 Review
Review

Affiliation:
¹Cellular Physiology and Translational Medicine, Department of Health Sciences, División de Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09310, Mexico
2Graduate Program in Experimental Biology, Department of Health Sciences, División de Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09310, Mexico
ORCID: https://orcid.org/0000-0002-6504-9724
Affiliation:
3Experimental Medicine and Carcinogenesis, Department of Health Sciences, División de Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09130, Mexico
ORCID: https://orcid.org/0000-0002-6963-2642
Affiliation:
¹Cellular Physiology and Translational Medicine, Department of Health Sciences, División de Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09310, Mexico
ORCID: https://orcid.org/0000-0002-3923-6278
Affiliation:
¹Cellular Physiology and Translational Medicine, Department of Health Sciences, División de Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09310, Mexico
ORCID: https://orcid.org/0000-0003-0501-7226
Affiliation:
3Experimental Medicine and Carcinogenesis, Department of Health Sciences, División de Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09130, Mexico
Email: legq@xanum.uam.mx
ORCID: https://orcid.org/0000-0002-5704-5985
Affiliation:
¹Cellular Physiology and Translational Medicine, Department of Health Sciences, División de Ciencias Biológicas y de la Salud, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, 09310, Mexico
Email: roxml@xanum.uam.mx
ORCID: https://orcid.org/0000-0001-8249-7257
Explor Dig Dis. 2025;4:100583 DOI: https://doi.org/10.37349/edd.2025.100583
Received: March 22, 2025 Accepted: June 24, 2025 Published: August 05, 2025
Academic Editor: Alfredo Caturano, Università degli Studi della Campania Luigi Vanvitelli, Italy
The article belongs to the special issue The Role of Gut Microbiota in the Pathogenesis and Management of Metabolic-Associated Steatotic Liver Disease (MASLD)
Metabolic dysfunction-associated steatotic liver disease (MASLD) is a leading cause of chronic liver disease worldwide. Its prevalence is increasing due to its close relationship with obesity, insulin resistance, and other metabolic disorders. In this context, the gut-liver axis has been identified as a fundamental regulator in the progression of MASLD, integrating metabolic, immunological, and inflammatory signals that influence hepatic homeostasis. This article reviews the interconnection between the intestine and the liver in the onset and progression of MASLD, highlighting the roles of cholesterol and its metabolism, intestinal barrier permeability, microbiota, and hepatic signaling pathways. We analyze how intestinal dysbiosis and alterations in the enterohepatic circulation of bile acids affect cholesterol absorption and metabolism. Furthermore, we address the influence of endotoxin translocation, activation of the innate immune system, and the interaction of key transcription factors on disease progression from steatosis to advanced fibrosis and hepatocellular carcinoma (HCC). Finally, therapeutic strategies, including pharmacological, dietary, and immunomodulation-based approaches, are discussed to regulate cholesterol metabolism, modulate the intestinal microbiota, and restore gut-liver axis homeostasis. Integrating this knowledge could open new perspectives for treating and preventing MASLD, addressing the disease from a broader and multidisciplinary viewpoint.
Metabolic dysfunction-associated steatotic liver disease (MASLD) is currently the most prevalent chronic liver disease worldwide and represents a growing public health problem. Its progression is closely linked to metabolic dysfunctions such as those presented in obesity, insulin resistance, dyslipidemia, and diabetes, contributing to a spectrum of liver alterations ranging from simple steatosis to metabolic dysfunction-associated steatohepatitis (MASH), fibrosis, cirrhosis, and, eventually, hepatocellular carcinoma (HCC) [1].
In recent years, medical liver terminology has evolved to more accurately reflect its close relationship with metabolic dysfunction and cardiovascular risk factors. Almost all patients previously diagnosed with non-alcoholic fatty liver disease (the former NAFLD acronym) are estimated to meet the established criteria for MASLD [2]. Obesity, metabolic syndrome, and white adipose tissue inflammation play a vital role in disease progression [3].
Beyond its clinical impact, MASLD has significant socioeconomic repercussions, representing a major challenge to global health [1]. In the United States alone, the economic burden of MASLD exceeds $100 billion, with liver transplantation being the most significant contributor to healthcare costs [4].
Globally, MASLD affects approximately 38% of adults and 7% to 14% of children and adolescents, and its prevalence in adults is projected to exceed 55% by 2040. However, notable regional variations exist, with the highest rates reported in Latin America and the Middle East, underscoring the need for culturally adapted interventions and a comprehensive global approach [5].
To manage the extensive and rapidly evolving body of literature on MASLD and the gut–liver axis, we adopted a structured literature search strategy. Initial searches were conducted in PubMed, Scopus, and Google Scholar using broad terms including “MASLD”, “MASH”, “NAFLD”, “NASH”, “gut–liver axis”, “cholesterol metabolism”, “bile acids”, “dysbiosis”, “hepatic inflammation”, and “hepatocellular carcinoma”. The inclusion of NAFLD and NASH was necessary to retrieve seminal studies published before the 2023 nomenclature update. The time frame was limited to publications from 2010 to 2024, with a particular focus on studies from the past five years. For each section of the manuscript, targeted sub-searches were carried out to refine the scope and ensure specificity. For example, we used: “intestinal permeability”, “tight junctions AND liver disease”, “bile acids AND gut microbiota”, and “cholesterol absorption AND NPC1L1” to support the gut–liver axis discussion; “cholesterol metabolism AND SREBP2”, “HMG-CoA reductase AND NAFLD”, and “statins AND HCC risk” to explore cholesterol homeostasis in MASLD; and “CRISPR/Cas9 AND CYP7A1”, “epigenetics AND cholesterol metabolism”, and “resveratrol OR berberine OR curcumin AND epigenetics” to address molecular regulation strategies. For sex-specific differences, terms like “estrogen signaling AND NAFLD”, “sex differences AND hepatic fibrosis”, and “female-specific HCC” were applied. To support the fibrosis and carcinogenesis section, focused searches included “hepatic stellate cells AND activation”, “mitochondrial ROS AND HSC”, “oxysterols AND HCC progression”, and “immune evasion AND cholesterol AND HCC.” Finally, to address extrahepatic complications, we used terms such as “MASLD AND cardiovascular disease”, “gut dysbiosis AND systemic inflammation”, and “MASLD AND gastrointestinal cancer”. Articles were included based on their originality, peer-review status, relevance to MASLD pathophysiology, and language (English only), with a preference for recent and high-impact publications. Through this process, we refined the reference list to include only the most relevant and representative articles in the field, ensuring scientific rigor, conceptual depth, and thematic coherence.
The gut-liver axis encompasses a bidirectional communication network that integrates anatomical, metabolic, and immune signals between the intestine and the liver. This interaction is mediated through multiple pathways, including the portal vein, biliary tract, and systemic circulation. The portal vein serves as the primary conduit for nutrients, microbial products, and xenobiotics absorbed in the gut, while the biliary tract and systemic circulation allow reciprocal signaling from the liver back to the intestine [6].
Given continuous exposure to luminal antigens, the intestinal barrier plays a critical regulatory role by selectively permitting the passage of essential molecules while restricting the translocation of potentially harmful compounds [7]. However, several environmental and dietary factors, such as dysbiosis, alcohol consumption, and high-fat diets—can compromise this barrier and promote increased intestinal permeability.
Cholesterol absorption constitutes a central physiological function within the gut-liver axis. In the small intestine, dietary cholesterol is solubilized into micelles by bile acids (BA) and subsequently absorbed by enterocytes, primarily via the Niemann-Pick C1-like 1 (NPC1L1) transporter. Once internalized, cholesterol is either esterified by acyl-CoA: cholesterol acyltransferase (ACAT) and incorporated into chylomicrons for lymphatic transport or transported in its free form directly to the liver via the portal vein. The liver then orchestrates its systemic distribution, storage, or conversion into BA. Under healthy conditions, this process is tightly regulated by nuclear receptors such as farnesoid X receptor (FXR) and receptors like liver X receptor (LXR), which control the expression of key transporters and enzymes to preserve lipid homeostasis [6]. However, in the context of dysbiosis and metabolic stress, as observed in MASLD, cholesterol absorption may become dysregulated, leading to hepatic lipid overload and chronic inflammation. Thus, impaired intestinal cholesterol handling emerges as a critical mechanistic link between gut barrier dysfunction and the progression of hepatic disease.
Importantly, the cholesterol imbalance observed in MASLD results not only from dietary intake but also from enhanced endogenous synthesis, driven by dysregulation of sterol regulatory element-binding protein 2 (SREBP2) and impaired AMP-activated protein kinase (AMPK) signaling [6].
Although cholesterol-deficient diets, such as veganism or other plant-based, low-cholesterol regimens, are naturally devoid of dietary cholesterol and are typically associated with lower serum low-density lipoprotein cholesterol (LDL-C) levels, this reduction is compensated mainly by an upregulation of endogenous cholesterol synthesis, primarily through increased activity of hepatic 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in the mevalonate pathway. Lütjohann et al. [8] demonstrated that individuals adhering to vegan diets may exhibit up to 35% higher rates of endogenous cholesterol synthesis compared to omnivores, despite significantly lower intestinal cholesterol absorption. However, this physiological adaptation does not result in hypercholesterolemia. On the contrary, serum cholesterol concentrations in vegans generally remain low or within normal ranges, largely due to the low intake of saturated fats and the high fiber content characteristic of plant-based diets, which enhance cholesterol clearance and BA excretion. This highlights the effectiveness of homeostatic regulatory mechanisms in maintaining lipid balance even in the absence of dietary cholesterol and underlines that cholesterol biosynthesis and intake must be interpreted in the context of overall metabolic and dietary profiles [8, 9].
The intestine plays a dual role by facilitating the digestion and absorption of nutrients while simultaneously serving as a selective barrier against pathogenic microorganisms and harmful substances. Due to its anatomical and functional proximity to the liver, the intestine is in constant communication with the hepatic system through portal circulation, allowing for the transfer of nutrients, microbial products, and metabolites. As the primary site of exposure to dietary components, the intestine has a critical influence on hepatic physiology by modulating both the quantity and quality of substrates that reach the liver. Therefore, hepatic function and systemic homeostasis are highly dependent on maintaining intestinal barrier integrity [10].
Additionally, the biliary tract facilitates the secretion of BA into the intestinal lumen, promoting the emulsification of lipids and shaping the composition of the gut microbiota. Conversely, systemic circulation allows liver-derived hormones, cytokines, and metabolites to modulate intestinal barrier function and local immune responses [6].
Despite the efficiency of this homeostatic system, several factors can compromise intestinal integrity and disrupt gut-liver communication. The intestinal barrier, composed of enterocytes, tight junction (TJ) proteins [e.g., zonula occludens-1 (ZO-1), occludin, and claudins], immune cells, and mucosal layers, selectively permits the passage of nutrients while preventing translocation of harmful substances [7]. However, chronic exposure to dietary fat, alcohol, bacterial endotoxins, and proinflammatory stimuli can increase intestinal permeability, a condition commonly referred to as “increased intestinal permeability”. This dysfunction permits microbial products, toxins, and excess lipids to reach the liver, triggering immune responses and hepatocellular injury.
High-cholesterol diets compromise intestinal barrier integrity through multiple mechanisms. Excessive luminal cholesterol promotes the generation of bioactive oxysterols—particularly 27-hydroxycholesterol (27-OHC) and 7-ketocholesterol—which compromise intestinal barrier integrity by altering the localization and membrane association of TJ proteins, such as claudins and occludin [11]. These effects are mediated through redox-sensitive pathways that destabilize epithelial TJs, enhance paracellular permeability, and trigger low-grade inflammation [6, 11]. This facilitates the translocation of luminal antigens and microbial products, thereby initiating hepatic immune activation and sustaining the progression of MASLD.
This effect is further exacerbated by the overactivation of nuclear receptors such as LXR, which modulates epithelial gene expression and contributes to enterocyte dysfunction [12]. Specifically, LXR activation induces ABCA1 and sterol regulatory element-binding protein-1c (SREBP-1c) expression, which promotes intracellular lipid accumulation and impairs epithelial renewal, thereby weakening barrier function [12]. Upon activation, LXR induces cholesterol efflux and lipogenesis, but also exerts antiproliferative effects by downregulating cell cycle regulators such as S-phase kinase-associated protein 2 (Skp2), c-Myc, CDKs, and cyclins, and inducing hypophosphorylation of the tumor suppressor retinoblastoma protein (Rb), thereby halting epithelial cell proliferation [13, 14]. This reduction in epithelial turnover impairs the renewal of the intestinal barrier, especially under metabolic or inflammatory stress [13, 14].
