promotes the polarization of pro-inflammatory M1-like macrophages [51]. Moreover, epithelial MUC1 induces CCL2 expression and mediates macrophage recruitment and activation [49]. Macrophages secrete the pro-inflammatory cytokine IL-1β, TNF-α, and IL-6, and may promote the transformation of cells into a stem cell state, leading to dysplasia and colitis-associated tumorigenesis [52]. After cell transformation, peripheral blood monocytes are attracted to the tumor site, secreting growth factors and chemokines (CCL2, CCL5, VEGF, and TGF-β) [50]. Moreover, CRC cells secrete EGF to alter macrophage polarization toward M2-like, which, at the late stages of tumor development, promotes tumor immune evasion and contributes to cancer progression [49, 50].
TAMs create a favorable environment for tumor cell growth, proliferation, invasion, and metastasis [11]. Accumulated experimental data demonstrated contradictory pro- and anti-tumor activities of TAMs in CRC [11]. The number of macrophages determined by the general marker CD68 positively correlated with a favorable prognosis in CRC [53–56]. Macrophages with M1-like and M2-like phenotypes were simultaneously present at the front of tumor invasion, and an increase in the total number of macrophages was associated with a better prognosis [57]. However, the predominance of M2-like TAMs (CD163+, CD206+, SPP1+, and Stabilin-1+) in CRC tumors, on the contrary, correlated with an unfavorable prognosis and disease progression [58–62]. In vitro studies showed that tumor factors derived from CRC cell lines did not affect the shift in the polarization of M1-like macrophages towards M2-like, but, on the contrary, increased the expression of the M1-like marker CD86 and decreased the expression of the M2-like marker CD163 [63]. Macrophages stimulated with conditioned media from CRC cell lines (HT-29 or HCT116) had higher levels of expression of both M2-like markers CD163 and IL-10, and M1-like markers IL-1β, interferon (IFN)-γ, and TNF-α [58]. Exosomes secreted by tumor cells can promote mixed polarization of macrophages. Exosomal vesicles from the SW620 cell line induced a mixed pattern of secretion of M1-like [C-X-C motif chemokine ligand 10 (CXCL10), IL-6, IL-23] and M2-like (IL-10) cytokines in inactive M0-like macrophages [64].
Thus, the high plasticity of macrophages and their complex role in tumor progression make them an ideal target for the development of immunotherapeutic approaches to increase the effectiveness of the existing CRC therapies [65].
The standard of care for CRC includes surgery, chemotherapy, radiotherapy, targeted therapy, and immunotherapy (Figure 1). Surgical intervention is the primary method of radical treatment for CRC; in early stages, it is used alone, while in locally advanced tumors it is combined with neoadjuvant or adjuvant therapy [66, 67]. In patients with RC, treatment tactics depend on the extent and location of the tumor process: if the upper third of the rectum is affected, radical surgery or neoadjuvant chemotherapy (FOLFOX/XELOX) followed by surgery is performed; for cancer of the mid and lower third of the rectum, radiation therapy (RT) or chemoradiotherapy (CRT) with total neoadjuvant therapy and surgical treatment are used. In patients with CC and locally advanced resectable tumors, neoadjuvant FOLFOX/XELOX chemotherapy and radical surgery are performed. For stages 2 and 3 CRC, immunotherapy with ICIs as neoadjuvant therapy is usually used for patients with dMMR or MSI-H tumors. For metastatic CRC (mCRC), various CRT, targeted therapy, or immunotherapy regimens are used depending on the patient`s condition and the effectiveness of previous treatment [66, 67].
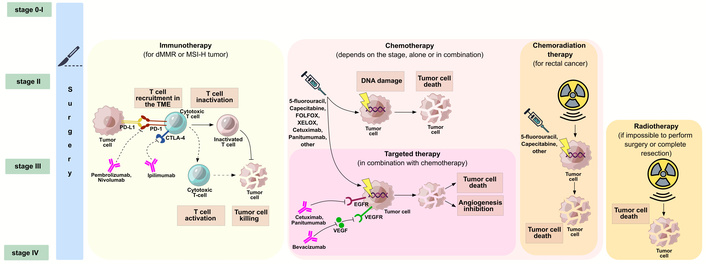
The standard of care for CRC. Cancer stage is one of the main, but not the only, factors determining the treatment strategy for CRC. Treatment options can be combined or used sequentially. CRC: colorectal cancer; CTLA-4: cytotoxic T lymphocyte-associated antigen-4; dMMR: deficient mismatch repair; EGFR: epidermal growth factor receptor; FOLFOX: folinic acid, fluorouracil and oxaliplatin; MSI-H: high microsatellite instability; PD-1: programmed cell death-1; PD-L1: programmed cell death ligand-1; TME: tumor microenvironment; VEGF: vascular endothelial growth factor; VEGFR: VEGF receptor; XELOX: capecitabine plus oxaliplatin.
Immunotherapy has revolutionized cancer treatment by introducing novel mechanisms that engage the host’s immune system against tumor cells [68, 69]. Immunotherapy efficacy largely depends on specific molecular features of the tumor, in particular on MMR status.
ICIs represent the most clinically established form of immunotherapy in CRC [70, 71]. Monoclonal antibodies targeting the PD-1/PD-L1 axis (e.g., pembrolizumab or nivolumab), as well as those targeting CTLA-4 (ipilimumab), are approved for dMMR/MSI-H mCRC [68, 69]. The high neoantigen burden in these tumors, comprised of mutant proteins unique to cancer cells, enhances their immunogenicity, making them more susceptible to ICIs [72]. For such tumors, ICIs can yield durable responses and are considered standard therapeutic options [69].
In 2014, the first anti-PD-1/PD-L1 drug pembrolizumab, was approved by the FDA for the second-line treatment of advanced malignant melanoma. Subsequently, it was approved for mCRC second-line treatment in 2017 [41]. Ipilimumab plus nivolumab was approved by the FDA for first-line treatment of unresectable or dMMR/MSI-H mCRC [73].
Pembrolizumab is a humanized monoclonal antibody that targets the PD-1 receptor on lymphocytes and has shown efficacy in several solid tumors [74]. Pembrolizumab has demonstrated durable anti-tumor activity as a first-line treatment for dMMR/MSI-H mCRC, with an objective response rate (ORR) of 45% and a complete response (CR) rate of 13% [75]. Pembrolizumab monotherapy represents a promising treatment option for untreated mCRC patients [76].
A case of locally advanced MSI-H RC that exhibited a remarkable response to pembrolizumab treatment was reported [76]. Pathological analysis revealed prominent infiltration of immune cells, predominantly CD8-positive T cells and clusters of PD-L1-expressing macrophages. Histopathological analysis demonstrated clusters of PD-L1-expressing macrophages within the tumor, suggesting the restoration of anti-tumor immunity through ICIs [76].
