 Mini Review
Mini Review

Affiliation:
Hepatogastroenterology Department, Theodor Bilharz Research Institute (TBRI), Giza 12411, Egypt
Email: maged_elghannam@yahoo.com
ORCID: https://orcid.org/0000-0002-3638-5286
Affiliation:
Hepatogastroenterology Department, Theodor Bilharz Research Institute (TBRI), Giza 12411, Egypt
ORCID: https://orcid.org/0000-0002-4638-0542
Affiliation:
Hepatogastroenterology Department, Theodor Bilharz Research Institute (TBRI), Giza 12411, Egypt
ORCID: https://orcid.org/0000-0002-3844-1122
Affiliation:
Hepatogastroenterology Department, Theodor Bilharz Research Institute (TBRI), Giza 12411, Egypt
ORCID: https://orcid.org/0000-0001-7498-6835
Affiliation:
Hepatogastroenterology Department, Theodor Bilharz Research Institute (TBRI), Giza 12411, Egypt
ORCID: https://orcid.org/0000-0003-3777-7668
Affiliation:
Hepatogastroenterology Department, Theodor Bilharz Research Institute (TBRI), Giza 12411, Egypt
ORCID: https://orcid.org/0000-0002-1214-6459
Affiliation:
Hepatogastroenterology Department, Theodor Bilharz Research Institute (TBRI), Giza 12411, Egypt
ORCID: https://orcid.org/0000-0002-4860-5231
Explor Dig Dis. 2026;5:1005122 DOI: https://doi.org/10.37349/edd.2026.1005122
Received: January 11, 2026 Accepted: April 13, 2026 Published: April 27, 2026
Academic Editor: Jose C. Fernandez-Checa, Institute of Biomedical Research of Barcelona (IIBB), CSIC, Spain
Janus kinase (JAK) inhibitors represent a major advancement in the management of immune-mediated inflammatory diseases. A balanced approach that carefully weighs therapeutic benefits against potential risks is essential. Through appropriate patient selection, close monitoring, and open physician–patient communication, the clinical potential of JAK inhibitors can be optimized while minimizing adverse outcomes. Nine JAK inhibitors have demonstrated utility in hepatogastrointestinal disorders; however, only two have FDA approval. JAK inhibitors are classified into reversible (competitive) and irreversible (covalent) inhibitors according to their chemical binding with amino acids. This review discusses the safety profile, adverse effects, and molecular selectivity of JAK inhibitors, and highlights their therapeutic roles in hepatogastrointestinal diseases, including inflammatory bowel disease, hepatic fibrosis, hepatocellular carcinoma, autoimmune diseases associated with cancer therapy in post-transplant patients, eosinophilic esophagitis, metabolic syndrome, and metabolic dysfunction-associated steatotic liver disease, and acute graft-versus-host disease following liver transplantation.
Janus kinases (JAKs) are intracellular, non-receptor tyrosine kinases (TYKs) that comprise four members: JAK1, JAK2, JAK3, and TYK2 [1, 2]. JAK3 expression is limited to immune cells, while JAK1, JAK2, and TYK2 are broadly expressed [3]. The cytokine signaling through the JAK–signal transducer and activator of transcription (STAT) pathway is responsible for the pathogenesis of many immune-mediated inflammatory disorders (IMIDs) in the gastrointestinal, respiratory, and dermatologic systems, such as inflammatory bowel disease (IBD), asthma, and pruritic dermatitis [4–7]. In addition, JAK signaling can shape tumor cell proliferation and angiogenesis [8], and JAK mutations are implicated in myeloproliferative disorders, lymphomas, and leukemia [9]. This article aims to present the recent advances in the therapeutic role of JAK inhibitors in hepato-gastrointestinal diseases.