Additionally, cholesterol overload upregulates NPC1L1 expression, which enhances intracellular lipid uptake and contributes to cholesterol accumulation in enterocytes [15]. Recent findings indicate that NPC1L1 overexpression not only increases intracellular cholesterol but also impairs epithelial antioxidant capacity by reducing levels of glutathione peroxidase and superoxide dismutase, thereby exacerbating local oxidative stress and inflammation [16].
These alterations are compounded by a reduction in goblet cells and mucin secretion, weakening the protective mucus layer [17]. Moreover, cholesterol-induced gut dysbiosis—characterized by an increased abundance of proinflammatory genera such as Desulfovibrio and Bilophila wadsworthia, along with a reduction of beneficial commensals including Lactobacillus and Faecalibacterium prausnitzii—exacerbates the production of endotoxins and epithelial injury [16, 18].
Collectively, these changes promote bacterial translocation, endotoxemia, and hepatic immune activation, ultimately driving the progression of MASLD [6, 12].
In MASLD, enhanced intestinal cholesterol absorption and impaired hepatic clearance further exacerbate lipid accumulation, mitochondrial dysfunction, and inflammation [18]. Altered BA homeostasis—resulting from impaired ileal reabsorption via apical sodium-dependent BA transporter (ASBT) and gut dysbiosis—compromises epithelial barrier integrity and facilitates the translocation of lipopolysaccharides (LPS) and other proinflammatory mediators, promoting hepatic stellate cell activation, fibrogenesis, and disease progression [6, 19].
The translocation of LPS also triggers the toll-like receptor 4 (TLR4)/NF-κB signaling pathway in Kupffer cells, promoting the secretion of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and IL-6, and sustaining the inflammatory milieu associated with liver injury and fibrosis [6, 16].
Cholesterol also facilitates intestinal endotoxin absorption by enhancing LPS translocation across the epithelial barrier through receptor-independent insertion into lipid membranes, destabilizing the TJ, and increasing permeability. High-cholesterol diets activate intestinal inflammasomes, leading to local myeloid cell recruitment and epithelial inflammation [20]. Additionally, dietary cholesterol promotes systemic dissemination of LPS via chylomicron-mediated transport, facilitating its delivery to the liver and peripheral tissues [21]. These processes are exacerbated by cholesterol-driven dysbiosis, which increases the abundance of gram-negative bacteria and proinflammatory metabolites [16]. Together, these mechanisms sustain low-grade systemic inflammation and hepatic immune activation, reinforcing the gut–liver inflammatory axis in MASLD [21].
Beyond liver pathology, MASLD is increasingly recognized as a multisystem disorder associated with heightened risk of extrahepatic complications, including major adverse cardiovascular events and gastrointestinal cancers. Gut–liver axis dysfunction contributes to these systemic outcomes by promoting chronic low-grade inflammation, dyslipidemia, altered glucose metabolism, and immune dysregulation. Emerging evidence suggests that intestinal dysbiosis, increased permeability, and the translocation of microbial products contribute to the pathogenesis of atherosclerosis and endothelial dysfunction [22]. Simultaneously, liver-derived proinflammatory and tumor-promoting mediators contribute to colorectal, pancreatic, and gastric carcinogenesis [19]. These findings underscore the importance of incorporating cardiovascular and oncologic risk assessment into MASLD management, suggesting that targeting gut–liver interactions may offer therapeutic opportunities to mitigate systemic disease progression.
Moreover, the gut–liver axis plays a pathogenic role in secondary steatotic liver disease (SLD) associated with endocrine disorders such as hypothyroidism, polycystic ovary syndrome (PCOS), and growth hormone deficiency. In these settings, gut dysbiosis and impaired intestinal barrier integrity lead to systemic inflammation and metabolic dysfunction, thereby exacerbating hepatic steatosis and fibrosis. Altered BA signaling, along with reduced activation of FXR and Takeda G protein-coupled receptor 5 (TGR5)—also known as G protein-coupled bile acid receptor 1 (GPBAR1)—further impairs glucose and lipid metabolism, contributing to disease progression [23]. These observations highlight gut–liver axis dysregulation as a shared mechanistic pathway in both primary MASLD and endocrine-associated SLD.
Finally, the gut–liver axis is increasingly recognized as a determinant of the pathogenic heterogeneity observed in MASLD. Interindividual differences in gut microbiota composition, intestinal barrier function, and BA signaling result in variable exposure to microbial products, inflammatory stimuli, and metabolic stressors. These differences influence hepatic immune responses, lipid metabolism, and fibrogenic activity, providing a plausible explanation for the divergence between simple steatosis and advanced fibrosis or HCC. This framework positions the gut–liver axis not only as a key driver of MASLD pathogenesis but also as a modulator of its clinical heterogeneity [24].
The Western diet, often characterized by its high content of carbohydrates, saturated fat, and cholesterol, plays a significant role in the development and progression of MASLD. Among these dietary components, cholesterol enrichment is particularly relevant, as it promotes lipid synthesis and accumulation in the liver. This, in turn, aggravates liver damage by inducing inflammation and oxidative stress (OxE), leading to DNA, protein, and lipid damage [25, 26].
Cholesterol consumption varies significantly worldwide, reflecting differences in dietary habits and lifestyles. In the United States, the average cholesterol intake is 293 mg/day, with an average in men of 348 mg/day, compared with a lower intake in women (242 mg/day); these levels have remained relatively stable over time [27]. In contrast, in Latin America and the Caribbean, average serum total cholesterol levels are around 193 mg/dL, without clear trends of change [28]. Notably, developed nations such as the United States and Japan generally report higher cholesterol levels than those in developing countries, a difference attributed mainly to distinct dietary patterns and lifestyles [29]. These variations in dietary cholesterol intake may contribute to differences in the prevalence and severity of MASLD worldwide, highlighting the importance of understanding its pathological consequences.
A growing body of evidence highlights the deleterious effects of high-cholesterol diets on liver health. Studies have demonstrated that cholesterol-rich diets can induce steatohepatitis, hepatocyte proliferation, angiogenesis, inflammation, and fibrogenesis, ultimately contributing to liver tumorigenesis [30]. In experimental models, these diets have been linked to a notable rise in liver tumor incidence, with increases ranging from 41% to 100% in transgenic mice, also promoting liver fibrosis and steatohepatitis [31].
Likewise, the use of alternative diets, such as high-fat diets, has been shown to induce steatosis associated with an increase in liver triglycerides. Mechanistically, these diets disrupt lipid metabolism by altering the expression of key metabolic genes, thereby worsening lipid accumulation and intensifying the inflammatory response [32, 33].
At the molecular level, excessive cholesterol intake induces mitochondrial dysfunction and OxE, which—rather than leading to cell death—paradoxically promotes apoptosis resistance and downregulation of DNA-repair enzymes. This altered cellular state prevents the elimination of damaged hepatocytes, allowing the accumulation of genetic mutations and the persistence of cells with dysfunctional metabolism. These alterations create a pro-carcinogenic environment, facilitating uncontrolled cell proliferation and the progression toward HCC. In addition, the impaired DNA-repair capacity increases genomic instability, a hallmark of cancer progression, further contributing to the malignant transformation of hepatocytes [26, 34].
Cholesterol accumulation can also impair the function of innate immune system cells, such as natural killer (NK) cells—cytotoxic lymphocytes of the innate immune system that recognize and eliminate stressed, transformed, or infected cells without prior sensitization—by reducing their cytotoxic capacity and increasing the risk of HCC in the context of MASLD [35, 36].
Therefore, cholesterol overload clearly establishes molecular and cellular conditions that favor the initiation and promotion of HCC. By inducing OxE, mitochondrial dysfunction, and apoptosis resistance, excessive cholesterol intake creates a permissive environment for hepatic tumorigenesis. These findings underscore the importance of dietary interventions and metabolic regulation strategies as potential approaches to mitigate the progression of MASLD and its transition to HCC.
Hepatic lipid deposition is a central mechanism in the pathogenesis of MASLD, which is strongly linked to obesity and metabolic syndrome, with a documented prevalence of 75.27% in the obese population [37]. The accumulation of lipids in hepatocytes results from an imbalance between lipid synthesis, uptake, storage, and degradation, which disrupts hepatic lipid homeostasis and promotes disease progression. This lipid overload impairs hepatocellular lipid metabolism, triggering OxE, mitochondrial dysfunction, and inflammation, all of which contribute to disease progression. In this context, the intricate regulation of lipid metabolism is dysregulated, promoting excessive triglyceride synthesis, the secretion of very low-density lipoproteins (VLDL), and the formation of lipid droplets (LD), exacerbating hepatic steatosis [30].
De novo lipogenesis is a crucial mechanism in hepatic lipid accumulation, primarily regulated by SREBP-1c and carbohydrate-responsive element-binding protein (ChREBP). Under normal conditions, insulin activates SREBP-1c, promoting the transcription of key lipogenic genes, including acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN), which catalyze the synthesis and elongation of fatty acids. However, in MASLD, insulin resistance-induced hyperinsulinemia exacerbates SREBP-1c activation, increasing triglyceride synthesis and promoting lipid accumulation in hepatocytes [38]. ChREBP, activated by glucose and fructose, further enhances lipogenesis. The hepatic metabolism of fructose, mediated by fructokinase, rapidly depletes ATP, activating ChREBP and inducing the expression of lipogenic genes. Unlike glucose, fructose bypasses phosphofructokinase regulation, allowing its uncontrolled conversion into lipogenic intermediates, intensifying hepatic steatosis [39].
In parallel, fatty acid oxidation (FAO) dysfunction exacerbates lipid accumulation. Peroxisome proliferator-activated receptor alpha (PPARα) is a key transcription factor regulating β-oxidation, inducing the expression of carnitine palmitoyl-transferase 1A (CPT1A), essential for long-chain fatty acid transport into mitochondria for degradation. However, in MASLD, PPARα expression is downregulated, diminishing β-oxidation efficiency and leading to hepatic lipid accumulation [40]. Additionally, AMPK, a key energy sensor, regulates lipid homeostasis by inhibiting ACC and thus suppressing de novo lipogenesis, while promoting FAO. Nevertheless, in MASLD, insulin resistance and excessive saturated fatty acids impair AMPK activity, further contributing to lipid accumulation [41].
Beyond synthesis and oxidation, fatty acid uptake and lipid droplet dynamics play critical roles in hepatic steatosis progression. CD36 (fatty acid translocase), a key fatty acid transporter, is overexpressed in hepatocytes in MASLD, increasing hepatic fatty acid influx and exacerbating lipid deposition [42]. The storage and mobilization of triglycerides within LD are regulated by perilipin 2 (PLIN2), also known as adipocyte differentiation-related protein (ADRP), which associates with LD surfaces, modulating lipid dynamics. Altered PLIN2 expression in MASLD disrupts LD homeostasis, leading to excessive triglyceride accumulation, which compromises cellular function and facilitates disease progression toward MASH and hepatic fibrosis [43].
Collectively, these mechanisms illustrate how altered hepatic fatty acid metabolism—driven by increased lipogenesis, impaired FAO, and excessive fatty acid uptake—promotes progressive lipid accumulation. This metabolic imbalance not only underpins hepatic steatosis but also establishes a pathological environment favoring inflammation, OxE, and fibrosis, accelerating MASLD progression [41].
Cholesterol is essential for hepatic and systemic homeostasis, contributing to cell membrane integrity, serving as a precursor for BA and steroid hormones, and acting as a covalent protein modifier in key metabolic pathways. Its homeostasis is tightly regulated by a complex interplay of synthesis, uptake, storage, and efflux mechanisms, ensuring a dynamic balance that maintains cellular and systemic function. However, excessive cholesterol accumulation disrupts this equilibrium and contributes to metabolic disorders, including cardiovascular disease, type 2 diabetes mellitus (T2DM), and MASLD [18].