Dual checkpoint inhibition has been proposed as a therapeutic strategy in patients with MSI-H CRC. Nivolumab is a humanized monoclonal antibody targeted at PD-1. Ipilimumab, another monoclonal antibody, was developed to target CTLA-4 [74].
Clinically significant efficacy of the combination of nivolumab and ipilimumab was demonstrated in patients with MSI-H mCRC [77]. Combination therapy increased progression-free survival (PFS) and ORRs compared with nivolumab monotherapy. However, the CR rate was similar in both groups (30% for combination therapy and 28% for monotherapy) [77].
A precise relationship between CMTM6 expression, PD-L1 levels, and M2-like macrophage infiltration was indicated in metastatic/refractory CRC treated with PD-1/PD-L1 inhibitor immunotherapy. CMTM6 expression was positively correlated with PD-L1 expression and CD163+ M2-like macrophage density in dMMR CRC [78].
Nivolumab disrupted the PD-1/PD-L1 inhibitory axis between cytotoxic T cells and TAMs, potentially reversing macrophage-mediated immunosuppression [68].
Despite the advances in immunotherapy, the development of resistance to immunotherapy remains a serious problem [68, 79, 80]. The mechanism of resistance to immunotherapy is extremely complex and is related to genetic factors and previous treatment of the patients. Immunotherapy resistance of CRC may be related to the following reasons: insufficient tumor antigen presentation, tumor antigen presentation damage, T cell exclusion, and immunosuppressive signaling in the TME [81]. Key immunosuppressive components in the TME include Tregs, IL-10, TAMs, MDSCs, and related cytokines [41].
Within the tumor immune environment, TAMs are increasingly considered a key element in the development of resistance to immunotherapy and the occurrence of relapses [82–84]. Macrophages enhance the development of tumor resistance through different mechanisms. TAMs express PD-L1 [85] and CTLA-4 ligands, which block the T lymphocytes’ adaptive immune response and reduce the anticancer effects of immunotherapy by binding to PD-1 and CTLA-4 on the surface of T cells. M2-like macrophages recruit mature Tregs via the secretion of cytokines, including CCL22 and CCL18. TGF-β and IL-10 secreted by M2-like macrophages promote the conversion of naive T cells to Tregs [86]. TAMs further contribute to immunotherapy resistance by secreting prostaglandin E2 and other cytokines that suppress T cell activity while inducing PD-L1 expression. As a result, PD-L1 binding to PD-1 on T cells diminishes cytotoxic function and promotes immune evasion in CRC [87]. Another mechanism of TAM-derived immunosuppression includes M2-like TAM activity by inhibiting the secretion of IFN-γ from CD8+ T cells. M2-like TAMs further produce excess levels of VEGF, creating a feed-forward loop, which maintains immunosuppression in the CRC tumor [88]. Chemorefractory CRC tumors recruit immunosuppressive macrophages through the CSF1/CSF1R axis, which promotes PD-L1 upregulation in tumor cells via TGF-β signaling, thereby establishing an immunosuppressive TME where high macrophage infiltration positively correlates with elevated PD-L1 levels [89].
As was mentioned above, the majority of CRC cases are MSS or pMMR and exhibit primary resistance to single-agent ICI therapy [90, 91]. This significant clinical challenge has driven extensive research into combination strategies designed to overcome the immunosuppression of the TME [80, 83].
Rational therapeutic approaches that are currently under investigation include a combination of ICIs with agents that alter the TME on multiple fronts:
Kinase inhibitors. Regorafenib can abolish immunosuppressive TAMs and Tregs infiltration, and in combination with anti-PD-1 therapy that boosts T cell activity, demonstrated synergistic anti-tumor effects in preclinical models [92].
Angiogenesis inhibitors. Bevacizumab, an anti-VEGF agent, normalizes tumor vasculature and increases immune cell infiltration, potentially sensitizing MSS tumors to ICIs, which is supported by emerging clinical data [90].
Macrophage-targeting agents. Direct strategies to deplete or repolarize TAMs are a promising area for research. They include blocking the CSF1/CSF1R axis (e.g., with PLX3397) to deplete macrophages, or using small molecules [e.g., SPHK1 (sphingosine kinase 1) or HDAC (histone deacetylase) inhibitors] to reprogram pro-tumor M2-like TAMs toward an anti-tumor M1-like phenotype [83, 93].
Inhibitors of other pathways: combining ICIs with inhibitors of immunosuppressive molecules—TREM2, CD73, A20, or macrophage migration inhibitory factor (MIF) [94–97].
Beyond checkpoint-based strategies, other immunotherapeutic modalities are being investigated. Adoptive cell therapy based on chimeric antigen receptor T (CAR-T) cells faces challenges in solid tumors, including CRC, but is being innovated through engineering cells to secrete immunomodulatory molecules [e.g., PD-1-TREM2 single-chain variable fragment (scFv)] that concurrently target tumor cells and other TME components [98]. Oncolytic virotherapy, which uses engineered viruses to lyse tumor cells and stimulate anti-tumor immunity, is also in early-phase clinical trials, as a monotherapy or in combination with systemic ICIs [99].
Below, we have collected data on the proposed novel immunotherapeutic approaches for the treatment of CRC that are under intensive investigation in pre-clinical models or in clinical trials.
The insufficient effectiveness of existing immunotherapy (ICIs) for treating CRC has prompted a search for novel therapeutic approaches based on either combination therapy or monotherapy. A number of studies are currently underway in preclinical in vitro and in vivo models (Table 1, Figure 2), as well as in clinical trials.
Effectiveness of combination therapy based on ICIs with TME inhibitors or plant extracts in pre-clinical animal tumor models.
