 Original Article
Original Article

Affiliation:
1Leeds Institute of Medical Research, University of Leeds, St James’s University Hospital, LS9 7TF Leeds, UK
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-7851-4533
Affiliation:
1Leeds Institute of Medical Research, University of Leeds, St James’s University Hospital, LS9 7TF Leeds, UK
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0001-5663-5548
Affiliation:
2Institute of Cancer and Genomic Sciences, University of Birmingham, B15 2TT Birmingham, UK
ORCID: https://orcid.org/0000-0001-5784-8705
Affiliation:
1Leeds Institute of Medical Research, University of Leeds, St James’s University Hospital, LS9 7TF Leeds, UK
3Department of Breast Medical Oncology, UT MD Anderson Cancer Center, Houston, TX 77030, USA
ORCID: https://orcid.org/0000-0001-9056-3453
Affiliation:
1Leeds Institute of Medical Research, University of Leeds, St James’s University Hospital, LS9 7TF Leeds, UK
ORCID: https://orcid.org/0000-0001-7414-1576
Affiliation:
3Department of Breast Medical Oncology, UT MD Anderson Cancer Center, Houston, TX 77030, USA
Affiliation:
4Department of Bioinformatics and Computational Biology, UT MD Anderson Cancer Center, Houston, TX 77030, USA
Affiliation:
5Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology and University of Tübingen, Auerbachstr. 112, D-70376 Stuttgart, Germany
6Germany iFIT Cluster of Excellence, University of Tübingen, Auerbachstr. 112, D-70376 Stuttgart, Germany
Affiliation:
3Department of Breast Medical Oncology, UT MD Anderson Cancer Center, Houston, TX 77030, USA
Affiliation:
5Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology and University of Tübingen, Auerbachstr. 112, D-70376 Stuttgart, Germany
6Germany iFIT Cluster of Excellence, University of Tübingen, Auerbachstr. 112, D-70376 Stuttgart, Germany
7German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ), Im Neuenheimer Feld 280, 69120 Heidelberg, Germany
ORCID: https://orcid.org/0000-0001-7531-2736
Affiliation:
4Department of Bioinformatics and Computational Biology, UT MD Anderson Cancer Center, Houston, TX 77030, USA
ORCID: https://orcid.org/0000-0002-5818-037X
Affiliation:
1Leeds Institute of Medical Research, University of Leeds, St James’s University Hospital, LS9 7TF Leeds, UK
8Institute of Medical Sciences, University of Aberdeen, Foresterhill, AB25 2ZD Aberdeen, UK
Email: speirs@abdn.ac.uk
ORCID: https://orcid.org/0000-0002-0602-4666
Explor Target Antitumor Ther. 2020;1:355–371 DOI: https://doi.org/10.37349/etat.2020.00021
Received: June 09, 2020 Accepted: September 18, 2020 Published: October 30, 2020
Academic Editor: Simon Langdon, University of Edinburgh, UK
Aim: Tamoxifen (TAM) resistance remains a clinical issue in breast cancer. The authors previously reported that 15-hydroxyprostaglandin dehydrogenase (HPGD) was significantly downregulated in tamoxifen-resistant (TAMr) breast cancer cell lines. Here, the authors investigated the relationship between HPGD expression, TAM resistance and prediction of outcome in breast cancer.
Methods: HPGD overexpression and silencing studies were performed in isogenic TAMr and parental human breast cancer cell lines to establish the impact of HPGD expression on TAM resistance. HPGD expression and clinical outcome relationships were explored using immunohistochemistry and in silico analysis.
Results: Restoration of HPGD expression and activity sensitised TAMr MCF-7 cells to TAM and 17β-oestradiol, whilst HPGD silencing in parental MCF-7 cells reduced TAM sensitivity. TAMr cells released more prostaglandin E2 (PGE2) than controls, which was reduced in TAMr cells stably transfected with HPGD. Exogenous PGE2 signalled through the EP4 receptor to reduce breast cancer cell sensitivity to TAM. Decreased HPGD expression was associated with decreased overall survival in ERα-positive breast cancer patients.
Conclusions: HPGD downregulation in breast cancer is associated with reduced response to TAM therapy via PGE2-EP4 signalling and decreases patient survival. The data offer a potential target to develop combination therapies that may overcome acquired tamoxifen resistance.
Due to its ability to bind to and modulate oestrogen receptor alpha (ERα) activity, Tamoxifen (TAM) was the first targeted therapy for breast cancer [1], with its widespread use in the clinic now extending to four decades. While aromatase inhibitors are now the preferred first line endocrine treatment in the post-menopausal setting [2], TAM is favoured in pre-menopausal breast cancer patients [3]. However, approximately 70% of patients initially respond to TAM, but most eventually acquire resistance [2]. Acquisition of TAM-resistance (TAMr) continues to be a major limitation for long-term management of breast cancer patients. However, the mechanism(s) responsible for TAM insensitivity is not yet fully understood.
In the last three decades, various groups have developed cell line models in order to help unravel the mechanistic basis of TAMr. Most of these have been achieved through long term culture of the hormone-sensitive MCF-7 human breast cancer cells in sub-lethal doses of TAM, during which resistant sub-clones develop after 3–6 months [4–8]. Using two independently-derived ER positive MCF-7 human breast cancer cell models of acquired TAMr developed in our laboratory [9], an Affymetrix® microarray study showed that 131 genes were upregulated and 156 were downregulated by at least 3-fold, in TAMr MCF-7 cells relative to isogenic control cells [10]. One of the genes that were downregulated was 15-hydroxyprostaglandin dehydrogenase (HPGD). HPGD (EC 1.1.1.141) is a member of the short-chain alcohol (OH) dehydrogenase family and is the key NAD+-dependent enzyme responsible for the biological inactivation of prostaglandins, including prostaglandin E2 (PGE2), which is synthesized via the cyclooxygenase (COX) pathway.