Endogenous cholesterol is primarily synthesized in the liver (~50%) and intestine (~24%), mainly within the endoplasmic reticulum (ER) [18]. The rate-limiting enzyme in cholesterol synthesis is HMG-CoA reductase, which is regulated by hormonal and metabolic signals. This enzyme is also the primary pharmacological target of statins [44, 45]. Cholesterol biosynthesis occurs via the mevalonate pathway, which requires acetyl-CoA, oxygen, and NADPH, and involves over 20 enzymes and nearly 30 enzymatic steps [46].
Once synthesized, cholesterol is incorporated into lipoproteins for systemic distribution, stored in LD, or used for BA synthesis. In hepatocytes, cholesterol is packaged into VLDL, which are secreted into the bloodstream and progressively hydrolyzed by lipoprotein lipase, forming LDL that deliver cholesterol to peripheral tissues and return it to the liver via receptor-mediated uptake. To maintain homeostasis, excess cholesterol is removed through reverse cholesterol transport, in which high-density lipoproteins (HDL) acquire cholesterol from peripheral cells and return it to the liver either directly via scavenger receptor class B type I (SR-BI) or indirectly through exchange with other lipoproteins. In the liver, cholesterol is either converted into BA or excreted via bile [47–49].
Unlike fatty acid metabolism, which is primarily controlled by SREBP-1c, cholesterol homeostasis is governed by SREBP2, a transcription factor that senses intracellular cholesterol levels and modulates cholesterol synthesis, uptake, and storage accordingly [50].
At the molecular level, cholesterol plays a crucial role in membrane organization and signaling by modulating the structure and function of lipid-protein microdomains, such as lipid rafts [51]. These platforms influence diverse processes, including receptor activity and intracellular signaling. One of the main pathways regulating cholesterol metabolism is the SREBP pathway, which coordinates cholesterol and fatty acid synthesis in response to lipid availability [50]. Under conditions of cholesterol depletion, SREBP2 is transported from the ER to the Golgi apparatus, where it undergoes proteolytic activation via site-1 and site-2 proteases (S1P and S2P). The cleaved active form of SREBP2 translocates to the nucleus and promotes the transcription of key genes such as HMG-CoA reductase and LDL receptor, thereby enhancing cholesterol biosynthesis and uptake [52, 53]. These steps are illustrated in Figure 1.
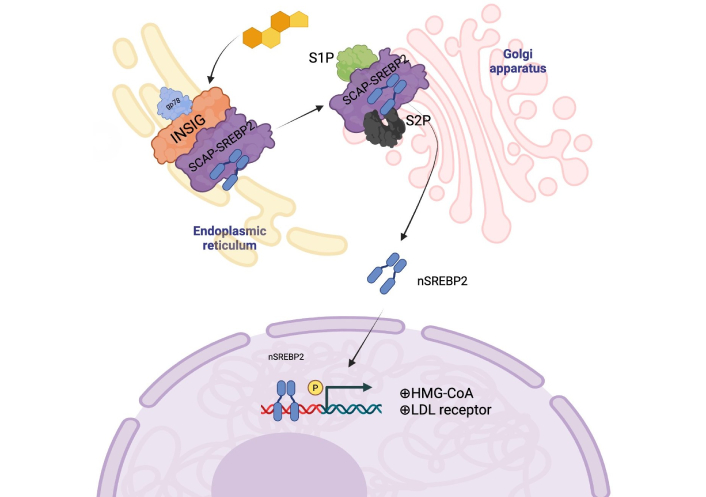
SREBP activation occurs when the cell requires more cholesterol or fatty acids. Under normal conditions, inhibitory proteins retain SREBP in the ER. Upon cholesterol depletion or stimuli such as insulin or sterols, the SREBP-SCAP complex translocates to the Golgi apparatus, where it undergoes a two-step proteolytic cleavage by S1P and S2P. The cleaved active SREBP then translocates to the nucleus, binding to SRE promoter regions and activating genes involved in cholesterol, fatty acid, and triglyceride biosynthesis—image created by BioRender. SREBP: sterol regulatory element-binding protein 2; ER: endoplasmic reticulum; SCAP: SREBP cleavage-activating protein. Created in BioRender. Gomez-Quiroz, L. (2025) BioRender.com/9cnov9m
This feedback mechanism ensures tight control of cholesterol homeostasis; however, under pathological conditions, such as insulin resistance and MASLD, dysregulated SREBP2 activity can promote excessive cholesterol synthesis and accumulation [54].
Cholesterol homeostasis is also modulated by the AMPK pathway, a central regulator of cellular energy status that integrates metabolic stress signals to maintain lipid and glucose balance [55]. Under physiological conditions, AMPK is activated via phosphorylation by liver kinase B1 (LKB1) and calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in response to reduced ATP levels [56]. Once activated, AMPK suppresses anabolic processes such as lipid and protein synthesis, while enhancing catabolic pathways, including FAO and autophagy, to restore energy homeostasis. However, cholesterol overload impairs AMPK activity, disrupting metabolic regulation. This inhibition leads to persistent activation of lipogenic enzymes like ACC and FASN, promoting lipid accumulation and hepatic steatosis. Moreover, AMPK suppression impairs mitochondrial function and autophagic flux, amplifying OxE and inflammation, which are key pathogenic mechanisms in MASLD progression [56, 57].
Unlike fatty acid accumulation, which is mainly driven by de novo lipogenesis and impaired β-oxidation, cholesterol excess induces metabolic inflexibility, mitochondrial dysfunction, and chronic inflammation, establishing a pro-injury hepatic microenvironment. In MASLD, intracellular cholesterol buildup leads to the generation of cytotoxic oxysterols that activate pro-inflammatory cascades and ER stress [11]. Concurrently, mitochondrial cholesterol accumulation destabilizes membrane integrity, impairs oxidative phosphorylation, and increases reactive oxygen species (ROS), ultimately triggering hepatocyte apoptosis [58]. These interrelated events contribute to the progression from MASLD to its inflammatory and fibrotic form, MASH, which is associated with increased risk of HCC [59].
To counteract cholesterol-induced cytotoxicity, hepatocytes activate adaptive mechanisms to preserve membrane integrity and viability. A pivotal response involves the esterification of free cholesterol by acyl-CoA: ACAT 1 and 2 (ACAT1 and ACAT2), facilitating storage in cytosolic LD. ACAT2 is primarily expressed in hepatocytes, whereas ACAT1 is enriched in hepatic macrophages [60]. This detoxification strategy prevents organelle membrane injury and attenuates lipotoxic stress. LD formation not only sequesters neutral lipids but also reflects broader metabolic reprogramming that enhances cell survival under lipid-overloaded conditions typical of MASLD [61, 62].
Collectively, these findings underscore the pivotal role of cholesterol dysregulation in hepatic metabolic dysfunction. Excess hepatic cholesterol perturbs BA homeostasis and gut microbiota composition, further affecting enterohepatic circulation and hepatic inflammation [63, 64]. A deeper understanding of these mechanisms may reveal novel therapeutic strategies to prevent cholesterol-driven liver injury and slow MASLD progression [65].
Exogenous cholesterol is obtained exclusively from animal-based foods, such as meat, dairy, and eggs, with higher concentrations in saturated fat-rich foods like egg yolks and seafood [66]. Its absorption occurs primarily in the duodenum and proximal jejunum, where dietary cholesterol is incorporated into mixed micelles composed of BA, phospholipids, and fatty acids, which facilitate transport across enterocytes. Inside enterocytes, cholesterol undergoes esterification before being incorporated into chylomicrons, which then enter the lymphatic system and eventually reach circulation [67].
Cholesterol uptake into enterocytes is mediated by NPC1L1, a transmembrane protein highly expressed in the brush border of intestinal epithelial cells. NPC1L1 functions as a sterol transporter, selectively recognizing cholesterol molecules and facilitating their incorporation into enterocytes. This process is influenced by the interaction of the LIM domain and actin-binding protein 1 (LIMA1), which regulates NPC1L1 recycling. Under cholesterol-deficient conditions, LIMA1 enhances NPC1L1 translocation to the apical membrane, increasing cholesterol uptake. Conversely, in cholesterol-sufficient states, NPC1L1 undergoes clathrin-mediated endocytosis, leading to internalization and reduced absorption [68].
Once in the enterocyte, free cholesterol is transported to the ER by Aster-B and Aster-C proteins, where it undergoes esterification by ACAT2. This esterification process enhances cholesterol hydrophobicity, preventing its premature efflux and promoting its incorporation into chylomicrons [69]. The newly formed cholesterol esters are packaged along with triglycerides, phospholipids, and apolipoproteins into nascent chylomicrons within the Golgi apparatus. These lipoprotein particles are then secreted into the lymphatic system via exocytosis and transported through the thoracic duct, thereby bypassing the hepatic portal circulation and directly entering the systemic circulation [67].
The gut microbiota also plays a key role in cholesterol absorption, influencing dietary lipid metabolism and systemic lipid homeostasis.
Under physiological conditions, primary BA are synthesized in the liver from cholesterol, predominantly via the classical CYP7A1-dependent pathway. These are later modified by the intestinal microbiota to produce secondary BA, thereby altering their biological properties [70]. Approximately 95% of BA are reabsorbed in the ileum and returned to the liver via enterohepatic circulation, a process governed by FXR and fibroblast growth factor 19 (FGF19) signaling [71, 72].
Excessive cholesterol intake alters gut microbiota composition, modifying BA biotransformation and hydrophobicity, which subsequently impacts FXR and TGR5 signaling—both of which regulate metabolic homeostasis and inflammation [63].
Cholesterol overload also alters the expression and function of BA transporters such as the ASBT, multidrug resistance-associated protein 3 (MRP3), and organic solute transporter α/β (OSTα/OSTβ), leading to the hepatic accumulation of toxic BAs that exacerbate liver injury [64].
Studies in MASLD models induced by high-cholesterol diets have demonstrated that this condition promotes intestinal dysbiosis, characterized by reduced microbial diversity and an altered abundance of key bacterial species. Specifically, increases in Escherichia and Prevotella, along with reductions in Akkermansia muciniphila and Faecalibacterium, correlate with a higher prevalence of pro-inflammatory and hydrophobic secondary BA [73–75]. These alterations contribute to intestinal barrier dysfunction, enhancing endotoxin translocation to the liver and exacerbating hepatic inflammation [74].
Chronic fructose consumption, which often coexists with high-cholesterol diets, further disrupts BA homeostasis by altering Clostridium species, leading to reduced levels of beneficial secondary BA such as lithocholic acid (LCA) and taurolithocholic acid (TLCA). This impairs intestinal FXR signaling, exacerbating metabolic dysfunction and promoting lipid accumulation and OxE in the liver [76].
Clostridium scindens, a prominent gut bacterium, catalyzes the 7α-dehydroxylation of primary BA, such as cholic acid, producing secondary bile acids (SBA) like deoxycholic acid (DCA). This microbial conversion substantially alters the composition of the BA pool and modulates FXR signaling, which plays a critical role in maintaining BA and lipid homeostasis. Activation of intestinal FXR by DCA stimulates FGF15/19 secretion, a pathway that reduces BA synthesis in the liver and exerts anti-inflammatory and antifibrotic effects, thereby attenuating key pathological features of MASLD [77]. Furthermore, C. scindens-mediated BA metabolism has been linked to colonization resistance against Clostridioides difficile, highlighting its role in maintaining gut homeostasis [78]. Modulation of C. scindens abundance or activity may thus represent a therapeutic strategy to restore FXR-FGF15/19 signaling and mitigate metabolic and inflammatory dysregulation in MASLD.
Conversely, excess cholesterol increases DCA levels, an SBA with pro-inflammatory and cytotoxic effects that accelerate MASLD progression [59]. However, certain SBA, such as hydroxydeoxycholic acid (HDCA), may partially counteract these effects by modulating gut microbiota composition and activating FXR to regulate lipid metabolism genes [65]. These interactions are summarized in Figure 2.