| Targets | Combination therapy | In vivo tumor model | Tumor growth and survival | Macrophage activity | Changes in the TME |
|---|---|---|---|---|---|
| Combination therapy of anti-PD-1/PD-L1 agents and TAM inhibitors | |||||
| MS4A4A [100] | Anti-MS4A4A mAb plus anti-PD-1 mAb | MC38-bearing C57BL/6 mice, CT26-bearing BALB/c mice (n = 5 per group) | Inhibited tumor growth (monotherapy); suppressed tumor growth and improved survival (combination) | An anti-MS4A4A monotherapy: decreased M2-TAM infiltration (F4/80+CD206+); downregulated M2-like markers (CD206) and immunosuppressive molecules (CD39, SIRPα); upregulated M1-like markers (iNOS, MHCII) | Reduced T cell exhaustion marker co-expression (PD-L1+LAG3+, PD-L1+TIM3+); enhanced CD8+ T cell effector function and proliferation (IFN-α, Ki67) |
| TREM2 [98] | CAR-T cells, secreted PD-1-TREM2 scFV | MC38-bearing C57BL/6J mice (n = 4) | Inhibited tumor progression; enhanced survival | Decreased percentages of M2-like TAMs (CD11b+Gr1+; F4/80+CD206+) | Increased CD8+ T cell infiltration; decreased MDSCs amount; enhanced perforin and granzyme B release; elevated intratumoral cytokines (IL-2, IL-15, TNF-α, IFN-γ) |
| SPHK1 [93] | SPHK1 inhibitors (PF543 or SKI II) plus anti-PD-1 Ab | MC38 and CT26 liver metastatic models in immunocompetent mice (n = 5–12 per group) | Partially repressed CRLM (monotherapy); enhanced metastasis inhibition; prolonged survival (combination) | Reduced total TAMs; decreased p-SPHK1+ TAMs | Increased intermediate exhausted CD8+ T cells with a moderate PD-1 level and PD-1lowCD8+ T cells expressing memory T cell markers (IL-7Ra and Ly6C) |
| CSF1R [102] | PLX3397 plus anti-PD-1 mAb plus anti-CTLA-4 mAb | C57BL/6J bearing subcutaneous MC38 mice | Compared with the monotherapy, PLX3397 showed a potent synergic effect when it was combined with an anti-PD-1 or an anti-CTLA-4 mAb | Decreased expression of macrophage markers CD206 and F4/80 (monotherapy PLX3397) | - |
| Combination therapy of anti-PD-1/PD-L1 agents and TME inhibitors | |||||
| A20 [95] | A20-knockdown plus, anti-PD-1 Ab | Metastatic BALB/c mouse model bearing A20-knockdown tumor cells (CT-26) (n = 4–8 per group) | Inhibited tumor growth in the lung, subcutaneous tumor growth; decreased number of tumor nodules in lung tissue; the overall survival was remarkably longer | Increased F4/80+ macrophages | Increased CD3+ and CD8+ T cells; increased amount of granzyme B+ immune cells |
| MIF [96] | anti-PD-1 Ab plus anti-MIF Ab | MC38-bearing (subcutaneously injected); C57BL/6J mice | Superior tumor rejection; reduction of tumor size | - | - |
| CD73 [97] | AB680 plus anti-PD-1 Ab | CT26-bearing BALB/c mice (n = 3 per group) | Tendency to reduce tumor growth compared to monotherapy | Increased the proportion of M1-like macrophages, no effect on M2-like macrophages (combination therapy); decreased the proportion of M2-like macrophages but had no effect on M1 macrophages (PD-1 blockade) | Trend towards increased CD8+/CD4+ ratio |
| Kinases [92] | Regorafenib plus anti-PD-1 Ab | Syngeneic murine MSS CT26 and MSI MC38 CC models | Sustained suppression of liver metastasis even after the end of treatment | Reduced F4/80+ and CD206+ macrophages; increased iNOS+ macrophages | Reduced number of Tregs under regorafenib treatment; increased intratumoral levels of IFN-γ, a pharmacodynamic marker of cytotoxic T cell activity under an anti-PD-1 therapy |
| WNT11/CAMKII [112] | KN93 plus anti-PD-1 Ab | C57BL/6 mouse liver metastases model with MC38 cells (n = 5 per group) | Decreased liver metastasis | Reduced CD206+ macrophage infiltration in liver metastasis | Increased infiltration of I-A/I-E+CD8+ T cells in liver metastasis; KN93 induced the expression of CXCL10 and CCL4 and reduced the expression of IL17D |
| Combination therapy of anti-PD-1/PD-L1 agents and epigenetic modulators | |||||
| HDAC [110] | Tucidinostat plus anti-PD-L1 Ab | CT26 subcutaneously injected into BALB/c or C57 BL/6 mice (n = 7 per group) | Tumor growth suppression; improved mice survival | Reduced the number of CD45+CD11b+F4/80+ TAMs; increased the proportion of CD45+CD11b+F4/80+/MHCII+ M1-like macrophages | Increased CD45+ lymphocytes count, proportion of tumor-infiltrating CD4+ T cells (CD45+CD3+CD4+) cells and CD8+ T cells (CD45+CD3+CD8+) cells; decreased expression of the exhaustion marker PD-1; increased serum level of CCL5 and IFN-γ |
| Class I HDACis (1, 2, 3, and 10), VEGFR [103] | Chidamide plus cabozantinib/regorafenib plus anti-PD-1 Ab | CT26-Bearing (subcutaneously injected) BALB/c mice | Inhibited tumor growth; increased survival rate; tumor shrinkage after discontinued treatment | Decreased amount of CD11b+ TAMs in tumors | Upregulated enrichment of IFN pathway gene signature and downregulated enrichment of the T cell gene signatures; reduced levels of polymorphonuclear-MDSCs in the tumor |
| Class I HDACis (1, 2, 3, and 10), VEGFR [103] | Chidamide plus cabozantinib/regorafenib plus anti-CTLA-4 Ab | CT26-Bearing (subcutaneously injected) BALB/c mice | Inhibited tumor growth; enhanced ORR | Suppressed macrophage/monocyte gene signatures | Enhanced T cell cytotoxicity |
| HDAC [111] | AVS100 plus anti-PD-1 Ab | CT26-bearing BALB/c mice (n = 6–7 per group) | An increase in responders from 35% in anti-PD-1 treatment alone to 80% in combination therapy; did not relapse after termination of the treatment; resistant to a subsequent tumor challenge | Increased IFN-TAMs; decreased regulatory and angiogenic TAMs | Up-regulation of genes involved in T cell activation, T cell effector function, and NF-κB signaling; increased expression of genes controlling IFN and NF-κB signaling (Stat4, Nfkb1, Rel, and Il12rb2), the inositol-3-phosphate/Akt pathway, and immune infiltration (Hcst, Itpr1, and Itgal1) with AVS100 treatment, independent of anti-PD-1 therapy |
| Combination therapy of anti-PD-1/PD-L1 agents and plant extract components | |||||
| [104] | Lobeline plus anti-PD-1 Ab | C57BL/6 mice bearing MC38 tumors (subcutaneously injected) (n = 6 per group) | Reduced tumor growth; upregulated SLURP1 expression in tumors; suppressed cancer cell proliferation | Increase in the number of intratumor CD45+CD11b+F4/80+ M1-like macrophages and decrease in the number of CD45+CD11b+CD206+ M2-like macrophages | Decreased Foxp3 protein levels; increased granzyme B levels |
| [113] | Astragaloside IV plus anti-PD-1 Ab | BALB/c mice bearing CT26 tumors (injected into the axillary fat pad) (n = 8) | Suppressed tumor growth more effectively than monotherapy | Astragaloside monotherapy: altered M2-like macrophage polarization toward M1-like (decreased CD11b+F4/80+CD206hi M2-like macrophages; increased CD11b+F4/80+MHCIIhi M1-like macrophages) | Increased the number of cytotoxic T cells; increased IFN-γ and IL12p70 secretion; decreased TGF-β secretion |
| [105] | SFIH plus anti-PD-1 Ab | MC38-bearing (subcutaneously injected), C57BL/6 mice (n = 7–8 per group) | Significant reduction in tumor volume compared with an anti-PD-1 Ab; no effect in the SFIH group | Increased CD11b+, F4/80+, MHCII+, CD206– M1-like macrophage infiltration; no changes in the CD11b+, F4/80+, MHCII–, CD206+ TAM population | Increased CD3+ and CD8+ T cells, release of granzyme B, expression of IFN-γ and CXCL9; decreased population of CD11b+, GR+ MDSCs, decreased expression of TGF-β1, TGF-β2, VEGFA, and IDO2; downregulated TGF-β signaling |
| [114] | CYP plus anti-PD-1 mAb | Xenograft models, subcutaneously injected: MC38-bearing C57BL/6 mice (n = 6) and CT26-bearing BALB/c mice (n = 3) | Combination therapy was more effective in reducing tumor volume and size (CYP alone had no significant anti-tumor effect); increased survival in mice | Decreased number of infiltrating macrophages, mainly CD206+ TAMs | Increased number of CD8+ T cells in the tumor; increased expression of granzyme B |
Ab: antibody; CAR-T: chimeric antigen receptor T; CCL: C-C motif chemokine ligand; CRLM: colorectal cancer liver metastasis; CTLA-4: cytotoxic T lymphocyte-associated antigen-4; CXCL: C-X-C motif chemokine ligand; CYP: Chinese yam polysaccharide; HDAC: histone deacetylase; ICIs: immune checkpoint inhibitors; IFN: interferon; IL: interleukin; mAb: monoclonal Ab; MDSCs: myeloid-derived suppressor cells; MHCII: major histocompatibility complex II; MIF: migration inhibitory factor; MSI: microsatellite instability; MSS: microsatellite stable; ORR: objective response rate; PD-1: programmed cell death-1; PD-L1: programmed cell death ligand-1; scFv: single-chain variable fragment; SFIH: sesquiterpene lactones derived from Inula helenium L.; SPHK1: sphingosine kinase; TAM: tumor-associated macrophage; TGF: transforming growth factor; TME: tumor microenvironment; TNF: tumor necrosis factor; Tregs: regulatory T cells; VEGF: vascular endothelial growth factor.