Downregulation of HPGD has been shown in several malignancies including lung, colon, bladder, endometrial, and gastric cancer and has been shown to have a tumour suppressor roles in some settings [11–16]. Studies in breast cancer are contradictory; higher HPGD expression has been reported in ERα-positive MCF-7 cell with reduced expression in ERα-negative MDA-MB-231 cells, where up-regulation was observed following treatment with demethylating agents [17]. In clinically more aggressive primary breast cancers, particularly those with a triple negative phenotype, and in breast cancer metastases, HPGD is overexpressed, with high HPGD expression associated with poor prognosis and reduced survival [18]. In the same study, HPGD silencing induced a mesenchymal-epithelial transition resulting in a less migratory phenotype. More recently, data mining from publicly available breast cancer gene expression datasets from the Cancer Genome Atlas (TCGA, https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) and Oncomine (https://www.oncomine.org/), revealed decreased expression of HPGD mRNA in breast cancer samples compared with normal, healthy breast tissue [19]. Furthermore, HPGD expression was lower still in more aggressive basal/triple negative and HER2-positive breast cancers. Conversely, in rare apocrine breast carcinomas, HPGD was highly expressed [20]. Application of a triclustering algorithm, δ-TRIMAX to microarray datasets characterized HPGD as one of ten so-called hub-genes associated with breast cancer [21], lending support for a role in breast carcinogenesis.
Building on our previous data [10], the aim of this study was to investigate a causal link between HPGD expression and function and TAMr, as well as explore the relationship between HPGD expression in breast cancer samples and clinical outcomes. The effects of HPGD expression on outcome were examined through in silico analysis and by immunohistochemistry in a breast cancer cohort with long term follow up, including breast cancer outcomes during adjuvant TAM.
The two stable TAMr MCF-7 human breast cancer cell lines have been described previously [9, 10]. All MCF-7 isogenic lines were cultured in phenol-red-free RPMI 1640 containing L-glutamine (Invitrogen, UK) supplemented with 5% charcoal-stripped steroid-depleted foetal calf serum (FCS; Harlan SeraLab, UK), 100 U/mL penicillin and 100 U/mL streptomycin and 100 nM 4-hydroxytamoxifen (TAM; Sigma-Aldrich, UK) for 12–24 months. Parental cells [termed wild type (WT) MCF-7] were cultured in the same medium, but with 0.01% (v/v) ethanol vehicle. Experiments were conducted in phenol-red-free RPMI 1640 supplemented with 5% charcoal-stripped steroid-depleted FCS + TAM (0.1–100 nM) or 17β-oestradiol (E2; 0.001-1 nM). Bi-monthly mycoplasma checks were consistently negative and annual short tandem repeat (STR) profiling confirmed cell provenance, both carried out as a service in Leeds. To minimize genetic drift, cell stocks were frozen at low passage and experimental cultures replaced from these stocks every 3–6 months. We also studied LCC1, -2 and -9 breast carcinoma cells, which were derived originally in the Clarke laboratory at Georgetown, Washington DC and cultured as described [7, 8, 22]. LCC1 cells are estrogen-independent and -responsive TAM-sensitive breast cancer cells, which are derived from an estrogen-independent variant of MCF-7 cells (MCF7/MIII) through in vivo selection in oophorectomized nude mice (with circulating estrogen levels similar to a postmenopausal woman), and subsequently re-cultured in vitro to become a stable cell line. LCC2 cells are stable, ER-positive, estrogen-independent, TAM-resistant, and respond to fulvestrant [7]. They were derived from the selection of LCC1 for TAM resistance in vitro. LCC9 cells exhibits cross resistance to TAM and are unresponsive to fulvestrant [7]. They were derived from the selection of LCC1 for fulvestrant resistance in vitro. MCF7:5C and MCF-7:WS8 are oestrogen-independent and dependent, respectively, ER-positive, progesterone receptor (PR)-negative, and TAM-resistant breast cancer cells [23]. They were derived from WT MCF-7 cells following long-term estrogen deprivation (LTED). The main characteristics of the cell lines used are detailed in Table 1.
Characteristics of TAM-resistant breast cancer cell lines derived from MCF-7
| Parental cell line | TAM resistant derivative | |
|---|---|---|
| Cell line | Method of induction of TAM resistance | |
| WT MCF-7(oestrogen-dependent, TAM-sensitive) | TAMr 1 | Chronic TAM exposure (12 months) |
| TAMr 2 | Chronic TAM exposure (21 months) | |
| MCF-7-5C | Long-term oestrogen deprivation | |
| MCF-7-WS8 | Long-term oestrogen deprivation | |
| MCF-7/LCC1 (oestrogen-independent, endocrine therapy responsive) | LCC2 | Stepwise selection against TAM |
| LCC9 | Stepwise selection against fulvestrant | |
Cells were seeded at 300, 000 cells per well in 6-well plates (Corning, UK). They were treated the next day with 1 nM E2 for the indicated durations. All experiments were performed in triplicate. Subsequently, cells were harvested in TRIzol (Life Technologies, UK) and total RNA was isolated using the Qiagen RNeasy RNA purification kit (Qiagen, UK). Isolated RNA (1 µg) was then processed on the Affymetrix Clariom S microarray platform (Affymetrix, UK) and used to determine the significance of differential expression of HPGD at basal levels.
Metabolic activity was used as a surrogate for cell proliferation, using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [9]. Cells were seeded into 96-well plates. After overnight attachment, TAM (0.1-100 nM), E2 (0.01–1 µM), PGE2 (1 µM), butaprost or PGE1-OH (both 0.1–1 µM) or ethanol vehicle was added. At 96 h post-treatment, cells were incubated with 1 mM MTT for 4 h. The purple formazan product was solubilised in propan-1-ol and read at 570 nm using an Opsys MRTM 15 (Dynex Technologies, UK). Chemicals were purchased from Sigma-Aldrich (UK) or Cayman Chemical (USA).
This was conducted as described previously [24], except that HPGD was detected using a rabbit polyclonal antibody (catalogue number NB200-179, Novus Biological, UK, 1:500). Bands were then visualised with Supersignal West pico ECL reagent (ThermoFisher, UK).