Cytokine signal transduction requires at least two JAK molecules within the receptor complex, either as homodimers or heterodimers [10]. JAKs contain seven domains (JH1–JH7) [11], with JH1 responsible for enzymatic kinase activity, while the remaining domains mediate receptor binding [12]. The receptor activation leads to phosphorylation of STAT proteins, STAT dimerization, and nuclear translocation, where they regulate gene transcription [4]. Among the JAK family, JAK1 and TYK2 are predominantly involved in inflammatory signaling, making them attractive therapeutic targets [13, 14]. Both autoimmune and malignant diseases were claimed to be a result of activation of JAKs and STATs mutants, while, on the other hand, inactivating mutations lead to immunodeficiency diseases [15]. The JAK–STAT pathway and protein members are shown in Figure 1.
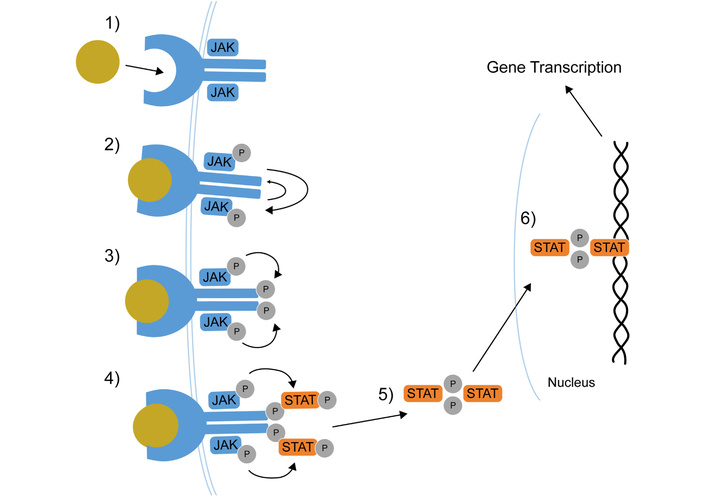
The JAK–STAT pathway and its members. Cytokine receptor binding; receptor-JAKs phosphorylation; JAKs phosphorylation; STATs phosphorylation; STATs dimerization; STAT dimers translocation to the nucleus for gene transcription. JAK: Janus kinase; P: phosphate group; STAT: signal transducer and activator of transcription. Reprinted from [16]. © Lin CMA, Cooles FAH, Isaacs JD 2020. Licensed under a Creative Commons Attribution 4.0 International License.
JAK inhibitors are small-molecule drugs that can be orally taken, with rapid action and low immunogenicity [17]. JAK inhibitors have strong action due to their ability to inhibit multiple cytokines at different cell signaling pathways. The preferential selectivity for JAK1 is the whole mark for JAK inhibitors’ efficacy, while the medication’s safety is related to the inhibition of JAK2- and JAK3-dependent pathways [13]. JAK inhibitors’ selectivity and unique mechanism of action allow more chances for personalized therapy [18]. However, the efficacy and safety profiles showed significant differences [5, 19]. Although twelve JAK inhibitors have been approved for clinical use [20], nine have demonstrated utility in hepatogastrointestinal disorders, with only two having FDA approval [21].
JAK inhibitors can be divided according to the selectivity against JAKs into non-selective first-generation, such as baricitinib and tofacitinib (TOFA), and second-generation selective drugs, such as filgotinib and upadacitinib (UPA). Another classification for JAK inhibitors based on their binding mode, whether reversible or irreversible, has been suggested.
They form reversible (non-covalent) binding interactions with the amino acids in the JAKs. The binding interactions include hydrogen bonds and hydrophobic interactions. These can be further subdivided into:
These agents compete with ATP at the catalytic binding site and include:
These agents bind to sites distinct from the ATP-binding domain. Examples include deucravacitinib (a selective TYK2 inhibitor), LS104, and ON044580.
These agents form covalent bonds with JAKs, particularly JAK3, targeting the unique Cys909 residue. Ritlecitinib is a notable example currently under clinical evaluation [24].