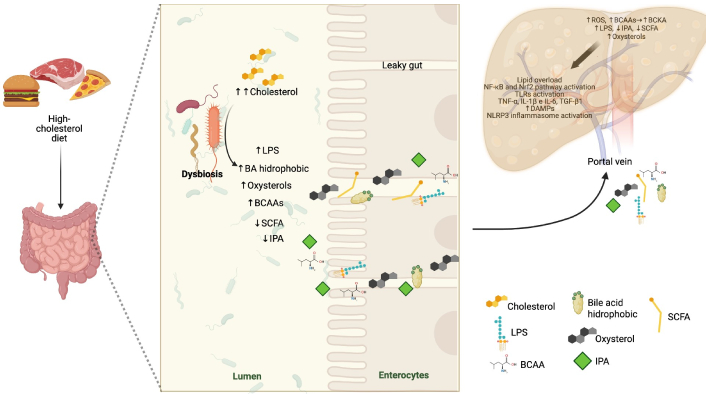
Schematic representation of the impact of a high-cholesterol diet on the gut-liver axis and its contribution to the progression of MASLD. Cholesterol intake induces intestinal dysbiosis, characterized by increased pro-inflammatory bacteria and decreased beneficial metabolites such as SCFA and indole-3-propionic acid (IPA). This microbial imbalance promotes increased intestinal permeability, allowing the translocation of lipopolysaccharides (LPS), hydrophobic bile acids, and oxysterols into the liver via the portal vein. In the liver, these compounds activate pro-inflammatory and OxE pathways, including NF-κB, Nrf2, and the NLRP3 inflammasome, leading to chronic inflammation, insulin resistance, and progressive liver damage. The figure illustrates the interplay between diet, gut microbiota, and hepatic homeostasis in MASLD—an image created with BioRender. MASLD: Metabolic dysfunction-associated steatotic liver disease; SCFA: short-chain fatty acids; OxE: oxidative stress; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; Nrf2: nuclear factor erythroid 2–related factor 2; NLRP3: NOD-like receptor family pyrin domain-containing 3; BCAAs: branched-chain amino acids; BCKA: branched-chain keto acids; ROS: reactive oxygen species; DAMPs: damage-associated molecular patterns; TNF-α: tumor necrosis factor-alpha; TGF-β1: transforming growth factor-beta 1; IL-1β: interleukin-1β. Created in BioRender. Gomez-quiroz, L. (2025) BioRender.com/kropro7
Importantly, SBA exert differential effects on hepatocyte physiology depending on their hydrophobicity and receptor interactions. Hydrophobic BA, such as DCA and LCA, are potent inducers of hepatocellular stress. DCA has been shown to activate c-Jun N-terminal kinase (JNK) signaling, leading to mitochondrial dysfunction, increased ROS generation, and apoptosis in hepatocytes. Additionally, DCA can inhibit FXR signaling, thereby impairing the regulation of BA synthesis and detoxification pathways, while simultaneously enhancing TGR5-mediated inflammatory responses in hepatic macrophages, further contributing to liver injury and fibrogenesis [59, 75].
In contrast, hydrophilic BA, such as ursodeoxycholic acid (UDCA) and HDCA have been associated with cytoprotective effects. UDCA stabilizes mitochondrial membranes, inhibits pro-apoptotic pathways, and reduces ER stress. Meanwhile, HDCA has been shown to restore gut-liver axis homeostasis by modulating the gut microbiota and by acting as an FXR agonist, promoting the expression of genes involved in BA transport and lipid metabolism, and reducing hepatic inflammation [65]. These opposing effects underscore how dysbiosis-driven shifts in BA composition—favoring accumulation of cytotoxic BA like DCA instead of protective ones like HDCA—can critically shape disease progression in MASLD.
Thus, the balance between hydrophobic and hydrophilic BA not only reflects microbial activity but also has direct implications for hepatocyte survival, immune activation, and fibrotic remodeling. Therapeutically, targeting BA composition or modulating FXR/TGR5 signaling represents a promising avenue for preventing the transition from steatosis to steatohepatitis and fibrosis in MASLD [70].
In advanced MASLD (MASH), cholesterol overload further contributes to mitochondrial dysfunction through the accumulation of CYP27A1-derived toxic cholesterol metabolites, intensifying OxE and hepatic inflammation. This process exacerbates liver damage and reinforces the pathological loop between cholesterol accumulation, disrupted BA signaling, and gut dysbiosis [58].
Given the intertwined effects of cholesterol on BA metabolism, gut microbiota composition, and hepatic signaling pathways, it is essential to distinguish the respective contributions of dietary cholesterol and intestinal absorption mechanisms. Dietary cholesterol has been shown to exacerbate liver pathology by reshaping gut microbial communities and increasing the production of lipotoxic metabolites. In murine models, high-fat/high-cholesterol diets promote the sequential development of steatosis, steatohepatitis, fibrosis, and ultimately HCC, frequently in conjunction with intestinal dysbiosis [79]. Furthermore, activation of intestinal liver X receptor alpha (LXRα) by dietary cholesterol has been implicated in accelerating hepatic carcinogenesis, highlighting the role of diet-driven signaling in the initiation of HCC [80].
Intestinal cholesterol absorption is primarily mediated by NPC1L1. Pharmacological inhibition of NPC1L1 with agents such as ezetimibe significantly reduces cholesterol absorption and has been shown to attenuate hepatic injury and tumor multiplicity in MASH-driven HCC models [15, 81]. Moreover, emerging evidence indicates that NPC1L1 expression may influence cancer prognosis, including in hepatic malignancies, positioning this transporter as a potential therapeutic target in cholesterol-associated liver disease [82].
While cholesterol is indispensable for essential physiological processes, its dysregulation can engage both tumor-promoting and tumor-suppressive pathways depending on the cellular and metabolic context. Therefore, acknowledging the divergent roles of dietary cholesterol and NPC1L1-mediated absorption is pivotal for understanding the full spectrum of cholesterol-driven liver pathophysiology and for identifying precise intervention points in MASLD-related hepatocarcinogenesis.
The intestinal barrier plays a crucial role in maintaining gut-liver homeostasis and consists of three main defense layers: the mechanical barrier, which provides structural integrity; the immune barrier, which regulates inflammatory responses; and the biological barrier, which involves the gut microbiota and its metabolic functions [3]. A high-cholesterol diet profoundly affects this barrier system, leading to increased intestinal permeability, bacterial translocation, BA dysregulation, immune activation, and hepatic inflammation—all of which contribute to the progression of MASLD.
The mechanical barrier, primarily composed of intestinal epithelial cells, goblet cells, Paneth cells, and the mucus layer, serves as a selective filter that regulates nutrient absorption while preventing the passage of harmful substances [12]. High-cholesterol diets compromise this barrier by altering TJ proteins, increasing paracellular permeability. Cholesterol-derived oxysterols induce structural and functional disruptions in key TJ components, including ZO-1, occludin, and junctional adhesion molecule-A (JAM-A), resulting in a weakened epithelial barrier that facilitates bacterial translocation and systemic inflammation [11, 83]. Murine models of cholesterol-rich diets show downregulation of claudins-1, -2, and -3, along with a reduction in goblet cells and mucin production, which further compromises the mucus layer and facilitates microbial invasion [84, 85]. Additionally, the activation of LXR, a key cholesterol sensor, has been linked to increased intestinal permeability, as it enhances sensitivity to cholesterol toxicity, thereby disrupting TJ integrity and epithelial function [86]. The upregulation of NPC1L1, the main cholesterol transporter in enterocytes, further exacerbates cholesterol absorption, leading to intracellular lipid accumulation and OxE, which weakens intestinal homeostasis [87].
The immune barrier, composed of gut-associated lymphoid tissue and specialized immune cells, is pivotal in regulating inflammatory responses to microbial and dietary stimuli [3]. Cholesterol-induced intestinal permeability allows the translocation of bacterial endotoxins, particularly LPS, into the portal circulation, triggering hepatic TLR4 activation, which promotes Kupffer cell activation, the release of inflammatory cytokines (TNF-α, IL-6, IL-1β), and insulin resistance [88, 89]. Dysbiosis associated with high-cholesterol diets leads to an overgrowth of endotoxin-producing bacteria, such as Enterobacteriaceae, Veillonellaceae, and Proteobacteria, which further amplify hepatic immune activation [90]. Moreover, the altered gut microbiota disrupts BA metabolism, increasing the abundance of toxic SBA that promote CD8+ T-cell-mediated ileitis, impair hepatic FXR signaling, and exacerbate MASLD progression [91]. Additionally, alcohol-producing bacterial strains, such as Klebsiella pneumoniae, contribute to hepatic injury through endogenous ethanol synthesis, which induces OxE, mitochondrial dysfunction, and impaired lipid metabolism [92].
The biological barrier, primarily represented by the gut microbiota, plays a critical role in cholesterol metabolism, BA homeostasis, and systemic lipid regulation. High-cholesterol diets are associated with reduced abundance of beneficial butyrate-producing bacteria, including Faecalibacterium prausnitzii, Oscillospira, and Ruminococcaceae, leading to impaired short-chain fatty acids (SCFA) production, which is essential for intestinal barrier integrity and lipid metabolism [90]. These microbial shifts affect BA profiles, impair enterohepatic circulation, and promote hepatic immune activation and metabolic dysregulation. The interconnected effects of dysbiosis on gut barrier function, BA signaling, and liver injury are schematically represented in Figure 3 [93]. Moreover, clinical evidence supports the role of intestinal dysbiosis in the progression of MASLD. Studies have shown that patients with MASLD exhibit specific alterations in gut microbiota composition, including increased abundance of Escherichia, Prevotella, Bacteroides, and Ruminococcus, along with reduced levels of A. muciniphila and Faecalibacterium prausnitzii—changes associated with hepatic inflammation and advanced fibrosis [94, 95]. Dysbiosis contributes to increased intestinal permeability, allowing translocation of microbial products that activate hepatic immune responses [96]. Additionally, altered microbial metabolism leads to elevated production of hepatotoxic SBA and dysregulated SCFA, which promote hepatic lipogenesis, OxE, and fibrosis [97]. These findings underscore the clinical relevance of microbiota-derived signals in MASLD pathogenesis and support the gut-liver axis as a therapeutic target.
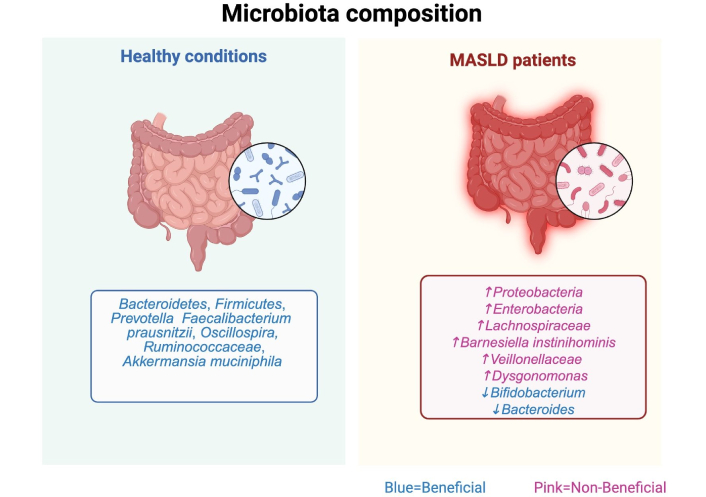
Comparative analysis of gut microbiota composition in healthy individuals and MASLD patients. MASLD is associated with reduced microbial diversity, an overrepresentation of pro-inflammatory taxa (e.g., Escherichia, Proteobacteria, Veillonellaceae), and depletion of beneficial microbes like Akkermansia muciniphila and Faecalibacterium prausnitzii. These shifts disrupt gut-liver axis homeostasis, enhance intestinal permeability, and promote LPS translocation, leading to TLR4-mediated hepatic inflammation. Altered secondary bile acid metabolism further impairs FXR signaling, exacerbating lipid accumulation and disease progression—image created by BioRender. MASLD: metabolic dysfunction-associated steatotic liver disease; TLR4: triggering hepatic toll-like receptor 4; LPS: lipopolysaccharides; FXR: farnesoid X receptor. Created in BioRender. Gomez-quiroz, L. (2025) BioRender.com/kug9ksk
The metabolic consequences of cholesterol-driven dysbiosis extend beyond microbial composition, affecting the availability of key bioactive metabolites essential for intestinal and hepatic homeostasis. SCFAs—particularly acetate, propionate, and butyrate—are significantly reduced in high-cholesterol diets due to the depletion of butyrate-producing bacteria such as Faecalibacterium prausnitzii and Ruminococcaceae [98]. SCFAs contribute to gut barrier integrity by promoting epithelial proliferation, enhancing TJ assembly, and limiting endotoxin translocation. Additionally, SCFAs act as histone deacetylase (HDAC) inhibitors, thereby modulating the transcription of pro-inflammatory genes and suppressing hepatic inflammation [99]. In the liver, SCFAs support mitochondrial function and stimulate FAO, which helps counteract hepatic lipid accumulation.