Total RNA was extracted, and reverse transcribed as described previously [24]. Real-time polymerase chain reaction (RT-PCR) for HPGD or prostaglandin E2 receptor 4 (EP4) mRNA was performed using a SYBR Green-based assay (ThermoFisher, UK) on an ABI 7700 sequence detection system using primers for HPGD (forward, 5’-TAGTTGGATTCACACGCTCAGC-3’; reverse, 5’-AAAGCCTGGACAAATGGCAT-3’) and EP4 (forward, 5’-TCTTACTCATTGCCACCTCCCT-3’; reverse, 5’-CTTGGCTGATATAACTGGTTGACG-3’). The ribosomal protein gene RPLPO was used as the reference gene (forward, 5’-GAAACTCTGCATTCTCGCTTCC-3’; reverse, 5’-GATGCAACAGTTGGGTGCCA-3’). Levels of HPGD or EP4 transcripts were quantified using the 2-ΔCt method [25].
A stab culture of human HPGD cDNA IMAGE clone ID 3638799 was obtained from MRC and HPGD was PCR-amplified from this using the following primers: forward, 5′-CCGGGATCCTGCACCATGCACGTGAAC-3′; reverse, 5′-CCCCAAGCTTTCATTGGGTTTTTGCTTG-3′. Products were cloned into pcDNA3.1(-)myc/his (gift from Dr Thomas Hughes, University of Leeds). Vectors containing HPGD or empty vector controls were transfected into TAMr cells using Lipofectamine 2000 (ThermoFisher, UK). Stable clones were selected and maintained in G418 (ThermoFisher, UK)-containing medium (250 µg/mL).
Both assays were performed using previously protocols published [26]. For PGE2, serum-free conditioned medium was collected from cells after overnight incubation, and PGE2 levels were measured using a competitive immunoassay (Amersham Biosciences, UK), and normalised to total cellular protein content as measured by Bradford assay. HPGD activity was measured in lysates of WT MCF-7 and TAMr cells by assessing the transfer of tritium from 3H-PGE2 to glutamate, catalysed by HPGD and glutamate dehydrogenase [27].
Small-interfering (si)-RNAs targeting EP4 or HPGD (ThermoFisher Scientific, UK) were reverse transfected into cells using Lipofectamine 2000 according to the manufacturer’s instructions. Briefly, MCF-7 cells were mixed with media containing small-interfering RNA (siRNA)/lipid (final siRNA concentration 10 nM) and seeded into 96 well plates at 0.4 x 104 cells/well and allowed to establish overnight before incubation with drug or vehicle control. Response to treatment was assessed by MTT assay. EP4 or HPGD knockdown was assessed by real-time quantitative polymerase chain reaction (qRT-PCR) as described previously [28].
Following ethical approval (06/Q1206/180) we investigated HPGD expression in 350 primary invasive breast cancers, all of which, had been surgically resected and received adjuvant TAM, and were represented on tissue microarrays (TMAs) [24, 29]. Out of the 350 cases, 108 cases experienced a relapse (TAMr) and 242 cases did not [tamoxifen-sensitive (TAMs)]. Mean follow-up was 80 months [range 1–229, standard deviation (SD) 44.2]. Tumour staining was achieved using previously described methods [24, 26].
The relationship between HPGD expression and cancer outcomes was analyzed using Kaplan-Meier Plotter (KMplot, https://kmplot.com/analysis/) [30]. Using the 2017 release of the database, a cohort of ERα-positive breast tumours, previously treated with TAM only and no adjuvant chemotherapy, was analyzed and the association between HPGD expression and overall survival (OS). Patients were dichotomized as high or low HPGD expression using lower tertile as a cut-off. Multivariate analysis was done using in-built software with Ki67, ER and HER2 as covariates.
We applied miRWalk version 3.0 [31] and TargetScan version 7.2 [32] to a previously described Affymetrix miRNA dataset of endocrine sensitive MCF-7:WS8 and resistant MCF-7:5C cells [33] in order to identify miRNA-target interactions sites for major HPGD transcripts using default settings. OncoLnc (http://www.oncolnc.org/) was used to retrieve HPGD and miR-3200-3p expression data from matching breast cancer cases in the TCGA database [34]. Kaplan-Meier Plotter [30] was used to investigate the significance of miR-3200-3p on survival in ERα+ breast cancer treated with any endocrine therapy (not limited to TAM). Cases were dichotomised as high or low miR-3200-3p expression using the median expression value as a cut-off.
For in vitro analyses, one-way ANOVA was performed. The log rank test was used to compare patient survival in the primary breast cancer cohort. Analyses were performed using GraphPad Prism version 7.03 (GraphPad Software, La Jolla California, USA). For Affymetrix analyses, ANOVA P-values were calculated using the Affymetrix Transcriptome Analysis Console (Affymetrix). The time course plots were plotted as fold-change with the baseline value set at 1.
We demonstrated that HPGD was downregulated in two independent TAMr MCF-7 cell lines, which had been cultured continuously in 100 nM TAM for 12 (TAMr 1) and 21 (TAMr 2) months [10], respectively by Western blot (Figure 1a) and real time PCR (Figure 1b) compared with parental MCF-7 cells. Microarray analysis of the independent LCC cell line series and MCF7:5C cells showed that only the oestrogen-independent but oestrogen-responsive TAMs cell line LCC1 showed increased expression of HPGD in response to E2 over time (Figure 1c).