TOFA in a 10 mg BID dose in 50-year-old or older patients induces significant cardiac risk, VTE, and pulmonary embolism (PE). This is more evident in those who had at least one pre-existing cardiovascular factor, such as smoking and/or atherosclerotic cardiovascular disease [25–27]. The risk for PE increases in older patients with prior VTE, obesity, and a history of chronic lung disease. In UPA trials, no additional risks were recorded [28]. Anticoagulants protect against thrombosis in high-risk patients [29]. An opposing opinion claims that IBD is a thrombogenic disease; this increase represents a false elevation [30, 31]. Both JAK inhibitors and anti-TNF therapies carry the same risk of cardiac and/or thrombotic events in IBD patients [32]. Recently, a meta-analysis study came to the same conclusion, minimizing the role of exposure time in amplifying the risk of cardiac and thrombotic events. Still, the lower dose of both TOFA 5 mg BID and UPA 30 mg QD slightly increased risks of thrombosis, signifying the pan-sensitivity of JAK at high doses [33].
It is mandatory for clinicians to screen patients for risk factors, such as smoking and obesity, history of previous thrombotic events, or hypercoagulable predisposition, to stratify patients prior to initiating JAK inhibitors, follow a healthy lifestyle, the lower effective dose of JAK inhibitors should be used for maintenance, and continue on anti-coagulants, especially for those with a history of VTE recurrence [34].
Dose-dependent, reversible, and within 1–2 months increases in serum lipids had been recorded during both the induction and maintenance phase that return to baseline levels during use of TOFA and UPA [35]. Both low-density lipoprotein:high-density lipoprotein-cholesterol (LDL:HDL-C) and total cholesterol:HDL-C ratios are stable without an increase. Lipid profile at baseline and half-yearly checking is mandatory. Lipid-lowering agents are a second option [34]. Filgotinib has no clinically relevant effects on lipid levels [36, 37].
Patients are at increased risk of pneumonia, fungal infections (e.g., Aspergillus, Pneumocystis jirovecii), histoplasmosis, cryptococcosis, nocardiosis, Clostridium difficile, and urinary tract infections [38]. Selective JAK1 inhibitors are safer than non-selective JAK inhibitors, like TOFA [39]. The risk of tuberculosis (TB) is higher in areas with high endemic regions [40]. The concomitant immunosuppressant use and/or underlying comorbidities increase the incidence of invasive fungal infections [41]. The risk of serious infection increases with the high 10 mg dose [25, 42]; the same as the UPA 30 mg dose, underscoring the importance of using the lowest effective dose for maintenance therapy [43]. The best example is the herpes zoster (HZ) reactivation [42, 44]. Testing for TB by serum QuantiFERON gold or T-spot should be performed in all patients before initiating a JAK inhibitor, and regularly thereafter. Latent TB should be treated prior to initiating a JAK inhibitor. No live or attenuated virus vaccines should be administered during JAK inhibitor therapy or immediately before it. Patients should follow the Centers for Disease Control (CDC) recommendations regarding COVID vaccination [45, 46]. Vaccination against influenza, pneumococcus, and varicella-zoster virus is recommended prior to therapy in addition to respiratory syncytial virus (RSV) if above the age of 50 [47].
In the IBD TOFA and UPA-treated patients, 8 cases with GI perforation had been recorded. All of them occur in risky, complicated patients with areas of deep ulcers, stricture, or fistula [42]. Concomitant use of NSAIDs or corticosteroids is an additional risk factor [48].
Lymphomas, lung cancer, melanoma, and non-melanoma skin cancer (NMSC) were recorded [49, 50]. TOFA recorded a higher incidence of malignancies compared to UPA, especially with a higher dose of 10 mg BID [51]. Sun protection should be encouraged. A regular skin examination is recommended prior to and annually after initiating JAK inhibitors [52].
It is the most common side effect and occasionally leads to treatment cessation. Higher doses and prior history of acne vulgaris are risk factors. Usually, it resolves with dose reduction; however, a few cases require pharmacological treatment and/or dermatology consultation [53].