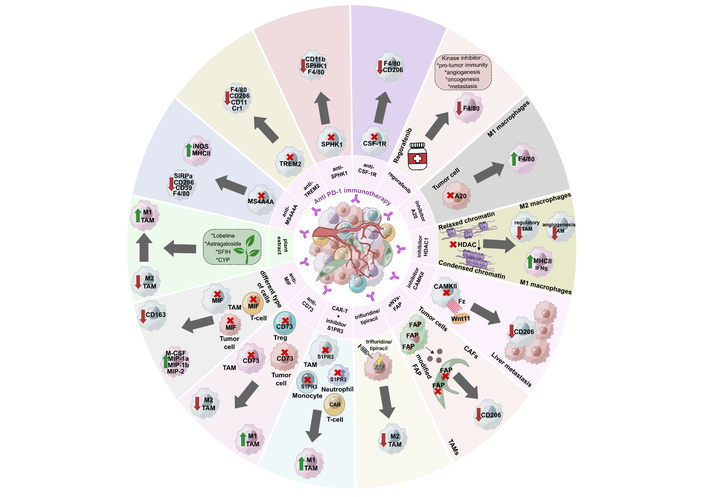
Combination therapy using ICIs and other inhibitors of the TME components. CAFs: cancer-associated fibroblasts; CAR: chimeric antigen receptor; CYP: Chinese yam polysaccharide; eNVs-FAP: FAP gene-engineered tumor cell-derived exosome-like nanovesicles; FAP: fibroblast activation protein-α; HDAC: histone deacetylase; ICIs: immune checkpoint inhibitors; MHCII: major histocompatibility complex II; MIF: migration inhibitory factor; S1PR3: sphingosine 1-phosphate receptor 3; SFIH: sesquiterpene lactones derived from Inula helenium L.; SPHK1: sphingosine kinase 1; TAM: tumor-associated macrophage; TME: tumor microenvironment; Treg: regulatory T cell.
The overwhelming majority of combination therapies presented in Table 1 demonstrated acceptable safety profiles in animal tumor models. No toxicity [100, 101], weight loss [93, 98, 101–105], and no histological, morphological, or functional organ changes [93, 101] were observed.
Both murine and human TAMs in CRC tumors express PD-1, which reduces the phagocytic activity of macrophages against tumor cells [106]. Furthermore, PD-1 expression in TAMs increases as the disease progresses [106]. A number of studies demonstrated a synergistic effect of an anti-PD-1 therapy with macrophage-targeting molecules such as MS4A4A, TREM2, SPHK1, and CSF1R. Combination therapy significantly increased mouse survival, decreased tumor growth, and decreased the metastasis rate (Table 1). Combination therapy resulted in the decreased expression of M2-like TAM markers (CD206, CD39, SIRPα) and increased expression of M1-like TAM markers [iNOS, major histocompatibility complex II (MHCII)].
MS4A4A+ macrophages are associated with M2-like polarization, T cell exhaustion, immunosuppression, and tumor progression [100]. MS4A4A blockade in combination with an anti-PD-1 therapy more effectively suppressed tumor growth, improved survival, and decreased relapse rates [100]. TREM2-positive macrophages were detected in CRC tumor infiltrates. TREM2 expression inversely correlated with overall and relapse-free survival [94]. Trem2 knockout mice exhibited slower tumor growth as well as a reduction in the number of tumor-infiltrating CD206+ TAMs compared to wild-type C57BL/6J mice [94]. CAR-T cells with the autocrine scFvs PD-1-TREM2 were engineered to target the PD-1/PD-L1 pathway and TREM2 on the surface of MDSCs and TAMs [98]. SPHK1 expression was found in TAMs from human CRC liver metastases compared to normal intestinal mucosa, primary CRC, and normal liver tissue [93]. High levels of stromal infiltration with SPHK1+ TAMs correlated with lymph node metastasis, TNM CRC stage, and poor overall survival in patients with CRC. The use of a SPHK1 inhibitor increased the efficacy of an anti-PD-1 therapy in a mouse model of CRC liver metastasis [93]. PLX3397 is a CSF1R inhibitor that significantly reduced tumor size, the metastasis rate, and led to M2-like TAM depletion (by decreased expression of CD206 and F4/80) in C57BL/6J mice bearing subcutaneous MC38 tumors [102]. PLX3397, in combination with anti-PD-1 therapy, exhibited a significant synergistic effect in vivo [102, 107].