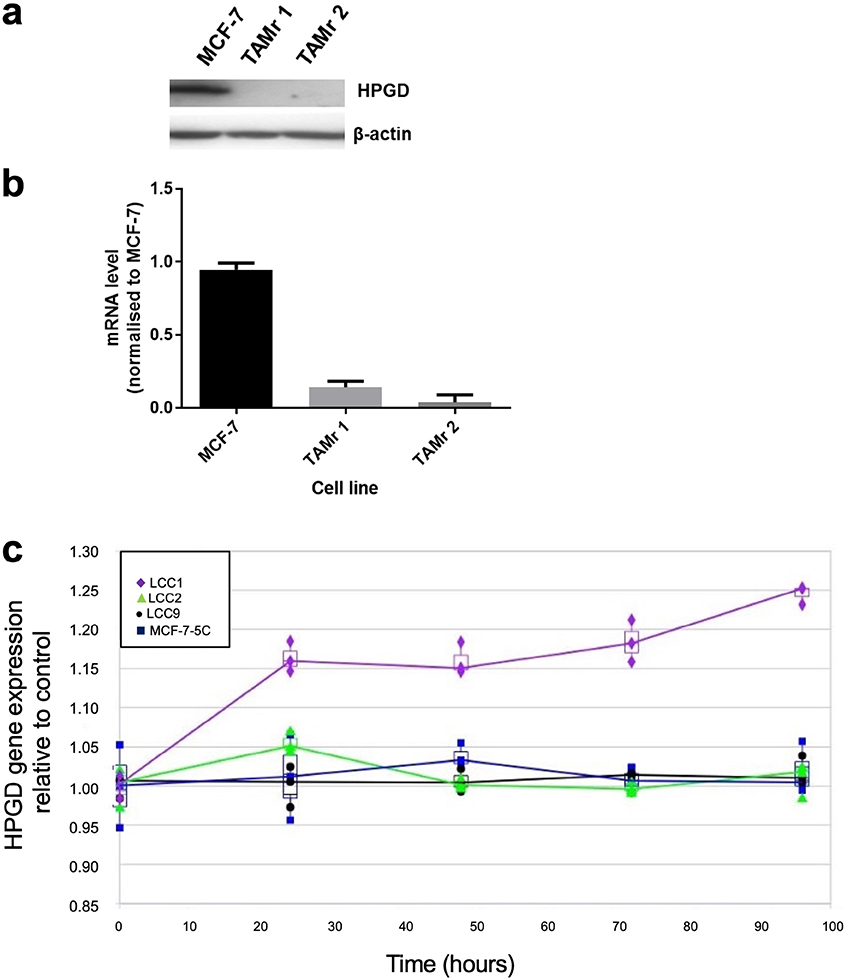
HPGD is downregulated in TAMr derivatives of MCF-7 cells. Western blot (a) showing HPGD protein expression in WT MCF-7 and its loss in two, independently derived, TAMr MCF-7 cell lines, TAMr 1 and TAMr 2; qRT-PCR (b) shows relative expression of HPGD mRNA in these cell lines. Expression was determined using RPLP0 as the reference gene and further normalized to baseline at time 0 h (± SD); in (c), expression of HPGD mRNA in LCC1 cells (TAMs) versus LCC2 cells (TAMr), LCC9 (TAMr and fulvestrant resistant), and MCF7:5C (TAMr) shows an upregulation over time in LCC1 cells only, with the resistant variants remaining largely unchanged during treatment with 1 nM E2
We then explored the effects of introduction of HPGD into MCF-7 TAMr 2 cells. Successful expression of HPGD protein following stable transfection was confirmed by Western blotting (Figure 2a). This restored activity of HPGD in each of the transfected cell lines to levels like WT MCF-7 (Figure 2a and b). This also sensitized these cells to the inhibitory effects of TAM to approximately 60% of the parental MCF-7 cell response (Figure 2c), whilst restoring sensitivity to E2 almost completely (Figure 2d).
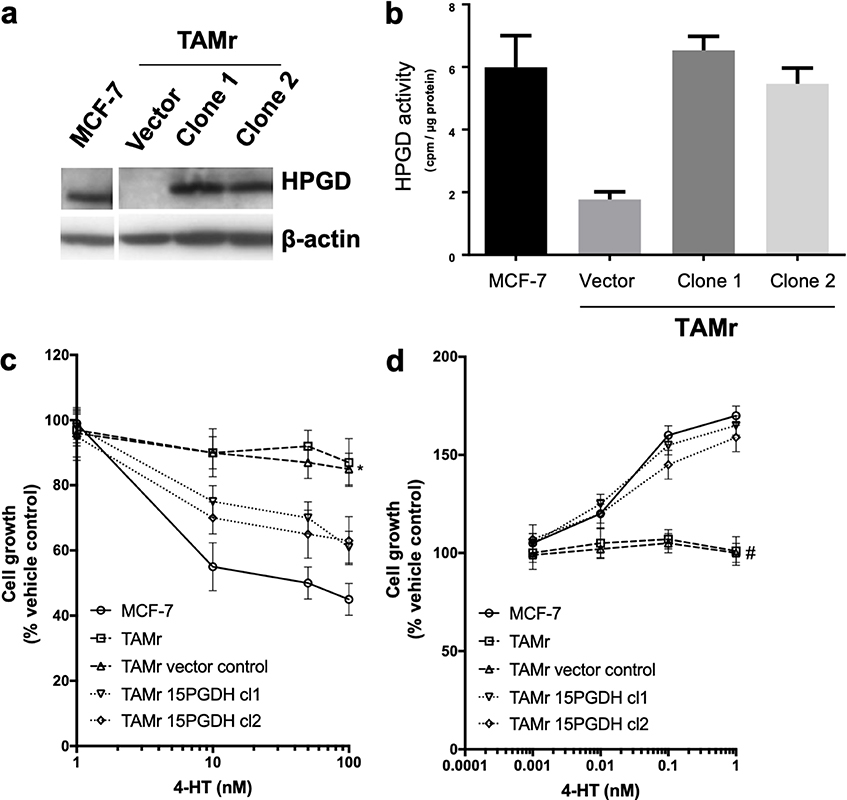
Effect of overexpressing HPGD in MCF-7 TAMr 2 cells. Western blot (a) showing HPGD expression in stably transfected clones of MCF-7 TAMr 2 cells compared to empty vector control and WT MCF-7; in (b), HPGD enzyme activity (cpm/μg protein) is restored to levels similar to WT MCF-7 following HPGD-transfection, sensitising cells to inhibitory effects of TAM (* P < 0.05 vs. MCF-7, Figure 2c) and restoring E2 sensitivity (# P < 0.004 vs. MCF-7, Figure 2d). Data are representative of triplicate experiments (± SEM)
Since HPGD is responsible for the biological inactivation of prostaglandins including PGE2, we tested whether increased PGE2 signaling might explain TAMr. We first examined PGE2 levels in serum-free conditioned medium collected from MCF-7 and MCF-7 TAMr 2 cells. TAMr cells released higher amounts of PGE2 compared with parental MCF-7 cells and this could be reduced to levels approaching that of MCF-7 by HPGD overexpression in these cells (Figure 3a). Next, we tested the effect of adding exogenous PGE2 (1 µM) on TAM response in two different ERα-positive breast cancer cell lines, MCF-7 and T47D. PGE2 reduced sensitivity of both cell lines to TAM (Figure 3b). As PGE2-signalling is mediated by G-protein coupled receptors, we examined if the stimulatory EP2 or EP4 receptors might be responsible for mediating the effects of PGE2 on TAM sensitivity. We demonstrated that PGE1-OH (an EP4 agonist) was able to mimic the effects PGE2 in decreasing TAM sensitivity (Figure 3c; P < 0.015), albeit to a lesser extent than PGE2. However, butaprost (EP2 agonist) had no effect at equivalent concentrations (Figure 3d). This suggested that the EP4 receptor might be responsible for mediating the effect of PGE2 on TAM sensitivity. Partial silencing of EP4 in WT MCF-7 cells was achieved using one siRNA (Figure 3e). EP4 silencing using siRNA 1 inhibited PGE2 induced TAM resistance in these cells (Figure 3f).