UPA exposure to adverse pregnancy outcomes was comparable to the general population [54]. However, JAK inhibitors are not recommended during pregnancy and/or breastfeeding [55, 56]. Contraceptive methods should be used in women of childbearing age and up to a month after discontinuation of the JAK inhibitor. The risks and benefits of therapy versus uncontrolled disease should be discussed with the patient [56].
All JAK inhibitors, except for UPA, have a potential impact on fertility in animal studies. However, randomized, placebo-controlled studies confirmed safety in humans. Filgotinib is the only JAK inhibitor without an impact on male fertility [57] (Figure 2).
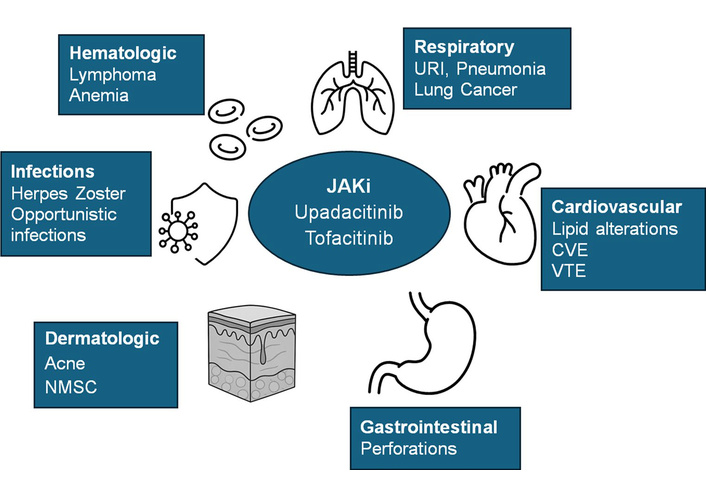
JAK inhibitors’ adverse effects. Reprinted from [45]. © The Author(s) 2025. Licensed under a Creative Commons Attribution 4.0 International License.
JAK inhibitors differ significantly in their pharmacokinetic and pharmacodynamic profiles. While no definitive biomarkers currently guide drug selection, emerging data suggest that JAK inhibitor selective agents may offer improved safety profiles [13]. Filgotinib, which is primarily metabolized in the intestine, may reduce the risk of hepatic drug-drug interactions and may be preferable in polytreated patients [58]. Filgotinib has also been shown to have no adverse impact on sperm parameters, making it a potential option for male patients planning to conceive [59].
Elderly patients with cardiovascular or malignancy risk factors should be observed closely [60]. Data from the FDA Adverse Event Reporting System and randomized trials have highlighted increased risks of gastrointestinal perforation, malignancy, and major adverse cardiovascular events, particularly with TOFA [48, 61]. Before initiating therapy, patients should undergo complete blood count, liver and renal function testing, lipid profile, TB screening, and viral hepatitis and HIV screening [62]. Live vaccines should be avoided during treatment [63].
The heterogeneity of responses to small-molecule therapies in IBD underscores the need for predictive biomarkers [64]. Advanced omics approaches have identified potential markers, including baseline mucosal phosphorylated STAT3 (pSTAT3) expression predicting response to filgotinib and serum proteins [human leukocyte antigen E (HLA-E), lipoteichoic acid (LTA), C-C motif chemokine ligand 21 (CCL21), and multiple epidermal growth factor-like domains protein 10 (MEGF10)], differentiating responders to ritlecitinib [65, 66]. Joustra et al. [67] reported low expression of fibroblast growth factor receptor 2 (FGFR2) and low-density lipoprotein receptor-related protein-associated protein 1 (LRPAP1), and a high expression of OR2L13 at baseline in responders. Another study identified a cluster of genes in the mucosa that was significantly correlated with endoscopic response. Within this cluster, the HUP gene “nucleotide binding domain” demonstrated a predictive accuracy of 100% [68].
JAK inhibitors represent an important therapeutic class in IBD and were first introduced in 2018 with the approval of TOFA, followed by the JAK inhibitor-selective agents filgotinib and UPA [69]. Still other small molecules are under clinical trials in phase I, II, and III (Table 1).