Similarly, indole derivatives such as indole-3-propionic acid (IPA) and indole-3-acetic acid (IAA), produced by microbial tryptophan metabolism, activate the aryl hydrocarbon receptor (AhR), a ligand-dependent transcription factor that regulates immune tolerance, OxE responses, and epithelial integrity. These compounds inhibit NF-κB signaling, reduce pro-inflammatory cytokine production, and promote cholesterol efflux, thereby mitigating steatosis and fibrosis [90, 100]. Reduced levels of these beneficial metabolites have been consistently reported in MASLD, contributing to disease progression.
From a therapeutic perspective, strategies aimed at restoring microbial metabolite profiles—such as the use of prebiotics, dietary fibers, and probiotic strains enriched in Faecalibacterium and Bifidobacterium—have demonstrated promising effects in experimental models. These approaches may enhance SCFA and indole production, strengthen gut barrier function, and attenuate hepatic inflammation, highlighting their potential in MASLD management [101].
Altogether, the interplay between intestinal dysbiosis, cholesterol overload, and BA imbalance creates a pathological cycle that reinforces hepatic inflammation, metabolic stress, and lipid accumulation in MASLD. The progressive disruption of gut barrier integrity, immune dysregulation, and microbial metabolism ultimately drives the transition from MASLD to advanced liver disease, highlighting the gut-liver axis as a critical target for future therapeutic interventions.
OxE, characterized by an imbalance between ROS production and antioxidant defense mechanisms, leads to oxidative damage of cellular components, including lipids, proteins, and DNA. This pathological condition is a major contributor to MASLD progression, particularly in the context of excessive cholesterol accumulation. A high-cholesterol diet exacerbates mitochondrial dysfunction, ER stress, and chronic inflammation, all of which contribute to hepatic injury and the transition from steatosis to fibrosis and HCC [26, 30]. Recently, ferroptosis has also been identified as an additional mechanism of cell death induced by OxE and cholesterol overload in the liver [102].
A growing body of clinical and epidemiological evidence supports the link between dyslipidemia and the progression of MASLD. Dyslipidemia, often characterized by elevated triglycerides and reduced HDL cholesterol, is a core component of the atherogenic lipid profile commonly found in MASLD patients [103, 104]. This dysregulated lipid state not only signals heightened cardiovascular risk but also correlates with liver disease severity.
Multicenter studies involving patients with MASH have identified serum cholesterol and triglycerides as independent predictors of advanced hepatic fibrosis [105]. Additionally, remnant cholesterol has emerged as a modifiable risk factor that contributes to hepatic fat accumulation and inflammation, thereby promoting disease progression [4].
Population-based analyses further support the strong association between MASLD and comorbid metabolic disorders such as obesity, T2DM, and dyslipidemia, which share overlapping pathophysiological mechanisms that exacerbate hepatic injury [5]. Novel clinical markers, such as the neutrophil-to-HDL cholesterol ratio (NHR), have also been proposed to capture the interplay between lipid metabolism and systemic inflammation, with elevated NHR values correlating with increased MASLD severity [106].
Moreover, clinical trials and observational studies suggest that lipid-lowering therapies—particularly statins—may offer hepatoprotective effects. In addition to improving serum lipid profiles, statins exhibit anti-inflammatory and antifibrotic properties, with agents like atorvastatin and rosuvastatin showing improvements in hepatic enzymes and fibrosis indices in MASLD patients [104]. However, additional large-scale randomized controlled trials are needed to confirm long-term safety and therapeutic efficacy in this population.
Together, these data underscore the clinical significance of lipid metabolism in MASLD and support the integration of epidemiological and clinical findings into the pathophysiological framework of the disease, setting the stage for exploring the underlying molecular mechanisms involved.
Mitochondria are central regulators of cellular energy metabolism and redox balance; however, excessive cholesterol accumulation can profoundly impair their function. Cholesterol-induced membrane rigidification alters mitochondrial membrane potential (ΔΨm), impairs electron transport chain activity, and leads to inefficient ATP production along with increased ROS generation [11, 34]. Cholesterol overload also affects mitochondrial biogenesis, promoting cytochrome c release and caspase activation, which triggers apoptosis and hepatocyte death [107]. Additionally, cholesterol oxidation products, known as oxysterols, worsen mitochondrial dysfunction by amplifying oxidative damage and inflammatory signaling [11]. This metabolic stress compromises hepatocellular resilience, leading to sustained tissue injury and greater susceptibility to hepatocarcinogenesis.
Recent clinical studies have demonstrated that plasma levels of specific oxysterols, such as 22R-OHC and 25-OHC, are significantly elevated in patients with MASLD, correlating with hepatic steatosis and inflammation severity [108, 109]. These oxysterols contribute to liver injury through several mechanisms, including pro-inflammatory signaling via LXRα, respiratory chain impairment, and enhanced OxE, ultimately promoting hepatocyte apoptosis [108, 110, 111]. Furthermore, the identification of elevated 26-OHC in steatotic hepatic organoids highlights the potential of oxysterols as diagnostic biomarkers and therapeutic targets in MASLD [109, 112, 113]. These findings underscore the translational relevance of oxysterol-mediated pathways in the human pathogenesis of MASLD.
The excessive accumulation of cholesterol within mitochondria also promotes lipotoxicity, a condition in which lipid metabolites induce OxE and inflammation. Lipid peroxidation products, such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), act as toxic mediators that further damage mitochondrial membranes, impair electron transport chain activity, and enhance the secretion of pro-inflammatory cytokines [114, 115].
Cholesterol-induced mitochondrial dysfunction not only disrupts oxidative phosphorylation and promotes ROS generation but also leads to mitochondrial membrane permeabilization and the release of mitochondrial DNA (mtDNA) into the cytosol. mtDNA acts as a potent damage-associated molecular pattern (DAMP) that activates pattern recognition receptors such as TLR9 and the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome. This dual activation triggers a pro-inflammatory cascade characterized by IL-1β and IL-18 release, further promoting hepatic inflammation and fibrogenesis. Notably, mtDNA-driven inflammasome activation has been implicated in the amplification of sterile inflammation in MASLD and represents a key mechanistic link between OxE and innate immune activation [26, 116].
Given its central role in bridging mitochondrial dysfunction with innate immune activation, targeting mtDNA-mediated inflammasome signaling may represent a promising therapeutic avenue in MASLD.
Recent findings indicate that cholesterol accumulation within mitochondria sensitizes hepatocytes to OxE and apoptotic signals, exacerbating liver damage in MASLD. This leads to increased mitochondrial fragmentation, bioenergetic failure, and resistance to apoptosis, which in turn accelerates disease progression and enhances susceptibility to fibrosis and HCC [34].
Excessive cholesterol accumulation disrupts ER homeostasis, leading to ER stress and the activation of the unfolded protein response (UPR), a cellular mechanism to restore proteostasis. Under chronic cholesterol overload, the UPR becomes maladaptive, contributing to hepatocyte apoptosis and inflammation [117]. Cholesterol-induced ER stress is primarily mediated through three major UPR pathways: the PERK-eIF2α-CHOP axis, which initially reduces protein synthesis to alleviate ER burden but ultimately promotes apoptosis [118]; the IRE1-XBP1 pathway, which enhances ER-associated degradation to clear misfolded proteins but, when persistently activated, leads to NF-κB-driven pro-inflammatory cytokine production [119]; and the activating transcription factor 6 (ATF6) pathway, which regulates genes involved in ER expansion and protein folding but also contributes to inflammation when stress persists [118].
Additionally, cholesterol overload disrupts ER calcium homeostasis, leading to excessive cytosolic calcium release, which exacerbates mitochondrial dysfunction by inducing mitochondrial permeability transition pore (mPTP) opening, further increasing ROS production and triggering hepatocyte apoptosis [120]. Cholesterol-driven ER stress also promotes Kupffer cell activation, resulting in increased secretion of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β, which perpetuate chronic hepatic inflammation [117, 121]. Additionally, cholesterol oxidation products, known as oxysterols, act as potent activators of the NLRP3 inflammasome, primarily by inducing lysosomal destabilization and ROS production, both of which serve as critical activation signals. Oxysterols promote lysosomal membrane permeabilization, leading to the release of cathepsins, which trigger inflammasome assembly. Simultaneously, mitochondrial dysfunction induced by oxysterols increases ROS levels, further amplifying NLRP3 activation. This cascade results in caspase-1 activation, promoting the maturation and secretion of IL-1β and TNF-α, key cytokines that exacerbate OxE and hepatocellular damage. The chronic inflammatory response driven by these cytokines enhances NF-κB signaling, sensitizing Kupffer cells and hepatocytes to inflammatory injury. Ultimately, these processes contribute to hepatic fibrosis and increased susceptibility to HCC by perpetuating inflammation and oxidative damage [11].
Furthermore, cholesterol accumulation in the ER impairs the hepatocyte growth factor (HGF)/c-Met signaling pathway, which plays a crucial role in liver regeneration. The inhibition of this pathway prevents effective tissue repair, thereby promoting fibrosis progression and accelerating MASLD toward cirrhosis and HCC. These interconnected processes create a pathological loop in which ER stress, calcium dysregulation, and inflammation exacerbate hepatocellular damage, increasing susceptibility to fibrosis and HCC [115, 122].
Ferroptosis has emerged as a pivotal mechanism of hepatocellular injury in MASLD, particularly in the setting of OxE, lipid peroxidation, and iron dysregulation. This regulated, iron-dependent form of cell death is driven by the accumulation of lipid hydroperoxides and the inactivation of glutathione peroxidase 4 (GPX4), a key antioxidant enzyme responsible for reducing toxic lipid peroxides into non-reactive lipid alcohols [123, 124].
In MASLD, the metabolic environment—marked by hepatic steatosis, increased ROS generation, and, in some cases, iron accumulation—creates a permissive context for ferroptotic processes to be initiated and sustained [125, 126].
A critical mediator in this pathway is 7α-hydroperoxycholesterol (7α-OOH), an oxysterol produced by the oxidation of free cholesterol. Under physiological conditions, GPX4 neutralizes lipid hydroperoxides derived from 7α-OOH; however, when GPX4 is deficient or inactivated—either genetically or due to sustained OxE—lipid peroxides accumulate, triggering membrane damage and ferroptotic cell death [102, 127]. The presence of redox-active iron further amplifies this cascade through the Fenton reaction, exacerbating lipid oxidation and cell injury.
Experimental studies have shown that hepatocytes in MASLD display reduced GPX4 expression and elevated levels of lipid peroxidation, reinforcing the pathological role of ferroptosis in the progression of liver damage [102]. The integration of ferroptosis into the mechanistic framework of cholesterol-induced hepatocellular injury enhances our understanding of hepatocyte vulnerability in MASLD and may offer novel therapeutic targets based on redox modulation and iron homeostasis.
Chronic OxE, mitochondrial dysfunction, and unresolved ER stress establish a self-perpetuating pathological loop that drives hepatic fibrogenesis, a critical turning point in the progression of MASLD toward cirrhosis and HCC. Persistent overproduction of ROS and lipid peroxidation not only cause direct hepatocellular injury but also function as profibrotic stimuli that activate hepatic stellate cells (HSC), the principal mediators of liver fibrosis. ROS-induced damage leads to the release of DAMPs, including mtDNA and oxidized lipids, which interact with TLRs and activate the NLRP3 inflammasome in Kupffer cells, amplifying the secretion of inflammatory cytokines (e.g., TNF-α, IL-6, IL-1β) and propagating fibrogenic signaling [30, 115].