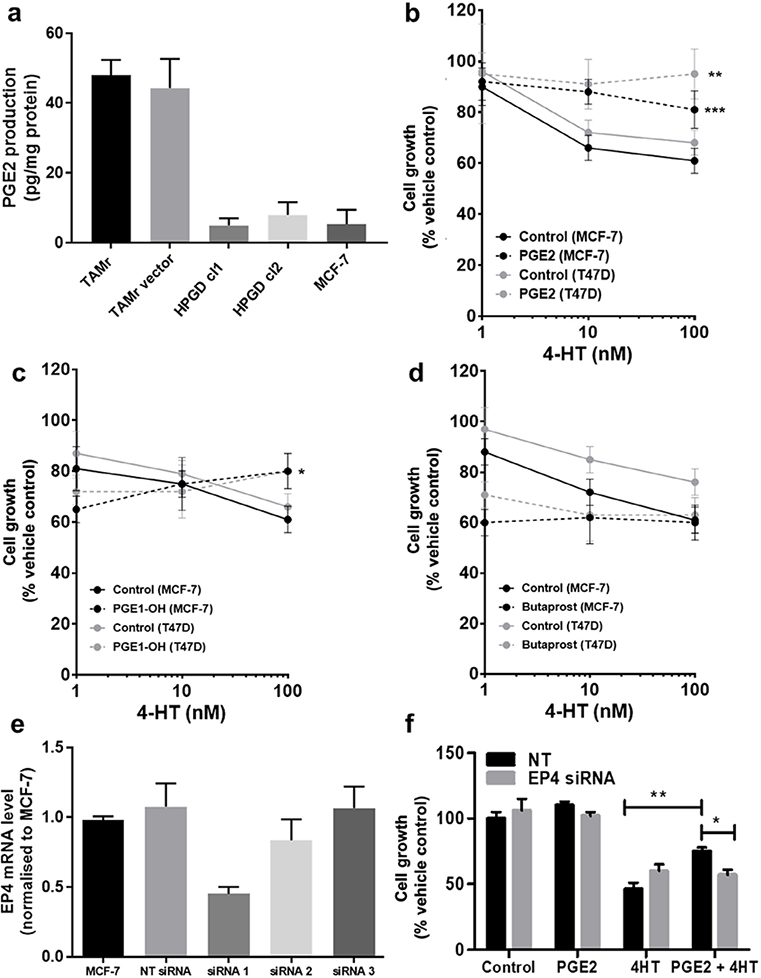
Modulating the PGE2 axis influences cellular response to TAM. (a) Overexpression of HPGD reduced PGE2 production in MCF-7 TAMr 2 cells to similar levels to basal production in WT MCF-7 cells; (b) exogenous PGE2 (1 μM, broken lines) reduced sensitivity of WT MCF-7 and T47D human breast cancer cells to TAM; (c) PGE1-OH mimicked the effects PGE2 in decreasing TAM sensitivity in WT MCF-7 and T47D cells, albeit to a lesser extent; (d) butaprost was ineffective at equivalent concentrations; (e) EP4 siRNA1 was selected for effective transient silencing of EP4 expression in WT MCF-7; (f) transient silencing of EP4 reduced sensitivity to TAM, only in the presence of PGE2. * denotes P < 0.05; ** denotes P < 0.002; *** denotes P < 0.005 normalised to respective vehicle controls
We analysed the relationship between HPGD expression and clinical outcome for breast cancer patients using the on-line resource Kmplot [30] and from our own cohort of breast cancer patient’s data in separate survival analyses. In the Kmplot cohort, we found that ER+ patients who received adjuvant TAM only and had ‘low’ expression of HPGD, had reduced OS by univariate analysis [hazard ratio (HR) = 0.28 (0.13–0.59), P = 0.0007] (Figure 4a). This remained significant on multivariate analysis (P = 0.001, Figure 4b). We then conducted a retrospective immunohistochemical study of HPGD expression in our own cohort of breast patients, who all received adjuvant TAM therapy and related HPGD expression to survival. As these TMAs have been used extensively in several other studies [24, 29, 35], the number of viable cores that could be reliably evaluated was only 144 out of 350 cases. The level of HPGD staining was generally weak to moderate (Figure 4c) in positive cases, however cases which completely lacked HPGD were associated with worse OS [HR = 0.3 (0.15–0.69), P = 0.047] (Figure 4d), in line with the in silico analysis (Figure 4a). The small number of TMA cases available precluded multivariate analysis.