Epigenetic mechanisms regulate TAM differentiation, recruitment, and functional activation, modulating their polarization to shape anti- or pro-tumor immune responses [108, 109]. Epigenetic modulation of TAM polarization represents a promising therapeutic strategy to reprogram the immunosuppressive TME toward an immunostimulatory state. DNMT inhibitors, HDAC inhibitors, and EZH2 inhibitors shift macrophage polarization from tumor-promoting M2-like toward anti-tumor M1-like phenotypes, and also potentiate immunotherapy by enhancing T cell activation and inflammatory cytokine release [108, 109]. Tucidinostat (chidamide), an oral benzamide-class HDAC inhibitor selective for HDAC1, HDAC2, HDAC3, and HDAC10 subtypes, improved the efficacy of an anti-PD-L1 therapy [110]. In vitro studies using Raw. 264.7 cells and bone marrow-derived macrophages (BMDMs) showed a dose-dependent increase in M1-like markers (iNOS, CD86, MHCII) under tucidinostat treatment. In vivo studies demonstrated that combination therapy reduced the number of CD45+CD11b+F4/80+ TAMs but increased the proportion of CD45+CD11b+F4/80+/MHCII+ M1-like macrophages in the tumor [110]. Different combination therapies with HDAC inhibitors and anti-PD-1/anti-CTLA-4/regorafenib treatment significantly inhibited tumor growth [103, 110, 111] (Table 1).
The WNT signaling pathway, enzyme A20, CD73, MIF, and multiple protein kinases play a role in the progression of cancer. The use of kinase inhibitors in combination with an anti-PD-1 therapy drastically suppresses tumor growth, increases tumor infiltration by F4/80+ macrophages, elevates the proportion of M1-like macrophages, and decreases the proportion of immunosuppressive TAMs (e.g., CD206+ TAMs) (Table 1).
Expression of the potent anti-inflammatory enzyme A20 was significantly higher in CRC tissues compared with normal tissues [95]. A20 was associated with poor response to an anti-PD-1 therapy and mediated inhibition of tumor cell phagocytosis by macrophages. A pronounced anti-tumor effect of an anti-PD-1 antibody was observed in the A20-deficient CT26 CC model in vivo. In immunodeficient CT26-bearing mice, the suppression of tumor growth, which was a result of A20 deficiency, could be restored by administering CSF1R antibodies due to inhibition of tumor cell phagocytosis by macrophages [95]. MIF has pro-oncogenic properties and is secreted by both immune and tumor cells [96]. In YUMMER1.7-bearing mice, combination treatment activated macrophages, as indicated by a significant increase in the expression of macrophage cytokines and chemokines (MIP-1β, MIP-2, M-CSF, and MIP-1α), and also increased the expression of cytokines in activated T cells (GM-CSF, IL-12p40, IL-12p70, IFN-γ, CXCL9, IL-1α). These effects were not observed in MC38-bearing mice [96]. CD73 is an immune system enzyme that plays a key role in tumor growth and metastasis as well as the formation of an immunosuppressive environment [97]. Combination therapy with anti-CD73 and anti-PD-1 agents showed a tendency to reduce tumor growth compared to monotherapy [97]. Combination therapy with regorafenib, a multikinase inhibitor, and an anti-PD-1 antibody demonstrated sustained suppression of liver metastasis even after the end of treatment, which was not observed with regorafenib alone [92]. WNT11 plays a crucial role in carcinogenesis, regulating both the malignant properties of tumor cells and modulating the TME [112]. In an MC38-bearing mouse with liver metastases, a significant reduction in metastases was observed under combination therapy using an anti-PD-1 antibody and a CAMKII (WNT11 downstream molecule) inhibitor compared to monotherapy [112].
Plant extract components contain substances with anti-tumor and immunomodulatory activities (Table 1). Their use in combination with an anti-PD-1 therapy was shown to reduce tumor progression in mouse models of CRC [104, 105, 113, 114]. In combination therapy, lobeline, astragaloside, sesquiterpene lactones derived from Inula helenium L. (SFIH), and Chinese yam polysaccharide (CYP) demonstrated an anti-tumor effect due to immunomodulation of the TME, in particular through modulation of the functional activity of macrophages [104, 105, 113, 114]. For example, lobeline (the active alkaloid of lobelia) directs macrophage polarization toward M1-like via the MAPK14/p53/Slurp1 signaling pathway, as evidenced by increased expression of M1-like macrophage-associated genes (Il1b, TNF, Il12a, CD86, CD80, CCR7, and CXCL9) and decreased expression of M2-like macrophage-associated genes (TGFβ1, VEGFα, EGF, Il6, Il10, Arg1, and CCL22) [104]. Combination therapy using CYP eliminated intestinal dysbiosis and reduced the production of detrimental metabolites such as deoxyguanosine. Deoxyguanosine was shown to significantly increase M2-like gene expression in macrophages [114].
Other components of the immunosuppressive TME may also provide targets for immunotherapy, effectively suppressing tumor growth, eliminating M2-like macrophages, and increasing M1-like macrophages [101, 115, 116]. CAFs are stromal cells that can overexpress fibroblast activation protein-α (FAP) in more than 90% of human tumor tissues [115]. FAP gene-engineered tumor cell-derived exosome-like nanovesicles (eNVs-FAP) can be used as a tumor vaccine to suppress tumor growth by inducing a pronounced tumor-specific cytotoxic T lymphocyte (CTL) response. eNVs-FAP reduced the number of CD206+ M2-TAMs, while the percentage of tumor-infiltrating CD45+ cells and CD3+ T lymphocytes increased [115]. Trifluridine/tipiracil (FTD/TPI) is an innovative antimetabolite agent with immunogenic cell death induction activity that was developed for the treatment of chemorefractory CRC [116]. The combination of FTD/TPI with oxaliplatin eliminated M2-type TAMs. Simultaneous administration of FTD/TPI and an anti-PD-1 therapy may provide a promising treatment option for patients with mCRC [116]. Inhibition of sphingosine 1-phosphate receptor 3 (S1PR3) facilitated the differentiation, activation, and survival of pro-inflammatory macrophages [101]. An S1PR3 inhibitor significantly enhanced CAR-T cell efficacy through multiple mechanisms. S1PR3 inhibition facilitated the differentiation, activation, and survival of pro-inflammatory macrophages, which was corroborated by increased M1-like macrophage infiltration in mouse tumors [101]. However, the proposed therapeutic approaches were not used in combination with ICIs in pre-clinical animal models.
Currently, nivolumab, pembrolizumab, dostarlimab, and ipilimumab are approved for the treatment of CRC [68, 69]. Other immunotherapy drugs, such as AMP-224, atezolizumab, avelumab, camrelizumab, durvalumab, envafolimab, sintilimab, spartalizumab, tislelizumab, and toripalimab, did not show significant anti-tumor effects for CRC treatment [68].
Ongoing clinical trials demonstrate that TAMs can be an indicator of response to immunotherapeutic treatment in CRC (Table 2).