JAK inhibitors with potential use in IBD.
| Drug | Target | Route of administration and doses | Clinical trial |
|---|---|---|---|
| Tofacitinib [70–72] | JAK1, 3 | Oral induction 10 mg BID; maintenance 5 mg BID (may increase to 10 mg BID in non-responders) | Phase III trials (OCTAVE Induction 1/2, Sustain, Open; RIVETING); FDA approved for UC |
| Filgotinib [73] | JAK1 | Oral 200 mg OD | Phase III, UC |
| Upadacitinib [74, 75] | JAK1 | Oral induction 45 mg OD; maintenance 15 mg or 30 mg OD | Phase III, FDA approved in 2022 for UC |
| Izencitinib (TD-1473) [76] | JAK 1, 2, 3, TYK2 | Oral gut specific 270 mg OD | Phase I, UC |
| Peficitinib (Smyraf) [77] | JAK 1, 2, 3, TYK2 | Oral 150 mg OD | Phase IIb, UC |
| Ritlecitinib (Litfulo) [78] | JAK3 | Oral 20 mg, 70 mg, or 200 mg OD | Phase II, umbrella study for UC |
| Brepocitinib [78] | TYK2, JAK1 | Oral 10 mg, 30 mg, or 60 mg OD | Phase II, umbrella study for UC |
| Deucravacitinib (Sotyktu) [79] | TYK2 | Oral 6 mg or 12 mg BID | Phase II, multiple immune-mediated disorders (including preclinical IBD models) |
| Ivarmacitinib [80] | JAK1 | Oral 4 mg OD, 4 mg BID, or 8 mg OD | Phase II, UC |
CD: Crohn’s disease; IBD: inflammatory bowel disease; JAK: Janus kinase; TYK2: tyrosine kinase 2; UC: ulcerative colitis. Adapted from [21]. © 2023 by the authors. Distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
TOFA is licensed for the treatment of moderate-to-severe ulcerative colitis (UC) and is considered a pan-JAK inhibitor with preferential activity against JAK1 and JAK3 [70]. In long-term extension studies including more than 1,100 patients, clinical remission rates at three years reached 59% with 5 mg twice daily and 34% with 10 mg twice daily. More than half of the initial non-responders achieved clinical response after extended induction [71]. Dose reduction from 10 mg to 5 mg twice daily was effective in maintaining remission in most patients [72]. Meta-analyses have confirmed its effectiveness even in highly refractory populations [81]. TOFA treats antibiotic-refractory pouchitis and Crohn’s disease (CD) inflammation [82, 83].
Filgotinib was approved in 2022 for moderate and severe UC after failure or intolerance of conventional or biologic therapy [73, 84]. It is a selective JAK1 inhibitor [85, 86] allowing for dose reduction, minimizing the side effects with maintenance of treatment efficacy [7]. Oral administration is followed by metabolism in the gut by carboxyl-esterase 2 (CES2) and CES1. The major metabolite exhibits similar preferential activity for JAK1 with higher systemic concentration compared to the parent drug [58].
The 200 mg once-daily dose, but not the 100 mg dose, was effective in inducing and maintaining remission [87]. Long-term data up to four years confirm sustained symptomatic remission and improved quality of life [88]. Filgotinib is an effective drug even for patients who have never received biological therapy [89]. The most frequently reported adverse effects include rhinitis and headaches [90] with a low risk of HZ infection. These side effects did not correlate with the dose of the drug [91].