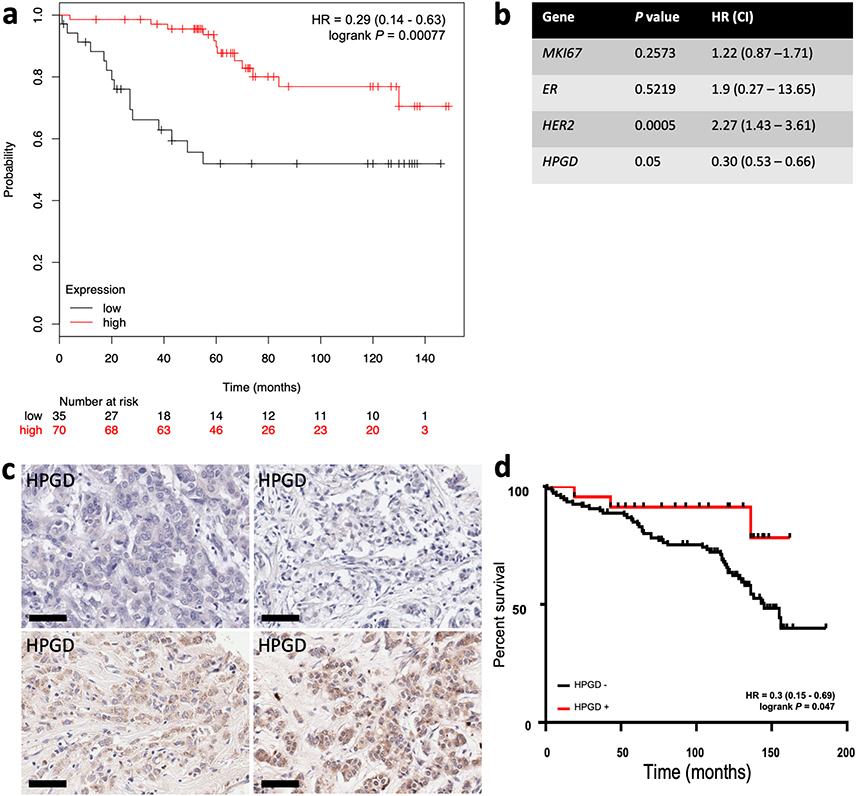
Kaplan-Meier survival analysis of the relationship between HPGD expression and breast cancer outcomes using in silico data mining and immunohistochemistry. Using KMplot, HPGD expression was categorised as ‘high’ (red) or ‘low’ (black), with the lower tertile expression used as cut-off. (a) Low HPGD expression is associated significantly reduced OS [HR 0.28 (0.13–0.59), P = 0.00041] in ERα breast tumours treated with TAM; (b) the association between HPGD expression and OS remained significant by multivariate analysis; (c) examples of HPGD staining (brown, DAB staining) in breast cancer tissue sections counterstained with haematoxylin (blue), the scale bar indicating 60 µm with some cases lacking any appreciable HPGD (top panels), and others showing weak to moderate staining (bottom panel); (d) a retrospective immunohistochemical analysis of HPGD expression in 130 breast cancers represented on TMAs showed HPGD-negative cases (black) had significantly worse OS [HR = 0.3 (0.15–0.69), P = 0.047] compared with HPGD-positive cases (red)
With growing evidence that HPGD is inactivated epigenetically, we sought to identify miRNAs that may post-transcriptionally modulate HPGD expression. We interrogated a global miRNA data set established in an independent TAM resistance model MCF-7:5C [33]. These TAMr cells were derived from WT MCF-7 cells (Table 1). In silico analysis revealed numerous miRNAs that were differentially expressed in TAMr MCF-7:5C cells compared with TAMs MCF-7:WS8 cells and that are predicted to interact with the three prime untranslated region (3′-UTR) of the HPGD transcripts (Table 2).
In silico prediction of miRNAs differentially expressed in TAMr MCF-7:5C vs. TAMs MCF-7:WS8 that are predicted to interact with 3’UTR of major HPGD transcripts
| miRNA | Fold change | Ensembl ida | Binding probabilityb | Longest consecutive pairingsb |
|---|---|---|---|---|
| hsa-miR-432-5p | 105.24 | ENST00000504433 | 0.85 | 7 |
| hsa-miR-382-5p | 54.71 | ENST00000296521, ENST00000542498 | 0.85 | 9 |
| hsa-miR-487a-3pc | 42.78 | ENST00000296522.6 | - | - |
| hsa-miR-31-5p | 28.67 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000510901, ENST00000541923, ENST00000542498 | 1.0 | 14 |
| hsa-miR-543 | 19.49 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000541923, ENST00000542498/ENST00000510901 | 0.85/0.92 | 17 |
| hsa-miR-493-3p | 13.35 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000541923, ENST00000542498/ENST00000510901 | 0.85/0.88 | 19 |
| hsa-miR-376c-3pc | 10.65 | ENST00000296522.6 | - | - |
| hsa-miR-337-5p | 6.46 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000541923, ENST00000542498/ENST00000510901 | 0.85/0.92 | 7 |
| hsa-miR-505-5p | 6.32 | ENST00000296522, ENST00000422112, ENST00000504433, ENST00000510901/ENST00000541923 | 1.0/0.85 | 6 |
| hsa-miR-1972 | 5.64 | ENST00000504433 | 0.92 | 7 |
| hsa-miR-410-3pc | 4.00 | ENST00000296522.6 | - | - |
| hsa-miR-154-5p | 3.39 | ENST00000504433 | 0.85 | 5 |
| hsa-miR-485-5p | 3.08 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000510901, ENST00000541923, ENST00000542498 | 1.0 | 7 |
| hsa-miR-199a-3pc | 2.91 | ENST00000296522.6 | - | - |
| hsa-miR-654-3pc | 2.89 | ENST00000296522.6 | - | - |
| hsa-miR-493-5p | 2.68 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000542498 | 0.85 | 10 |
| hsa-miR-381-3p | 2.66 | ENST00000510901 | 0.85 | 8 |
| hsa-miR-330-3p | 2.54 | ENST00000504433/ENST00000510901/ENST00000542498 | 0.92/0.85/1 | 9/7/7 |
| hsa-miR-154-3pc | 2.35 | ENST00000296522.6 | - | - |
| hsa-miR-30a-3p | 1.88 | ENST00000504433 | 0.92/0.85 | 10/8 |
| hsa-miR-4298 | 1.83 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000510901, ENST00000541923, ENST00000542498 | 1.0 | 8 |
| hsa-miR-3200-3pc | 1.82 | ENST00000296522.6 | - | - |
| hsa-miR-106a-5pc | 1.75 | ENST00000296522.6 | - | - |
| hsa-miR-20b-5pc | 1.74 | ENST00000296522.6 | - | - |
| hsa-miR-425-5pc | 1.71 | ENST00000296522.6 | - | - |
| hsa-miR-17-5p | 1.69 | ENST00000504433 | 0.85 | 10 |
| hsa-miR-500a-5p | 1.66 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000541923, ENST00000542498, ENST00000510901 | 0.92 | 8 |
| hsa-miR-1293 | 1.60 | ENST00000296522, ENST00000296521, ENST00000422112, ENST00000541923, ENST00000542498, ENST00000510901 | 1.0 | 9 |
| hsa-miR-20a-5p | 1.56 | ENST00000504433 | 0.92 | 10 |
Listed are miRNAs with fold change > 1.5; miRNAs underlined refer to the DLK-DIO3 locus on chromosome 14; miRWalk 3.0 [31] and TargetScan 7.2 [32] tools have been used to identify miRNA-target interactions; a major targeted transcripts are listed with respective Ensembl id; b binding probability and longest consecutive pairings are listed according to miRWalk 3.0 predictions; c additional miRNAs predicted only with TargetScan 7.2 tool (on most prevalent ENST00000296522.6 transcript)
We then tested the association between each of the listed miRNAs and HPGD mRNA levels in 987 breast cancer samples from the TCGA database using OncoLnc [34] where both miRNA and mRNA expression data were available. Only one miRNA, namely miR-3200-3p, had an inverse relationship between its expression levels and HPGD mRNA levels in this dataset (Spearman, ρ = –0.25, P < 0.0001). We used the Kmplot online resource [30] to analyse the association between miR-3200-3p expression and OS in ER+ breast cancer patients treated with any endocrine therapy, irrespective of grade, molecular subtype, and lymph node status. High miR-3200-3p expression was associated with significantly reduced OS (Figure 5a) and a weak negative correlation (R2 = 0.25) was observed between miR-3200-3p and HPGD (Figure 5b) using data from TCGA analysed using the OncoLnc platform [34].