Ongoing clinical trials based on combination immunotherapy for CRC treatment.
| Therapy | Targets | Type of cancer, sample size | Efficiency of therapy | Macrophages as indicators of response to therapy | ID clinical trials | Adverse events (rates) |
|---|---|---|---|---|---|---|
| Regorafenib plus nivolumab [117, 124] | Kinase, PD-1 | MSS/pMMR CRC, n = 22 | ORR in 36% of cases (in clinical trial) | CD206+CD11b+ M2-like macrophages within the tumor were significantly higher in responders before treatment | Phase Ib (NCT03406871) | Liver transaminase increased, creatinine increased, and platelet count decreased (the most common, 70% in the clinical trial) |
| Pimitespib plus nivolumab [117, 125] | Hsp90, PD-1 | MSS CRC, n = 23 | ORR in 16% of cases (in clinical trial) | CD206+CD11b+ M2-like macrophages within the tumor were significantly higher in non-responders before treatment | Phase Ib (UMIN000032801) | Rash, proteinuria, palmar-plantar erythrodysesthesia (the most common, grade 3 or worse, 7% in clinical trial) |
| Pembrolizumab (KEYNOTE 177 clinical trial) or nivolumab (group with first-line therapy) [118, 123] | PD-1 | hypermutated CRC, n = 16 | Persistent response in 56% of cases | CD68+CD74+ cells were observed in CRC tumors with a durable response. 80% of CD68+CD74+ cells expressed markers HLA-ABC, HLA-DR, CD40, CD16, and CD163 | Phase 3 (NCT02563002) + patients with first-line therapy | Diarrhea, fatigue, nausea, abdominal pain, anemia, hypertension (97% patients in clinical trial) |
| Oncolytic virotherapy plus LP002 [99] | Tumor cells, PD-1 | MSS CRC with liver metastasis, n = 4 | One out of four patients demonstrated a durable response | Responder had elevated levels of both M1-like and M2-like macrophages | Phase I (NCT04755543) | Fever, nausea, fatigue, constipation, dry mouth (100% patients) |
| Pembrolizumab with or without XL888 [119] | PD-1, Hsp90 | Advanced CRC with liver metastasis, n = 18 | No ORR with combination therapy. 25% of patients had stable disease | Decreased CD68+ and CD68+IL6+ in liver metastasis with combination therapy | Phase Ib/II (NCT03095781) | Diarrhea, fatigue, abdominal pain, constipation, nausea, vomiting, eye disorders, anorexia, hypomagnesemia, cough, increased liver enzymes (grade 3–4, 12.5% patients in combination therapy) |
| Pembrolizumab plus maraviroc [121] | CCR5, PD-1 | pMMR CRC, n = 20 | One patient achieved a partial response, 94.7% patients had disease progression (ORR in 5.3% of cases) | Anti-tumor macrophage activation | Phase I (NCT03274804) | Hyperglycemia (1 case, grade 4) |
| Sintilimab plus chidamide with or without bevacizumab [122] | PD-1, HDAC, VEGF | Unresectable chemotherapy-refractory locally advanced or MSS/pMMR CRC, n = 48 | 18-week PFS of triple combination therapy vs. double combination (64.0% vs. 21.7%) ORR of triple combination therapy vs. double combination (44.0% vs. 13.0%) | Increased the number of monocytic lineage cells in triple therapy responders | Phase 2 (NCT04724239) | Proteinuria, thrombocytopenia, neutropenia, anemia, leukopenia, diarrhea (96% patients in triple combination therapy, 100% patients in double combination therapy) |
CCR5: C-C motif chemokine receptor 5; CRC: colorectal cancer; HDAC: histone deacetylase; MSS: microsatellite stable; ORR: objective response rate; PD-1: programmed cell death-1; PFS: progression-free survival; pMMR: mismatch repair proficient; VEGF: vascular endothelial growth factor.
The Phase Ib REGONIVO and Phase Ib TASNIVO clinical trials for MSS or pMMR CRC evaluated the clinical efficacy of regorafenib plus nivolumab and pimitespib (HSP90 inhibitor TAS-116) plus nivolumab, respectively [117]. Objective tumor response was observed in 36% of cases in the REGONIVO group and 16% of cases in the TASNIVO group [117]. Analysis of pre-treatment tumor immune infiltrates showed that in the REGONIVO group, the density of CD206+CD11b+ M2-like macrophages was significantly higher in responders compared to non-responders. In the TASNIVO group, the opposite results were observed: the density of M2-like macrophages was significantly lower in responders than in non-responders [117]. Another study comprehensively evaluated tumor tissue from patients with hypermutated CRC treated with pembrolizumab (KEYNOTE 177 clinical trial) or nivolumab (non-clinical trial patients) to identify tumor cell and immune cell interactions in response to immunotherapy [118]. Higher numbers of CD68+CD74+ cells were observed in CRC tumors with durable response (DB-CRC) than in CRC tumors without durable response (nDB-CRC). Interestingly, 80% of CD68+CD74+ cells expressed markers of antigen presentation (HLA-ABC, HLA-DR, CD40, CD16, and CD163) [118]. Responsive tumors contained abundant infiltration of PD-L1+ macrophages in close proximity to PD-1+ cytotoxic T cells. CD68+CD74+ macrophages with antigen-presenting ability served as a key predictor of treatment response, highlighting their critical role in mediating pembrolizumab’s therapeutic effect in the TME [118]. In a phase I clinical trial evaluating combination therapy with oncolytic virotherapy and anti-PD-1 antibodies, only one out of four patients demonstrated a durable response [99]. A comprehensive multi-omics analysis revealed that the patient who responded to the combination therapy had low infiltration of T cells and NK cell subsets, but a higher number of macrophages compared to non-responders. Interestingly, the responder had elevated levels of both M1-like and M2-like macrophages [99].
In a Phase Ib/II clinical trial evaluating the efficacy of pembrolizumab in combination with XL888 (an Hsp90 inhibitor) in patients with CRC, metastatic liver biopsies were analyzed [119]. Using multiplex immunohistochemistry, the authors found that patients receiving the combination therapy, compared with those receiving pembrolizumab alone, had a trend toward decreased numbers of CD68+ and CD68+IL6+ macrophages. However, no ORR was observed in the group treated with combination therapy. The best result was achieved in 25% of patients, who had stable disease [119]. In advanced MSS/pMMR CRC (N = 110) after first-line treatment failure, treatment with sintilimab (a PD-1 inhibitor) plus bevacizumab (the study was not registered) increased CD8+ T cell infiltration and reduced the numbers of TAMs and CAFs. At the same time, PFS rates in the sintilimab plus bevacizumab treatment group were significantly higher than in the FOLFIRI plus bevacizumab treatment group. The experimental group also had statistically significantly higher rates of partial responses [90, 120]. The Phase 1 PICCASSO study investigated the efficacy of a combination of pembrolizumab and maraviroc (a CCR5 inhibitor) in refractory pMMR CRC [121]. CCR5 regulates macrophage polarization toward the pro-tumor M2-like phenotype, and CCR5 inhibition led to anti-tumor macrophage activation. Only one patient achieved a partial response; in the majority of patients (94.7%), the best response was disease progression during the treatment, but after the treatment, patients demonstrated superior response rates [121]. The phase II CAPability-01 trial in unresectable locally advanced or MSS/pMMR mCRC demonstrated that chidamide combined with sintilimab (anti-PD-1) with or without bevacizumab (anti-VEGF) converted immunosuppressive microenvironments into immunoactive states [122]. Patients receiving triple combination therapy (chidamide, sintilimab, and bevacizumab) compared with patients receiving double combination therapy (chidamide, sintilimab) showed a higher 18-week PFS (64.0% vs. 21.7%) and ORR (44.0% vs. 13.0%). At the same time, in patients who responded to triple therapy, an increase in the number of monocytic lineage cells was observed in their tumors [122].