Mitochondrial ROS generated under conditions of cholesterol overload further aggravate hepatocellular damage and serve as key activators of HSC [34]. These ROS, in conjunction with lipid peroxidation products such as 4-HNE and MDA, facilitate the release of mtDNA, which activates TLR9 and the NLRP3 inflammasome in Kupffer cells [11]. The resultant cytokine secretion creates a profibrotic microenvironment that perpetuates HSC activation. Once activated, HSCs undergo a phenotypic transformation into myofibroblast-like cells, initiating extracellular matrix (ECM) deposition and driving the transition from steatosis to hepatic fibrosis in MASLD [30].
Recent studies have further demonstrated that dietary cholesterol directly contributes to HSC activation through intracellular accumulation and inflammatory signaling pathways. Free cholesterol buildup sensitizes HSC to transforming growth factor-beta (TGF-β) signaling, amplifying profibrotic gene expression [e.g., collagen type I alpha chain (COL1A1)], alpha-smooth muscle actin (α-SMA) even in the absence of overt hepatocellular injury [30]. Simultaneously, cholesterol-induced activation of TLR4 in Kupffer cells and HSC promotes the production of TNF-α and IL-1β, reinforcing the fibrogenic microenvironment and sustaining chronic inflammation [11]. These findings indicate that cholesterol functions not only as a metabolic substrate but also as an active regulator of fibrosis and a driver of disease progression [81].
Beyond its intracellular effects, cholesterol promotes HSC activation via receptor-mediated pathways that contribute to fibrogenesis and the establishment of a tumor-permissive niche. LDL receptor-mediated uptake leads to intracellular cholesterol accumulation in HSC, inducing their transdifferentiation into myofibroblast-like cells. These activated cells express fibrogenic markers such as α-SMA and COL1A1 and actively remodel the ECM. Concurrently, oxidized LDL (oxLDL) binds to lectin-like oxidized LDL receptor-1 (LOX-1), a scavenger receptor expressed on HSC, triggering OxE and NF-κB signaling. This pathway enhances the secretion of fibrogenic and inflammatory cytokines, particularly TGF-β1, exacerbating matrix deposition, promoting liver stiffness, and impairing immune cell function—hallmarks of fibrotic progression toward HCC [81, 128].
Mitochondrial cholesterol accumulation further intensifies fibrogenic responses by impairing mitochondrial membrane fluidity, disrupting electron transport chain function, and increasing ROS production through mechanisms such as ROS-induced ROS release involving mPTP opening [129]. These mitochondrial ROS act as intracellular messengers that activate quiescent HSC, stimulating their proliferation, contractility, and ECM synthesis [130, 131]. Under ER stress conditions, mitochondrial dysfunction converges with the inositol-requiring enzyme 1α (IRE1α)/NADPH oxidase 4 (NOX4) axis: activation of IRE1α upregulates NOX4, an important ROS source in HSC [132]. Mitochondrial ROS also contribute to NLRP3 inflammasome activation, leading to IL-1β secretion and reinforcing profibrotic responses [133, 134]. These interrelated pathways represent promising therapeutic targets. For instance, the mitochondria-targeted antioxidant peptide SS-31, which stabilizes mitochondrial membranes and reduces ROS levels, has shown efficacy in suppressing HSC activation and slowing fibrosis progression [133]. Similarly, pharmacological inhibition of the IRE1α/NOX4 axis has demonstrated antifibrotic effects in preclinical models [132]. While hepatic fibrosis is multifactorial, the mechanistic role of mitochondrial cholesterol in HSC activation offers crucial insights into MASLD pathogenesis and supports the development of targeted antifibrotic interventions [135].
Clinically, this pathological cascade culminates in progressive hepatic fibrosis [121], which is typically evaluated using histological scoring or non-invasive imaging and remains a principal contributor to cirrhosis and HCC development in MASLD. Cholesterol overload also amplifies TGF-β and platelet-derived growth factor (PDGF) signaling—two pivotal regulators of HSC activation. TGF-β, largely secreted by Kupffer cells and damaged hepatocytes, binds to TGF-β receptor type I, initiating Smad2/3-dependent transcriptional programs that drive COL1A1 synthesis and ECM deposition. Cholesterol enrichment in lipid rafts stabilizes these receptors, prolonging Smad activation and sustaining fibrogenic signaling. Meanwhile, PDGF-BB isoform (a homodimer of PDGF-B subunits) binds to PDGF receptor beta (PDGFR-β) on HSC, activating RhoA/ROCK signaling that enhances HSC migration and contractility, contributing to scar formation and hepatic stiffness [30, 115].
Furthermore, cholesterol-induced OxE and inflammation contribute to the establishment of an oncogenic microenvironment that facilitates HCC initiation and progression. ROS-mediated DNA damage induces genomic instability and activates oncogenic pathways, including NF-κB and STAT3, which enhance the survival and proliferation of pre-malignant hepatocytes [6, 11]. Additionally, cholesterol metabolites, particularly oxysterols, modulate LXR and SREBP2 signaling, leading to altered lipid metabolism and metabolic reprogramming that further favors tumorigenesis [6, 11].
Among oxysterols, 27-OHC, produced via CYP27A1 activity, has emerged as a key mediator connecting cholesterol metabolism with hepatocarcinogenesis. Acting as a selective estrogen receptor modulator, 27-OHC binds estrogen receptor alpha (ERα) in hepatocytes, activating STAT3 signaling—a central pathway promoting cell proliferation, survival, and migration [136–138]. This interaction induces conformational changes in ERα, facilitating coactivator recruitment and STAT3 phosphorylation, thereby promoting hepatic inflammation, fibrogenesis, and carcinogenesis [128, 139]. The dual role of 27-OHC as both a metabolic intermediate and signaling effector positions it as a critical driver of hepatocarcinogenesis in cholesterol-overloaded livers. Moreover, its context-specific effects suggest modulation by sex hormones and genetic background, offering opportunities for sex-specific therapeutic interventions [140].
Emerging studies have also implicated 27-OHC in HCC metastasis through the SULT2A1 axis. Downregulation of SULT2A1, a key enzyme responsible for oxysterol sulfation, increases intracellular 27-OHC accumulation and correlates with enhanced metastatic potential and poor prognosis in HCC patients [141]. Furthermore, chronic exposure to elevated 27-OHC levels disrupts redox balance via GRP75 activation, promoting mitochondrial dysfunction and metabolic reprogramming in tumor cells [142]. Although studies directly quantifying serum 27-OHC in relation to HCC risk are limited, data from large epidemiological cohorts such as EPIC-Heidelberg support a key role for cholesterol metabolism alterations in liver cancer development, underscoring the translational relevance of oxysterol-related pathways in early hepatocarcinogenesis [143, 144]. Additional clinical studies further highlight the dual contribution of hypercholesterolemia and oxysterol accumulation to both HCC initiation and progression [145].
These findings suggest that 27-OHC may serve not only as a mechanistic contributor to HCC but also as a biomarker for disease progression and a potential target for therapeutic intervention in cholesterol-driven liver malignancies.
A pivotal mechanism by which cholesterol promotes hepatocarcinogenesis is immune evasion. Cholesterol impairs NK cell functionality by suppressing the expression of ligands for natural killer group 2 member D (NKG2D) on hepatocytes and altering lipid raft composition, thereby reducing NK cell-mediated tumor surveillance [146]. Additionally, cholesterol accumulation upregulates programmed death ligand-1 (PD-L1) on hepatocytes, engaging PD-1 on T cells, which fosters an immunosuppressive tumor microenvironment that allows malignant hepatocytes to evade immune clearance [146]. Hypercholesterolemic conditions also impair NK cell cytotoxicity by disrupting granzyme B (GrB) exocytosis due to lipid raft disorganization, as evidenced by reduced CD107a surface expression without changes in intracellular GrB content [147]. Moreover, GrB cleaves NOTCH1, diminishing its transcriptional activity; paradoxically, cholesterol accumulation can also promote NOTCH1 nuclear translocation, contributing to cellular senescence and dysfunction in immune and hepatic cells [148, 149]. These findings highlight a complex cholesterol–GrB–NOTCH1 axis that disrupts NK cell surveillance and facilitates oncogenic progression, particularly in MASLD contexts where chronic inflammation, lipid overload, and gut-derived endotoxemia converge.
Thus, cholesterol-driven immune dysregulation complements its metabolic and fibrogenic effects, reinforcing HCC progression within a microenvironment characterized by chronic injury, immune escape, and impaired tissue barrier integrity.
Finally, Simoni-Nieves et al. [30] demonstrated that cholesterol overload not only amplifies HSC activation but also modulates ECM composition, increasing fibronectin and laminin deposition, which creates a scaffold supporting tumor cell adhesion, invasion, and metastasis. This sustained fibrotic, inflammatory, and immunosuppressive microenvironment establishes a permissive niche for HCC initiation and progression.
Beyond representing a reparative response to chronic liver injury, fibrosis actively contributes to hepatocarcinogenesis by disrupting liver architecture, increasing parenchymal stiffness, and inducing hypoxia, which promotes epithelial-to-mesenchymal transition (EMT), angiogenesis, and malignant transformation. Activated HSC within this fibrotic niche secrete cytokines (e.g., TGF-β, IL-6), chemokines, and ROS, amplifying STAT3 and NF-κB signaling in hepatocytes, while ECM remodeling enzymes and deposition of fibronectin and laminin generate a scaffold that facilitates tumor cell adhesion, migration, and invasion [26, 30, 107]. Clinically, the degree of fibrosis remains the strongest predictor of liver-related outcomes in MASLD, even in the absence of classical cirrhosis, highlighting fibrosis as an active pathogenic driver that facilitates the accumulation, survival, and clonal expansion of genetically unstable hepatocytes [59, 115].
Thus, fibrosis should be recognized not merely as a histological endpoint but as a dynamic and transitional state that bridges chronic metabolic injury to malignant transformation, representing a critical target for both antifibrotic therapies and cancer prevention strategies in MASLD.
Building upon this framework, the research group led by Geoffrey Farrell has made seminal contributions to understanding MASLD progression, particularly implicating cholesterol crystallization within hepatocyte LD as a potent trigger of hepatocellular injury and inflammation in both murine and human MASH and identifying crystalline cholesterol as a driver of disease progression [150, 151]. They also demonstrated that activation of innate immune receptors such as TLR9 and the NLRP3 inflammasome in response to lipotoxic stimuli exacerbates liver inflammation and fibrosis, identifying these as key therapeutic targets [152, 153]. These findings have been extended to pharmacological studies, where reversal of MASH pathology was achieved through PPAR-δ agonism, supporting the therapeutic potential of modulating lipid-induced inflammation [154].
Complementing these findings, Gutiérrez-Ruiz MC, and collaborators have demonstrated that cholesterol overload promotes mitochondrial dysfunction, ROS-mediated hepatocyte apoptosis, and HSC activation, while also altering ECM composition and contributing to immune evasion and tumor cell invasion. Together, these studies reinforce the concept of MASLD as a progressive disease spectrum in which cholesterol-driven lipotoxicity, immune dysregulation, and fibrotic remodeling play central roles in hepatocarcinogenesis [30, 34].
The increasing prevalence of MASLD underscores the urgent need for novel therapeutic strategies to mitigate the deleterious effects of cholesterol accumulation, OxE, and immune dysregulation. Despite advancements in understanding the molecular mechanisms underlying cholesterol-induced hepatic dysfunction, current treatment options remain limited. Future research must focus on targeting key metabolic pathways, restoring cellular homeostasis, and exploring microbiota-based interventions to prevent MASLD progression toward fibrosis and HCC.
Given cholesterol’s pivotal role in MASLD pathogenesis, developing strategies to regulate its metabolism and maintain hepatic lipid homeostasis is of primary importance. FXR agonists, such as obeticholic acid (OCA), have demonstrated potential in reducing hepatic lipid accumulation by enhancing BA metabolism and modulating cholesterol transport [6]. OCA, a potent FXR agonist, has shown promise in the treatment of MASLD and its progressive forms, including MASH. Through FXR activation, OCA modulates BA synthesis, cholesterol metabolism, glucose homeostasis, and hepatic inflammation—key mechanisms implicated in MASLD progression. In the REGENERATE trial, OCA improved liver fibrosis by ≥ 1 stage in 22.4% of patients versus 9.6% in the placebo group [155]. However, these benefits were accompanied by adverse effects such as dose-dependent pruritus and increased LDL cholesterol levels, raising concerns regarding cardiovascular risk and necessitating lipid-lowering co-therapy. Moreover, emerging data suggest that FXR-SHP signaling may exert context-dependent pro-fibrotic effects in hepatic and biliary cells [156]. As such, newer non-steroidal FXR agonists are under investigation to preserve efficacy while mitigating these limitations. Long-term studies are needed to evaluate their efficacy in preventing disease progression and reducing fibrosis risk.