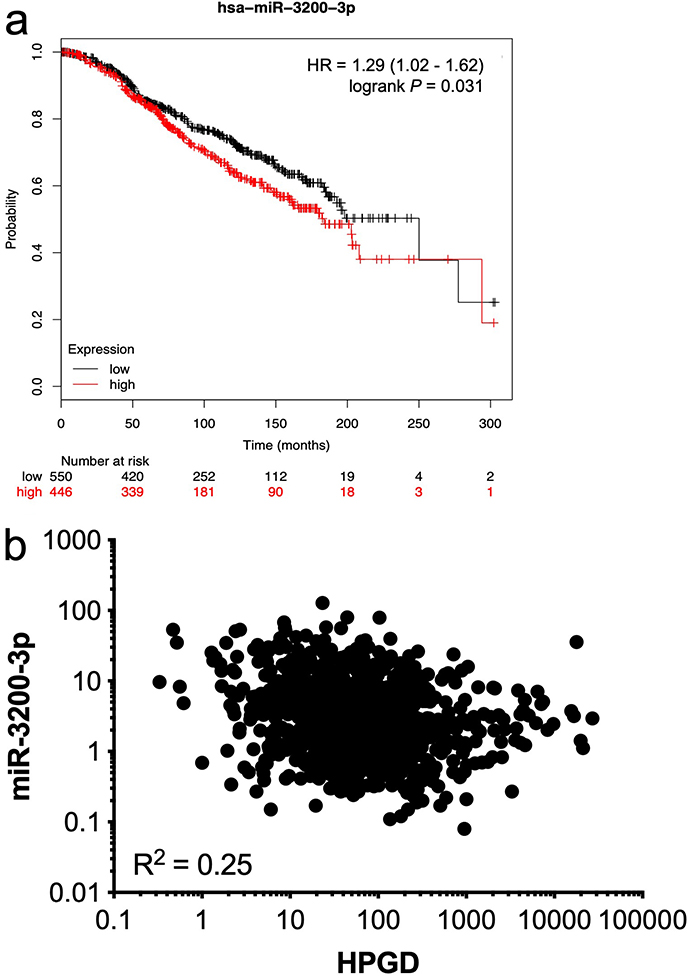
Relationship between HPGD and miR-3200-3p expression in breast cancer. Using KMplot, miR-3200-3p expression was categorized as high or low, with median expression used as cut-off. Kaplan-Meier survival analysis demonstrated a statistically significant effect of miR-3200-3p on the OS of ERα-positive breast cancer patients treated with any endocrine therapy (a); correlation analysis of matched gene expression levels of HPGD and miR-3200-3p in 987 breast cancer cases obtained from the TCGA revealed a weak negative correlation [R2 = 0.25; (b)]
We have demonstrated a downregulation of HPGD expression in TAM-resistant breast cancer both in vitro in multiple series of TAMs and TAMr isogenic cell line pairs and in clinical samples using retrospective immunohistochemistry studies and in silico approaches. We have also shown that TAM resistance is associated with PGE2 signalling via the EP4 receptor in vitro.
A key role of HPGD is to inactivate PGE2. Having confirmed its downregulation in TAMr cells using various experimental approaches and in different cell lineages, we showed that stable transfection of human HPGD into MCF-7 TAMr 2MCF-7 cells restored HPGD enzyme activity, at the same time significantly sensitising these cells to TAM, and suggesting a functional role of HPGD downregulation in acquired TAMr. Our hypothesis that HPGD mediated its effects via this axis was strengthened by the observation that treatment of 2 different ER+ TAM-sensitive cell lines with exogenous PGE2 increased the resistance of both cell lines to the growth-inhibitory effects of TAM.
PGE2 exerts its effects through binding to cell surface G-protein coupled EP receptors. Four subtypes have been identified, EP1, -2, -3 and -4, with each coupled to different intracellular signalling pathways. All are stimulatory except for EP3, which exerts inhibitory effects. We used a pharmacological approach to target EP2 and EP4, treating parental MCF-7 and T47D cells with PGE1-OH (EP4 receptor agonist) or with butaprost (EP2 receptor agonist). PGE1-OH was able to mimic the effects of PGE2 (albeit to a lesser extent), whereas butaprost had no effect at equivalent concentrations. We suggest that the EP4 receptor is responsible for mediating the TAMr effect of PGE2. While our knockdown experiment implied a role for EP4, we were only able to achieve partial knockdown with a single siRNA, which limits the conclusions from our data. Consistent with our data, a study has shown that EP4 gene expression was upregulated in TAMr MCF-7 cells, also generated from long term oestrogen deprivation, and in patients resistant to aromatase inhibitor therapy. Furthermore, the exposure to two separate EP4-specific antagonists, GW627368X and ONO-AE3-208, decreased estrogen-independent cell growth in LTED MCF-7 cells [36]. These authors proposed a molecular mechanism by which EP4 signalling may drive ligand-independent ERα activation by driving the binding of the coactivator-associated arginine methyltransferase 1 (CARM1), an ERα co-factor, to the receptor, leading to its activated status [36].