All types of therapy demonstrated an acceptable safety profile. The most common adverse events were grade 3, and in studies with pembrolizumab [123], pembrolizumab plus XL888 [119], and pembrolizumab plus maraviroc [121]—grades 4 and 5. The incidence of some adverse event grade ≥ 3 did not exceed 28% when regorafenib plus nivolumab [124], pimitespib plus nivolumab [125], pembrolizumab [123], pembrolizumab plus maraviroc [121], pembrolizumab in combination with XL888 [119], and sintilimab plus chidamide with or without bevacizumab [122] were used.
Accumulating evidence from immunopathological studies, pre-clinical models, and ongoing clinical trials highlights the key role of TAMs in shaping the response to immunotherapy in CRC. However, the multifaceted phenotype and plasticity of TAMs across patients and disease stages hinder the identification of effective immunotherapeutic targets and the optimization of current treatment regimens. In this context, advanced omics technologies, combined with state-of-the-art bioinformatics approaches, provide a powerful framework for dissecting functional programs in TAMs and prioritizing novel immunotherapeutic targets.
In particular, advances in the omics era have enabled whole-transcriptome profiling of TAMs. Differential gene expression analysis of bulk RNA-seq data allows identification of dozens of differentially expressed genes (DEGs) [126, 127]. Gene set enrichment analysis (GSEA) facilitates functional dissection of DEGs based on their known involvement in distinct biological pathways [128]. GSEA helps prioritize TAM-associated genes involved in immunoregulatory processes using functional annotations from public databases such as Gene Ontology [129], KEGG [130], Reactome [131], and CellMarker 2.0 [132]. Gene network analysis provides a framework for further prioritization of DEGs. The WGCNA method [133] constructs gene co-expression networks using correlation patterns among a list of variable genes. Further analysis of the resulting co-expression networks identifies gene modules—clusters of co-expressed genes—and their corresponding hub genes, defined as the most highly interconnected genes within each module. The relevance of WGCNA is highlighted by its ability to identify disease-associated gene modules and hub genes related to clinical outcomes, immune states, and metabolic dysfunction across diverse pathological contexts, including cancer [134–136]. Importantly, functional perturbation of WGCNA-identified hub genes represents a promising strategy for target discovery, as silencing of such central regulatory nodes enables assessment of their causal contribution to disease-associated programs and highlights candidates with potential therapeutic relevance [137, 138].
Several studies have identified gene modules associated with immunosuppression in TAMs, demonstrating that WGCNA-derived networks capture functionally relevant immune programs. For example, immunosuppressive modules enriched for hypoxia- and angiogenesis-related genes were linked to poor prognosis in glioblastoma and characterized by elevated expression of TAM-associated regulators such as TREM1, whose functional perturbation attenuated tumor-promoting properties of TAMs [139]. Similarly, module-based immune scoring approaches in glioma and hepatocellular carcinoma revealed coordinated expression of immune checkpoint molecules, inflammatory mediators, and M2-like macrophage markers, e.g., CD163, S100A9, and SPP1, highlighting modular immune states associated with tumor progression, immune evasion, and unfavorable patient outcome [140, 141]. Furthermore, several independent studies have reported immune-related modules in circulating monocytes. In triple-negative breast cancer, an immunosuppressive module composed of genes such as CD163, S100A9, TREM1, and FCN1 was found to be correlated with an unfavorable response to neoadjuvant chemotherapy [142]. We previously identified diverse co-expression modules in high-grade ovarian cancer following neoadjuvant chemotherapy, including circulating monocyte modules characterized by activated antigen processing and presentation, as well as suppression-related gene programs involving RUNX1 and TREM1 [143]. Altogether, WGCNA-derived co-expression modules indicate a significant skewing of monocytes toward a suppressive state already at the circulating stage after chemotherapy.
Single-cell omics approaches greatly expand our understanding of the complex phenotype of TAMs [144]. Single-cell multi-omics technologies have enabled reaching an unprecedented level of profiling, extending the reconstruction of gene co-expression networks to gene regulatory networks. The SCENIC method integrates transcriptome and chromatin accessibility to construct gene regulatory networks that facilitate the detection of key TFs [145]. SCENIC has been applied across diverse disease contexts to infer TF-driven macrophage programs. For example, SCENIC-based analysis revealed regulatory similarities between TAMs and cirrhosis-associated macrophages, enabling construction of a macrophage-naive CD4+ T cell-related transcriptional score derived from target genes such as BOD1, SEC61A1, RHEB, CFL1, PTMA, C1orf109, and E2F5 [146]. In oral squamous cell carcinoma, SCENIC further resolved heterogeneous CD163+ macrophage subpopulations with distinct immunoregulatory programs, identifying CMKLR1+ macrophages as a key subpopulation that inhibits tumor progression [147]. Further coupling of gene regulatory networks with AI-based approaches has enabled in silico TF perturbation analysis. The CellOracle method enables the identification of transcriptional shifts and the prediction of alterations in differentiation and activation trajectories of cells in response to in silico inactivation of key TFs [148]. In silico perturbation analysis using CellOracle has shown consistency with subsequent experimental silencing, as illustrated by predictions that disruption of FOSL1 attenuates migratory transcriptional programs, which were experimentally validated by reduced keratinocyte motility upon FOSL1 siRNA knockdown [149]. Moreover, in macrophages, CellOracle in silico perturbation was supported by a preprint demonstrating that RUNX1 silencing reprograms cells toward a reparative phenotype and promotes cardiac recovery [150]. Collectively, the above-mentioned findings highlight that the profound plasticity of TAMs necessitates integrative single-cell multi-omics and computational bioinformatics frameworks to reliably identify and functionally validate new immunoregulatory targets.
Accumulated data demonstrate the undoubted role of TAMs in tumor progression and have established TAMs as important determinants of the response to immunotherapy in CRC. However, most studies to date have focused on correlative associations rather than mechanisms, leaving a gap in our understanding of how specific TAM subsets directly modulate ICI responses in CRC.
Key findings obtained using omics technologies and state-of-the-art molecular genetic methods have expanded our understanding of the functional diversity of TAMs in human tumors. The most recent studies identified between 6 and 23 macrophage subsets of TAMs, each with its own functional activity: immunoregulatory TAMs, lipid-associated TAMs, pro-angiogenic TAMs, inflammatory TAMs, immunostimulatory macrophages, and others [144, 151–153]. Nonetheless, the functional relevance of many of these subsets remains largely inferred from transcriptomic signatures, with limited validation at the protein level or in functional models, raising concerns about whether they represent sustainable or transient activation states.