UPA is a second-generation, JAK1-selective inhibitor and the only JAK inhibitor approved for both UC and CD [74, 75]. It is indicated in patients with inadequate response or intolerance to conventional or biologic therapy. Clinical trials demonstrated superiority over placebo for both induction and maintenance of remission [92, 93]. UPA is administered orally at a dose of 45 mg for 8 weeks, followed by 30 mg or 15 mg for maintenance therapy [43]. Reduction in the induction dose is recommended for patients with hepatic or renal impairment [94]. Rapid clinical improvement had been reported within a few weeks [95]. Prolonged induction therapy up to 16 weeks benefited a significant proportion of initial non-responders [96]. UPA ranked high in terms of clinical response, achievement and maintenance of remission, and endoscopic improvement [94, 97], efficacy in resolving extra-intestinal manifestations such as perianal fistula closure [98]. Adverse effects were generally mild to moderate, including acne, upper respiratory tract infections and nasopharyngitis, headaches, and increased creatine kinase levels [99, 100].
Esophageal fibroblasts express eotaxin-3 via STAT6 signaling in response to Th2 cytokines. Unlike epithelial cells, eotaxin-3 expression in fibroblasts is not suppressed by proton pump inhibitors (PPIs), limiting the impact of PPIs on subepithelial fibrosis. To the contrary, Th2 cytokine-induced eotaxin-3 expression in both epithelial cells and fibroblasts can be blocked by JAK–STAT6 inhibitors. A potential role in treating both inflammation and fibrosis in eosinophilic esophagitis has been suggested for JAK inhibitors [101].
The safety of combining JAK inhibitors with post-transplant immunosuppression has been explored in patients with IBD following solid organ transplantation. In small cohorts, TOFA in combination with tacrolimus achieved high rates of clinical remission without significant infectious, thromboembolic, or cardiovascular complications. These findings suggest that JAK inhibitors may be a safe and effective option in this challenging population, although larger studies with longer follow-up are required [102].
Hypothalamic neurons regulate food intake, energy expenditure, and glucose homeostasis with high expression of JAK2 and STAT3 [103]. Leptin, an appetite regulator, acts through signaling through the JAK2–STAT3 pathway. Hyperactivation of this JAK–STAT–SOCS axis in leptin resistance contributes to obesity and metabolic dysfunction [104].
JAK–STAT signaling also regulates metabolic target organs, including the liver, muscle, adipose tissue, and pancreas. Overactivation of this pathway contributes to insulin resistance, inflammation, and metabolic syndrome [105, 106]. Selective JAK inhibitors may therefore offer therapeutic potential in obesity, type II diabetes, MASLD, and cardiovascular risk reduction [107].
There is significant crosstalk between the JAK–STAT pathway and insulin signaling. Hyperactivation of JAK–STAT signaling impairs Akt phosphorylation and disrupts glucose homeostasis [108]. Elevated IL-6 levels in obesity increase SOCS1 and SOCS3 expression, which inhibit insulin receptor substrates and promote insulin resistance [109]. Inhibition of JAK–STAT signaling may reduce inflammation and improve insulin sensitivity [110]. Animal studies suggest tissue-specific effects of JAK loss, highlighting the complexity of this pathway [111–113]. Further clinical studies are required to define the risk–benefit profile of JAK inhibitors in metabolic disease (Figure 3).
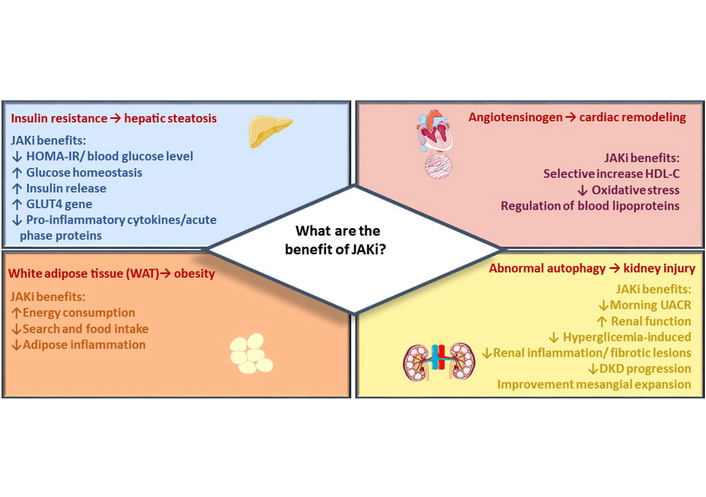
JAK inhibitor metabolic effects. The figure summarizes the main beneficial effects of JAK inhibitors across target organs of metabolism. Reprinted from [110]. © 2023 Collotta, Franchina, Carlucci and Collino. Distributed under the terms of the Creative Commons Attribution License (CC BY).