Other nuclear receptor agonists, including PPAR modulators, have emerged as potential candidates due to their ability to enhance FAO, reduce lipid accumulation, and improve insulin sensitivity [11]. Combining FXR and PPAR agonists may provide synergistic effects in controlling cholesterol-driven metabolic dysfunction and MASLD-related inflammation [40].
Beyond nuclear receptor modulation, targeting cholesterol absorption and transport pathways presents another promising avenue for MASLD management. NPC1L1 inhibitors, such as ezetimibe, have been proposed to reduce intestinal cholesterol absorption, while ATP-binding cassette (ABC) transporters such as ABCA1 (transporter A1) and ABCG5/G8 (transporter G5 and G8) play crucial roles in regulating cholesterol efflux from hepatocytes [34]. Enhancing the function of these transporters through pharmacological or gene-based approaches could facilitate cholesterol clearance and prevent hepatic lipid overload. Additionally, BA sequestrants, which bind BA in the intestine and prevent their reabsorption, could be further explored as potential MASLD therapies [30].
Furthermore, a more nuanced understanding of the clinical use of these pharmacological agents is required. Despite its documented efficacy in improving fibrosis in MASLD and MASH, the clinical use of OCA is limited by dose-dependent pruritus and increased LDL-cholesterol levels, which have raised concerns regarding cardiovascular risk and necessitate lipid-lowering co-therapies [156–158]. These limitations have prompted the development of newer non-steroidal FXR agonists aimed at preserving therapeutic benefits while minimizing adverse effects [156].
In parallel, NPC1L1 inhibitors such as ezetimibe, which reduce intestinal cholesterol absorption, have demonstrated hepatoprotective effects in preclinical models of steatosis and insulin resistance, including reduced hepatic lipid accumulation [159]. While clinical studies in MASLD are still limited, ezetimibe’s favorable metabolic profile supports further investigation. However, the variability in treatment response highlights the need for more robust clinical trials to confirm long-term efficacy and safety in this population.
Building on the evidence discussed in the previous section regarding ferroptosis as a key mechanism of hepatocellular injury in MASLD, therapeutic strategies aimed at modulating this pathway have gained increasing attention. Inhibition of ferroptosis has been shown to reduce liver damage in experimental MASLD models, particularly by preserving GPX4 activity and limiting lipid peroxidation [102].
While GPX4 is widely recognized as the principal enzymatic barrier against ferroptosis, emerging evidence supports the therapeutic potential of targeting additional, GPX4-independent antioxidant systems to protect hepatocytes from lipid peroxidation-induced cell death. Among these, ferroptosis suppressor protein 1 (FSP1) has been identified as a potent inhibitor of ferroptosis through its ability to reduce coenzyme Q10 (ubiquinone) to ubiquinol, thereby neutralizing lipid peroxides at the plasma membrane independently of glutathione [160]. Likewise, dihydroorotate dehydrogenase (DHODH) exerts a similar protective function within mitochondria, reducing coenzyme Q and limiting oxidative damage in cells with high mitochondrial activity [161].
In parallel, immune-mediated mechanisms also contribute significantly to hepatic inflammation and fibrosis in MASLD. TLRs, particularly TLR4 and TLR9, are activated by microbial components such as LPS and unmethylated CpG motifs, leading to Kupffer cell activation, cytokine release (e.g., TNF-α, IL-6, IL-1β), and insulin resistance [88, 89]. Pharmacological inhibition of TLR signaling has shown potential to attenuate hepatic inflammation and may represent a viable therapeutic avenue. Moreover, the use of IL-15 agonists has gained attention due to their capacity to restore NK cell function. NK cells play a critical role in limiting fibrosis by inducing apoptosis in activated HSC and secreting antifibrotic cytokines such as interferon gamma (IFN-γ) [162, 163]. IL-15 superagonists enhance NK cell proliferation and cytotoxicity, even in immunosuppressive environments driven by TGF-β1, suggesting a novel immunotherapeutic strategy. However, systemic toxicity associated with excessive NK cell activation remains a concern, underscoring the need for controlled dosing and further clinical evaluation [164]. These immunomodulatory strategies complement metabolic interventions and may offer new directions to modulate inflammation and fibrosis in MASLD.
In addition to these defined pathways, GPX4-independent enzymatic activity capable of reducing phosphatidylcholine hydroperoxides (PC-OOH) has been identified, suggesting the existence of additional, yet uncharacterized, protective systems that could be exploited for therapeutic purposes [165]. Exploiting these alternative ferroptosis defense mechanisms could offer new strategies to complement GPX4-dependent approaches, especially in cases where GPX4 expression is impaired, as observed in subsets of MASLD patients. Targeting FSP1, DHODH, or yet-unknown reductive pathways could therefore represent a promising avenue to reduce lipid peroxidation, preserve hepatocyte viability, and slow disease progression.
Beyond OxE and ferroptosis regulation, pharmacological interventions that directly modulate hepatic lipid metabolism continue to emerge. GLP-1 receptor agonists, such as semaglutide, have gained attention not only for their glucose-lowering and weight-reducing effects in T2DM, but also for their broader metabolic benefits, including modulation of lipid homeostasis. Recent evidence suggests that these agents can reduce hepatic cholesterol levels, potentially through activation of AMPK, which in turn inhibits ACC, a central regulator of fatty acid synthesis and oxidation [166, 167]. Through this AMPK/ACC signaling pathway, GLP-1 agonists may attenuate hepatic lipid accumulation and promote cholesterol efflux. Moreover, emerging GLP-1 analogues with cholesterol-targeted activity support a direct role in hepatic cholesterol metabolism [168]. These findings highlight the potential of GLP-1 receptor agonists as adjunctive therapies in conditions characterized by hepatic lipid dysregulation, including MASLD.
Emerging evidence suggests that the gut microbiota plays a crucial role in cholesterol metabolism and MASLD progression. Dysbiosis, characterized by an imbalance in gut microbial composition, has been linked to altered BA metabolism, increased cholesterol absorption, and systemic inflammation. Strategies targeting the gut microbiota have shown promise in modulating hepatic lipid homeostasis and mitigating MASLD progression.
Probiotic and prebiotic supplementation has been explored as a potential therapeutic approach. Specific probiotic strains, such as Lactobacillus plantarum and Bifidobacterium longum, have demonstrated the ability to reduce intestinal cholesterol absorption, enhance BA metabolism, and improve gut barrier integrity [169]. Additionally, fecal microbiota transplantation (FMT) has emerged as a promising strategy to restore a healthy microbiome and counteract cholesterol-driven hepatic dysfunction [91].
Among the most studied next-generation probiotics, A. muciniphila has garnered interest for its potential to modulate the gut-liver axis in MASLD. Its administration has been shown to restore intestinal barrier function, reduce systemic inflammation, and shift BA composition in animal models. However, while A. muciniphila influences BA metabolism through the intestinal FXR–FGF15 endocrine axis; a direct mechanistic link with hepatic CYP7A1 expression remains poorly elucidated. Studies report reduced levels of SBA, such as deoxycholic and lithocholic acids, following A. muciniphila supplementation, yet these effects appear to be secondary to broader microbial and signaling shifts [170, 171]. This highlights the complexity of enterohepatic regulation and the need for further research into A. muciniphila’s molecular impact on hepatic gene expression and BA synthesis.
Another approach involves targeting microbial-derived metabolites, such as SCFA, which play a critical role in modulating host metabolism and inflammation. SCFA, including butyrate and propionate, have been shown to enhance mitochondrial function, reduce OxE, and regulate immune responses in the liver [172]. Future studies should further explore the synergistic effects of microbiota-targeted therapies in combination with lipid-lowering agents to develop personalized interventions for MASLD patients.
Considering these findings, personalized dietary strategies designed to restore gut microbial balance and increase beneficial metabolites such as SCFA and indole derivatives are gaining traction as adjunctive interventions for MASLD. High-cholesterol diets are known to reduce the abundance of butyrate-producing bacteria such as Faecalibacterium prausnitzii and Ruminococcaceae, impairing intestinal barrier integrity and promoting hepatic inflammation [90, 98]. SCFA not only support epithelial TJ and suppress endotoxemia but also act as HDAC inhibitors to reduce pro-inflammatory gene expression [79, 99]. Similarly, microbial indole metabolites modulate hepatic cholesterol metabolism and immune responses via activation of the AhR signaling pathway [100]. Interventions incorporating dietary fibers, prebiotics, and next-generation probiotics such as Faecalibacterium and Bifidobacterium have shown promise in restoring these metabolites and mitigating hepatic injury [101, 173]. Integrating these personalized approaches into MASLD therapy could offer a more comprehensive and sustainable management strategy.
These advances in microbiota-targeted and dietary interventions underscore the importance of systemic metabolic regulation and gut-liver signaling in MASLD pathogenesis. Nevertheless, intracellular organelles such as mitochondria and the ER are also pivotal mediators of lipid homeostasis, OxE, and cell survival. Targeting these organelles offers additional therapeutic avenues aimed at restoring hepatocellular integrity and preventing MASLD progression.
Cholesterol-induced mitochondrial dysfunction is strongly correlated with hepatic OxE, suggesting that therapeutic strategies to restore mitochondrial homeostasis may be key to MASLD treatment. Mitochondrial protective agents, including AMPK activators (e.g., metformin) and mitochondrial biogenesis enhancers (e.g., PGC-1α agonists), have shown potential in reducing cholesterol-induced ROS generation and preventing bioenergetic failure [174]. Further studies should investigate the combined effects of mitochondrial-targeted therapies with lipid-lowering agents to determine their potential in providing enhanced metabolic protection against MASLD progression.
Since cholesterol accumulation disrupts ER homeostasis, leading to prolonged ER stress and inflammation, modulating UPR signaling and protein folding mechanisms could provide an effective therapeutic approach. Chemical chaperones such as the tauroursodeoxycholic acid (TUDCA) and 4-phenylbutyrate (4-PNB) have been investigated for their ability to alleviate cholesterol-induced ER stress and improve hepatocyte survival [175, 176]. Further research is needed to assess their clinical applicability and long-term effects on MASLD-associated fibrosis and inflammation.
Among emerging mitochondrial-targeted strategies, MitoQ, a mitochondria-accumulating antioxidant, has shown promise in mitigating OxE and improving mitochondrial function. By reducing mitochondrial ROS and activating the Nrf2 antioxidant response, MitoQ may attenuate hepatic inflammation and fibrosis in preclinical models of metabolic liver disease [177, 178]. Given the role of OxE in HCC development, MitoQ has also been proposed as a potential adjunct therapy to prevent MASLD progression toward malignant transformation.
In parallel, CYP27A1, a mitochondrial enzyme involved in cholesterol hydroxylation and BA synthesis, has been identified as a potential therapeutic target in certain cancers due to its role in producing 27-OHC—a cholesterol-derived oxysterol with immunomodulatory and pro-tumorigenic properties. While CYP27A1 inhibition has not yet been explored specifically in MASLD or HCC, its contribution to 27-OHC accumulation and downstream inflammatory signaling suggests a plausible role in liver disease progression. Experimental inhibition of CYP27A1 has shown efficacy in reducing tumor burden and enhancing immunotherapy in extrahepatic malignancies [179]. Therefore, targeting CYP27A1 may represent a promising avenue for modulating cholesterol-driven hepatic injury and tumorigenesis, warranting further investigation in MASLD-related HCC.
Chronic inflammation is a key driver of MASLD progression, making hepatic immune modulation a promising therapeutic avenue. Cholesterol overload impairs NK cell function, facilitating hepatic fibrosis and carcinogenesis. Strategies aimed at restoring NK cell cytotoxicity, such as IL-15 agonists, are under investigation as potential immunotherapeutic approaches for MASLD and liver cancer [122, 180].