Moreover, current research is still limited by the high heterogeneity of TAMs in tumors, necessitating the search for at least a unified method for macrophage typing in tumors. Critically, most biomarker studies rely on single-time point biopsies, ignoring the dynamic plasticity of macrophages under therapeutic pressure. This static view may explain why candidate biomarkers such as CD163 or CD206 have failed to consistently predict immunotherapy outcomes in CRC. In this context, many earlier studies examined the functional activity of TAMs using a limited number of markers, such as F4/80, CD206, CD163, MHCII, IFN-γ, and iNOS, underscoring the need for more comprehensive approaches. What is more important, a novel insight emerging from recent studies is that TAM heterogeneity does not simply exist between diverse tumor types, but is also intratumoral and spatially organized. It means that the location of a TAM subset relative to tumor-immune interfaces may be as critical as its polarization state. Future biomarker search must therefore consider spatial resolution and dynamic changes in TAM functional states to capture clinically relevant TAM phenotypes.
The complexity of the mechanisms regulating the formation of functional phenotypes of macrophages requires the development of a multi-target approach aimed at TAMs. That is why the clinical translation of TAM-targeting strategies (e.g., CSF-1R inhibitors, CCR2 antagonists) has demonstrated modest results in CRC, largely due to compensatory recruitment of macrophages in the TME, off-target effects, and the absence of predictive biomarkers for patient selection. Moreover, the high plasticity of macrophages in response to various stimuli contributes to the formation of heterogeneity between patients [154], which requires patient stratification based on macrophage biomarkers, coupled with the development of an individualized macrophage-based immunotherapy approach. Combination therapy is the future direction, but clinical translation still requires more precise stratification. A critical inference is that combination therapies should move beyond simple “reprogramming” or “depletion” paradigms. Instead, they must be tailored to the dominant TAM subset present in each patient, guided by real-time immune monitoring. Without such stratification, even rationally designed combinations are likely to fail.
To achieve such a level of precision, appropriate preclinical models are essential. The use of mouse models for research purposes has a number of advantages: small size, rapid reproduction, low cost, ease of carrying out genetic modifications, and, as a result, the possibility of conducting numerous studies in a relatively short period [155]. However, differences in size, physiology, and target homology between humans and mice lead to limitations in the use and interpretation of specific therapies [155]. When using mouse models, a number of factors must be considered to effectively evaluate therapy. Pre-clinical models involve subcutaneous, orthotopic, and xenograft mouse tumors. The choice of mouse model is important for assessing marker expression and the effectiveness of drug therapy [156–159]. Tumor cell morphology, drug uptake, and gene expression may differ depending on the model chosen [156–159]. Orthotopic human tumor xenografts are considered more suitable for predicting drug response. In contrast, genetically modified mouse strains are more suitable for studying the role of genes in tumor development and progression [160]. Furthermore, differences in the outcomes of pre-clinical versus clinical trials of immunotargeted drugs may stem from differences in the TME, particularly immune cells. Today, the optimal approach is to use humanized mouse models, which are physiologically and pathologically the best to mimic human tumors [161]. Such models most accurately reflect the interactions between immune and tumor cells. However, even humanized models often fail to recapitulate the full spectrum of TAM heterogeneity seen in patients, as they are typically engrafted with a limited set of immune lineages. Future models should integrate patient-derived tumor fragments with autologous immune cells to preserve the native TAM diversity and spatial architecture. Taken together, integrating multi-omics analyses with functionally annotated preclinical models, including humanized mice, will be essential to develop effective strategies for TAM programming at the transcriptional, epigenetic, and metabolic levels.
In summary, macrophage plasticity, the choice of study model, the choice of markers of therapy effectiveness, differences in TME, and differences in physiology between mice and humans—these factors may be the reason why immunotherapy shows effective results in preclinical in vitro and in vivo models, but the results in clinical trials are still unsatisfactory. This translational gap emphasizes the need for a paradigm shift: from viewing TAMs as a homogeneous myeloid population to targeting them as a dynamic ecosystem that adapts to both therapy and the evolving TME.
BMDMs: bone marrow-derived macrophages
CAFs: cancer-associated fibroblasts
CAR-T: chimeric antigen receptor T
CC: colon cancer
CCL22: C-C motif chemokine ligand 22
CCR2: C-C motif chemokine receptor 2
CIMP: CpG island methylation phenotype
CMSs: consensus molecular subtypes
CR: complete response
CRC: colorectal cancer
CRT: chemoradiotherapy
CTL: cytotoxic T lymphocyte
CTLA-4: cytotoxic T lymphocyte-associated antigen-4
CXCL10: C-X-C motif chemokine ligand 10
CYP: Chinese yam polysaccharide
DB-CRC: colorectal cancer tumors with durable response
DEGs: differentially expressed genes
dMMR: deficient mismatch repair
EMT: epithelial-mesenchymal transition
eNVs-FAP: fibroblast activation protein-α gene-engineered tumor cell-derived exosome-like nanovesicles
FAP: fibroblast activation protein-α
FTD/TPI: trifluridine/tipiracil
GSEA: gene set enrichment analysis
HDAC: histone deacetylase
ICIs: immune checkpoint inhibitors
IFN: interferon
IL: interleukin
mCRC: metastatic colorectal cancer
MDSCs: myeloid-derived suppressor cells
MHCII: major histocompatibility complex II
MIF: migration inhibitory factor
MSI: microsatellite instability
MSI-H: high microsatellite instability
MSI-L: low microsatellite instability
MSS: microsatellite stable
nDB-CRC: colorectal cancer tumors without durable response
ORR: objective response rate
PD-1: programmed cell death-1
PD-L1: programmed cell death ligand-1
PFS: progression-free survival
pMMR: mismatch repair proficient
RC: rectal cancer
S1PR3: sphingosine 1-phosphate receptor 3
scFv: single-chain variable fragment
SFIH: sesquiterpene lactones derived from Inula helenium L.
SPHK1: sphingosine kinase 1
TAMs: tumor-associated macrophages
TF: transcription factor
TGF-β: transforming growth factor β
TME: tumor microenvironment
TNF: tumor necrosis factor
Tregs: regulatory T cells
VEGF: vascular endothelial growth factor
TS: Writing—original draft, Visualization. KS: Writing—original draft. ES: Writing—original draft, Visualization. PI: Writing—original draft. AD: Writing—review & editing. IL: Conceptualization, Project administration, Writing—review & editing. JK: Conceptualization, Project administration, Writing—review & editing. All authors read and approved the submitted version.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The study was supported by the Russian Science Foundation, grant [RSF 25-25-00904]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 552
Download: 23
Times Cited: 0
Mariam Rojas ... Joan Maurel
Anastasia Rays ... Аlexey Tryakin