TOFA was reported to be safe and effective in a liver transplant recipient with UC [114]. Recently, Con et al. [102] in 2024 reported eight liver transplant recipients with IBD, seven of whom received a first-line JAK inhibitor with TOFA. All failed one or more biologic therapies prior to commencing JAK inhibitor, including six patients who had failed two or more biologic agents. JAK inhibitor was initiated in the outpatient setting. No serious adverse events were documented, nor were any interactions with transplant medications observed. Combining JAK inhibitors with post-transplant immunosuppression was a safe and clinically effective approach for the management of IBD in liver transplant recipients in both biologic-naïve and biologic-experienced patients. Larger cohorts with a longer duration of follow-up are needed [102].
aGVHD-LT is rare but associated with high mortality [115, 116]. JAK inhibitors disrupt immune cell communication and induce apoptosis in activated immune cells [117]. Ruxolitinib, a JAK1/2 inhibitor, modulates T-cell responses and cytokine signaling, attenuating the aberrant immune response [118]. Clinical studies and case reports indicate that ruxolitinib combined with corticosteroids can reduce steroid requirements and improve outcomes in steroid-refractory GVHD [119]. Further clinical trials are needed to establish optimal dosing, duration, and long-term safety in this population [120].
The JAK–STAT pathway is dysregulated in many autoimmune, inflammatory, and metabolic diseases. JAK inhibitors have emerged as a powerful therapeutic class offering rapid and targeted immunomodulation. Treatment decisions should be individualized, taking into account the comprehensive risk–benefit profile, patient comorbidities, and long-term safety considerations. IBD, eosinophilic esophagitis, metabolic syndrome, MASLD, IBD following liver transplantation, and aGVHD-LT represent key hepatogastrointestinal conditions in which JAK inhibitors may play an important therapeutic role.
aGVHD-LT: acute graft-versus-host disease after liver transplantation
CCL21: C-C motif chemokine ligand 21
CD: Crohn’s disease
CDC: Centers for Disease Control
CES2: carboxyl-esterase 2
FGFR2: fibroblast growth factor receptor 2
HDL-C: high-density lipoprotein-cholesterol
HLA-E: human leukocyte antigen E
HZ: herpes zoster
IBD: inflammatory bowel disease
IMIDs: immune-mediated inflammatory disorders
JAKs: Janus kinases
LDL: low-density lipoprotein
LRPAP1: low-density lipoprotein receptor-related protein-associated protein 1
LTA: lipoteichoic acid
MACE: major cardiovascular event
MASLD: metabolic dysfunction-associated steatotic disease
MEGF10: multiple epidermal growth factor-like domains protein 10
NMSC: non-melanoma skin cancer
PE: pulmonary embolism
PPIs: proton pump inhibitors
RSV: respiratory syncytial virus
STAT: signal transducer and activator of transcription
TB: tuberculosis
TOFA: tofacitinib
TYKs: tyrosine kinases
UC: ulcerative colitis
UPA: upadacitinib
VTE: venous thromboembolic event
MTE: Conceptualization, Data curation, Methodology, Supervision, Writing—original draft, Writing—review & editing. MHH: Conceptualization, Formal analysis, Visualization, Writing—original draft, Writing—review & editing. YAA: Validation, Writing—original draft, Writing—review & editing. EAT: Methodology, Validation, Writing—original draft, Writing—review & editing. GME: Data curation, Supervision, Writing—original draft, Writing—review & editing. AAE: Formal analysis, Visualization, Writing—original draft, Writing—review & editing. MDE: Data curation, Supervision, Validation, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1921
Download: 27
Times Cited: 0