Additionally, therapies that target hepatic macrophage activation and Kupffer cell-mediated inflammation may help reduce cholesterol-driven hepatic damage. Inhibition of TLR signaling, particularly TLR4 and TLR9, has been proposed as a mechanism to block pro-inflammatory cascades [121]. Monoclonal antibodies targeting TNF-α and IL-6 may also prove beneficial in reducing cholesterol-induced inflammation and preventing fibrosis progression [30].
Recent advances in genome editing have introduced CRISPR/Cas9 as a precise and powerful tool to modulate genes involved in hepatic cholesterol regulation. Two key targets of interest are CYP7A1, which encodes the rate-limiting enzyme in the conversion of cholesterol to BA, and SREBP2, a master transcription factor that governs cholesterol biosynthesis and uptake. Experimental studies have demonstrated that CRISPR/Cas9-mediated modulation of CYP7A1 can enhance BA synthesis and promote hepatic cholesterol catabolism, thereby reducing lipid accumulation in the liver [181, 182]. Conversely, targeted suppression of SREBP2 has been shown to lower cholesterol synthesis under hyperlipidemic conditions, helping to restore lipid balance [183]. These gene-level interventions offer mechanistic specificity that could benefit MASLD patients with persistent dysregulation of cholesterol metabolism. However, challenges remain regarding off-target effects, control over gene expression, and the complex interplay between lipid pathways. Further research is essential to optimize these approaches for clinical application and to ensure safe, long-term modulation of hepatic lipid homeostasis [184, 185].
In parallel, recent evidence has highlighted the intricate role of epigenetic regulation in cholesterol metabolism and its implications for MASLD progression and hepatocarcinogenesis. Epigenetic mechanisms, including DNA methylation, histone modifications, and non-coding RNAs, influence the expression of key metabolic genes such as PPARα, SREBP2, and CYP7A1. For instance, PPARα silencing via DNA hypermethylation contributes to dysregulated lipid metabolism and hepatic fibrosis through pathways such as ferroptosis and pyroptosis [186]. Likewise, histone acetylation dynamics regulate cholesterol transport and storage, with disruptions in these mechanisms promoting metabolic imbalance [187].
These epigenetic disruptions not only alter lipid homeostasis but also foster a microenvironment conducive to tumorigenesis. Notably, LXR downregulation—a nuclear receptor central to cholesterol homeostasis—has been associated with poor survival in HCC patients [80]. Moreover, sex-specific epigenetic regulation appears to underlie distinct susceptibilities to liver cancer; for example, repression of CYP51 leads to female-prevalent HCC via transcriptional network remodeling [188].
In terms of therapeutic opportunities, natural compounds such as curcumin, berberine, and resveratrol have shown potential to modulate cholesterol metabolism through epigenetic pathways [54]. Similarly, statin use has been associated with reduced HCC risk in MASLD patients, particularly at higher cumulative doses of lipophilic statins [189], suggesting that pharmacological modulation of cholesterol metabolism may confer protection beyond lipid lowering.
While substantial progress has been made in elucidating epigenetic drivers of MASLD, broader environmental and metabolic factors—such as diet, lifestyle, and genetic predisposition—also modulate disease trajectory. This underscores the need for personalized medicine approaches, integrating epigenetic and metabolic profiling to tailor interventions and prevent progression to HCC.
Emerging evidence indicates that MASLD is a sexually dimorphic disease, with distinct differences in metabolic regulation, gut microbiota composition, and fibrotic progression between males and females [190]. Estrogen has been shown to exert protective effects by maintaining hepatic lipid homeostasis, promoting TJ integrity, and modulating immune responses. The loss of estrogen signaling in postmenopausal women is associated with increased gut permeability, dysbiosis, and heightened susceptibility to fibrosis. In contrast, androgen signaling in males has been linked to greater hepatic cholesterol accumulation and proinflammatory gene expression. These sex-related differences also intersect with epigenetic regulation. For instance, chronic repression of CYP51, a key enzyme in cholesterol biosynthesis, has been associated with female-predominant hepatocarcinogenesis through transcriptional network remodeling [188]. Additionally, LXR downregulation, which negatively impacts cholesterol efflux and anti-inflammatory responses, correlates with poor prognosis in HCC and may operate differently between sexes [80]. These findings underscore the importance of incorporating sex as a biological variable in the study of MASLD pathogenesis and in the development of targeted therapeutic strategies.
While multiple promising therapeutic targets have been identified, challenges remain in translating these findings into clinically effective treatments. One major limitation is the heterogeneity of disease progression, as MASLD can develop at varying rates depending on genetic, metabolic, and environmental factors. Future studies should aim to develop personalized therapeutic approaches, integrating genomic, lipidomic, and metabolomic analyses to tailor treatments based on individual metabolic profiles [122].
Despite encouraging results from preclinical studies, many experimental treatments have yet to demonstrate long-term efficacy and safety in clinical trials. The complexity of MASLD requires multifactorial therapeutic strategies that combine lipid-lowering agents, mitochondrial protectants, ER stress modulators, microbiota-based interventions, and immunotherapies. Future clinical trials should focus on evaluating combination therapies to determine the most effective treatment regimens for MASLD patients.
The treatment of MASLD from a gut-liver approach focuses on personalized modulation of the intestinal microbiota, optimization of dietary interventions, development of new technologies, and implementation of innovative therapeutic agents. Identification of specific microbial profiles will allow the design of targeted treatments using probiotics, prebiotics, and synbiotics. At the same time, FMT could be refined with precision approaches to maximize metabolic benefits and reduce risks. Furthermore, developing selective antimicrobials will optimize microbial composition without affecting beneficial bacteria [191].
Regarding dietary interventions, personalizing diets based on the patient’s individual microbiota composition and metabolic profile will play a key role in managing MASLD. Focusing on functional foods and bioactive compounds, such as SCFA, will offer new therapeutic opportunities. Integrating artificial intelligence and multi-omics analysis will allow the design of dietary strategies tailored to each patient [173, 192].
Emerging technologies, such as advanced SHIME® (simulator of the human gut microbial ecosystem) models, will enable the testing of therapeutic interventions prior to their application in clinical studies. Multi-omics approaches will aid in identifying novel therapeutic targets and biomarkers of MASLD progression. Similarly, the development of synthetic microbiomes aimed at restoring intestinal homeostasis in patients with severe dysbiosis represents a promising avenue for future intervention [193, 194].
Using novel therapeutic agents, such as glucagon-like peptide-1 receptor agonists (GLP-1 RaS), will expand to modulate the microbiota and improve liver health [173]. Integrating safe phytocompounds from Traditional Chinese Medicine with conventional therapies could enhance anti-inflammatory and hepatoprotective effects. In contrast, developing postbiotic metabolites with beneficial effects on inflammation and hepatic metabolism will open new treatment possibilities [193].
However, these advances encounter significant challenges, including the need for long-term studies to validate the safety and effectiveness of microbiota-targeted interventions, the regulation and standardization of microbiota-based therapies to ensure their reproducibility and safety, and the exploration of the interaction between host genetics and microbiota to develop truly personalized approaches. As research advances, incorporating these insights into therapeutic strategies could profoundly transform the management of MASLD and facilitate a more precise and practical approach based on the modulation of the gut-liver axis.
MASLD is a multifactorial disease characterized by the convergence of metabolic, inflammatory, and immunological alterations, with cholesterol overload emerging as a central driver of hepatic injury and disease progression. Dysregulation of cholesterol metabolism—via enhanced intestinal absorption, increased endogenous synthesis, and impaired hepatic clearance—contributes to lipotoxicity, OxE, and unresolved ER stress, ultimately promoting fibrogenesis and progression to HCC. The gut-liver axis plays a pivotal role in integrating dietary, microbial, and metabolic cues, where dysbiosis, altered BA signaling, and increased intestinal permeability exacerbate hepatic inflammation and immune dysfunction.
Therapeutic strategies targeting cholesterol handling and restoring gut-liver homeostasis are evolving rapidly. Approaches such as dietary modulation, FXR agonists, NPC1L1 inhibitors, and microbiota-directed therapies—including prebiotics, probiotics, postbiotics, and FMT—offer avenues for early intervention. The incorporation of redox-active compounds (e.g., MitoQ), ER stress modulators (e.g., TUDCA), and GLP-1 receptor agonists further underscores the therapeutic potential of targeting mitochondrial dysfunction and systemic inflammation.
Emerging concepts such as ferroptosis regulation (via GPX4, FSP1, DHODH), IL-15-based immunotherapies, and CRISPR/Cas9-mediated gene modulation expand the landscape of intervention beyond traditional metabolic control. Importantly, the inclusion of sex-specific, epigenetic, and immunometabolic factors reinforces the need for precision-based approaches tailored to the diverse phenotypes of MASLD.
Altogether, an integrative strategy that encompasses metabolic, microbial, redox, immune, and genetic dimensions will be essential to disrupt the pathological trajectory of MASLD and reduce the burden of advanced liver disease and HCC.
27-OHC: 27-hydroxycholesterol
4-HNE: 4-hydroxynonenal
ACC: acetyl-CoA carboxylase
AMPK: AMP-activated protein kinase
ASBT: apical sodium-dependent bile acid transporter
BA: bile acids
ChREBP: carbohydrate-responsive element-binding protein
COL1A1: collagen type I alpha chain
DAMP: damage-associated molecular pattern
DCA: deoxycholic acid
ECM: extracellular matrix
ER: endoplasmic reticulum
FAO: fatty acid oxidation
FASN: fatty acid synthase
FGF19: fibroblast growth factor 19
FMT: fecal microbiota transplantation
FXR: farnesoid X receptor
GPX4: glutathione peroxidase 4
GrB: granzyme B
HCC: hepatocellular carcinoma
HDAC: histone deacetylase
HDCA: hydroxydeoxycholic acid
HDL: high-density lipoproteins
HMG-CoA: 3-hydroxy-3-methylglutaryl-coenzyme A
HSC: hepatic stellate cells
LCA: lithocholic acid
LD: lipid droplets
LDL-C: low-density lipoproteins cholesterol
LIMA1: LIM domain and actin-binding protein 1
LPS: lipopolysaccharide
LXR: liver X receptor
LXRα: liver X receptor alpha
MASH: metabolic dysfunction-associated steatohepatitis
MASLD: metabolic dysfunction-associated steatotic liver disease
MDA: malondialdehyde
mPTP: mitochondrial permeability transition pore
mtDNA: mitochondrial DNA
NHR: neutrophil-to-high-density lipoprotein cholesterol ratio
NK: natural killer
NLRP3: NOD-like receptor family pyrin domain-containing 3
NPC1L1: Niemann-Pick C1-like 1
OCA: obeticholic acid
OxE: oxidative stress
PDGF: platelet-derived growth factor
PLIN2: perilipin 2
PPARα: peroxisome proliferator-activated receptor alpha
ROS: reactive oxygen species
SBA: secondary bile acids
SCFA: short-chain fatty acids
SREBP2: sterol regulatory element-binding protein 2
T2DM: type 2 diabetes mellitus
TGF-β: transforming growth factor-beta
TGR5: Takeda G protein-coupled receptor 5
TJ: tight junction
TLR4: toll-like receptor 4
TNF-α: tumor necrosis factor-alpha
UDCA: ursodeoxycholic acid
UPR: unfolded protein response
VLDL: very low-density lipoproteins
ZO-1: zonula occludens-1
During the preparation of this work, the authors used ChatGPT to improve readability, enhance translation, and assist in text organization. The tool was not used to generate scientific content, analyze, or interpret data, or draw conclusions. After using the tool, the authors carefully reviewed and edited the content as needed and assume full responsibility for the accuracy and integrity of the publication.
LARR: Investigation, Writing—original draft. LEGQ: Conceptualization, Funding acquisition, Writing—review & editing. LBO: Formal analysis, Visualization. VSA: Formal analysis, Visualization, Data curation. MCGR: Conceptualization. RUML: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
Prof. Luis E. Gómez-Quiroz serves as an Editorial Board Member for Exploration of Digestive Diseases. He was not involved in the peer review or decision-making process for this manuscript. The other authors declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was partially funded by a [SECTEI-037-2024] grant and Universidad Autónoma Metropolitana-Iztapalapa (UAM-I). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 12325
Download: 85
Times Cited: 0