There is evidence that HPGD is post-transcriptionally regulated via miRNAs, with miR-21 identified as targeting HPGD in oral tongue squamous cell carcinoma [37]. Silencing of miR-21 expression in human breast cancer cell lines has been shown to increase TAM sensitivity [38]. miR-21 was found to be overexpressed in TAMr cell line compared to TAMs cell lines [39], which may contribute to HPGD downregulation in TAM resistant breast cancer. However, miR-21 was not identified as differentially expressed in TAMr MCF-7:5C cells compared with TAMs MCF-7:WS8 cells. We identified several differentially expressed miRNAs predicted to interact with the 3’UTR of HPGD transcripts. Of these, miR-3200-3p expression was found to be inversely correlated with HPGD mRNA level, suggesting a possible role of miR-3200-3p in the regulation of HPGD expression. Using an in silico approach we found that increased miR-3200-3p levels were significantly associated with decreased OS in ER positive breast cancer patients treated with endocrine therapy. There are no data available thus far establishing a direct interaction between miR-3200-3p and HPGD mRNA, however correlation between HPGD and miR-3200-3p in breast cancer cases in the TCGA database using the OncoLnc platform [34] revealed a weak negative correlation. A key next step would be to validate that the 3′-UTR of HPGD is a target of miR-3200-3p in a luciferase reporter assay in vitro, with in vivo validation also warranted. Interestingly, while this work was under review, miR-3200-3p was identified using data from TCGA as a so-called miRNA master regulator (MMR) in breast cancer and was one of 61 MMRs which displayed high oncogenic activity [40].
The immunohistochemistry on human breast cancer samples and the in silico analysis demonstrated that reduced expression of HPGD was associated with worse patient outcomes, consistent with a study demonstrating that low levels of HPGD predicted reduced recurrence free survival and OS in a cohort of breast cancer patients, split into normal weight, overweight and obese groups, in which few received endocrine therapy (15%) [41]. In our study, all cases received adjuvant TAM, with HPGD downregulation associated with reduced response to endocrine treatment and worse clinical outcomes.
Some studies of HPGD expression in breast tissues have demonstrated higher expression in normal breast tissue than breast cancer [19], supporting a tumour suppressive role [17, 42]. However, this is an inconsistent finding. One study showed that high, rather than low, HPGD expression was significantly associated with poor outcome in triple negative breast cancer [18]. However, tumours lacking expression of either ERα, PR and HER2 are recognised to have worse prognosis. Our cohort was predominantly ER-positive (77%) and the in silico analysis was restricted to ER-positive cases only. Such context-dependency has been supported through a data mining approach where HPGD expression was found to be differentially expressed across the various molecular subtypes of breast cancer [19]. However, in this study [19], HPGD expression was significantly decreased in triple negative breast cancer compared to other molecular subtypes.
Data extrapolated from clinical trials is beginning to indicate that the risk of breast cancer relapse is reduced by extending time on endocrine therapy from 5 to 10 years [43]. While this is good for patient outcome, there will likely be a lag period before these benefits are seen in the clinic. Understanding the mechanisms which underline endocrine therapy resistance remains an important biological question. This study demonstrated that HPGD downregulation in ER-positive breast cancer is associated with reduced response to adjuvant TAM therapy via PGE2-EP4 signalling and decreased patient survival. These data offer a potential target to develop combination therapies that may overcome TAM acquired resistance.
3′-UTR: three prime untranslated region
CARM1: coactivator-associated arginine methyltransferase
COX: cyclooxygenase
E2: 17β-oestradiol
EP4: prostaglandin E2 receptor 4
ERα: oestrogen receptor alpha
FCS: foetal calf serum
HPGD: 15-hydroxyprostaglandin dehydrogenase
LTED: long-term estrogen deprivation
MMR: miRNA master regulator
MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
OH: alcohol
OS: overall survival
PGE2: prostaglandin E2
PR: progesterone receptor
qRT-PCR: real-time quantitative polymerase chain reaction
RT-PCR: real-time polymerase chain reaction
SD: standard deviation
siRNA: small-interfering RNA
TAM: Tamoxifen
TAMr: tamoxifen-resistant
TAMs: tamoxifen-sensitive
TCGA: the cancer genome atlas
TMA: tissue microarray
WT: wild type
We thank Werbena Hamilton-Burke, Loaie Maraqa and Mark Peter for providing clinical data on the breast tumours which populated the TMAs used in this study.
Conception and design: MAH, VS. Acquisition of data: AMS, BA, PYM, BMB, RH, PF, HB, MV, MC, VCJ, VS. Analysis and interpretation of data: AMS, BA, PYM, BMB, RH, PF, HB, MV, MC, VCJ, VS. Writing and/or revision of the manuscript: MV, MC, MAH, VS. All authors read and approved the manuscript before submission.
The authors declare that they have no conflicts of interest.
Access to histopathology archives for TMA construction and immunohistochemistry was approved by the Leeds (East) Research Ethics (06/Q1206/180).
The informed consent to participate in the study was obtained from all participants.
The informed consent to publication was obtained from relevant participants.
Part of the results described is based upon data generated by the TCGA Research Network (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga), the online resources of KMplot (https://kmplot.com/analysis/) and OncoLnc (http://www.oncolnc.org/).
This work was supported in part by Cancer Research UK grant C13432/A9047; Yorkshire Cancer Research grant L314; the Robert Bosch Foundation, Stuttgart, Germany; the NIH; MD Anderson’s Cancer Center support grant CA016672; the George and Barbara Bush Foundation for Innovative Cancer Research; and the Dallas/Fort Worth Living Legend Chair of Cancer Research. The study sponsor(s) played no part in the study design; in the collection, analysis and interpretation of the data; in the writing of the report; and in the decision to submit the paper for publication.
© The Author(s) 2020.
Copyright: © The Author(s) 2020. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

