 Review
Review

Affiliation:
1R&D Department, Kintsugi Therapeutics S.L., 08007 Barcelona, Spain
ORCID: https://orcid.org/0000-0001-6091-8225
Affiliation:
1R&D Department, Kintsugi Therapeutics S.L., 08007 Barcelona, Spain
ORCID: https://orcid.org/0000-0002-2801-8603
Affiliation:
2Program of Hepatology, Centre for Applied Medical Research (CIMA), University of Navarra, 31008 Pamplona, Spain
3National Institute for the Study of Liver and Gastrointestinal Diseases (CIBERehd, Carlos III Health Institute), 28029 Madrid, Spain
4IdiSNA, Navarra Institute for Health Research, 31008 Pamplona, Spain
ORCID: https://orcid.org/0000-0002-4830-1924
Affiliation:
2Program of Hepatology, Centre for Applied Medical Research (CIMA), University of Navarra, 31008 Pamplona, Spain
3National Institute for the Study of Liver and Gastrointestinal Diseases (CIBERehd, Carlos III Health Institute), 28029 Madrid, Spain
4IdiSNA, Navarra Institute for Health Research, 31008 Pamplona, Spain
ORCID: https://orcid.org/0000-0001-7075-2476
Affiliation:
1R&D Department, Kintsugi Therapeutics S.L., 08007 Barcelona, Spain
ORCID: https://orcid.org/0000-0002-9509-8033
Affiliation:
2Program of Hepatology, Centre for Applied Medical Research (CIMA), University of Navarra, 31008 Pamplona, Spain
3National Institute for the Study of Liver and Gastrointestinal Diseases (CIBERehd, Carlos III Health Institute), 28029 Madrid, Spain
4IdiSNA, Navarra Institute for Health Research, 31008 Pamplona, Spain
Email: maavila@unav.es
ORCID: https://orcid.org/0000-0001-6570-3557
Affiliation:
2Program of Hepatology, Centre for Applied Medical Research (CIMA), University of Navarra, 31008 Pamplona, Spain
3National Institute for the Study of Liver and Gastrointestinal Diseases (CIBERehd, Carlos III Health Institute), 28029 Madrid, Spain
4IdiSNA, Navarra Institute for Health Research, 31008 Pamplona, Spain
Email: magarfer@unav.es
ORCID: https://orcid.org/0000-0003-0375-6236
Explor Dig Dis. 2022;1:80–96 DOI: https://doi.org/10.37349/edd.2022.00007
Received: June 02, 2022 Accepted: July 15, 2022 Published: October 31, 2022
Academic Editor: Fabio Marra, University of Florence, Italy
Caspases are key factors in the regulation of the apoptotic and/or inflammatory responses, both crucial in the pathogenesis of diverse diseases. Caspase-2 is the most evolutionary conserved albeit functionally poorly defined member of the caspase family. The precise role of caspase-2 as an initiator or effector caspase is still unknown, but it has been involved in a wide variety of functions, from apoptosis to genomic stability, oxidative stress, metabolism, and cancer. However, many conflicting results render the exact function of this protease still unresolved. Although caspase-2 has several hundred substrates, the activation, processing, and activity on specific substrates remain poorly described. Recent evidence indicates that caspase-2 has a role in metabolic homeostasis and is required for lipotoxicity-induced apoptosis in hepatocytes, contributing to non-alcoholic steatohepatitis (NASH) progression towards hepatocellular carcinoma (HCC). Caspase-2 protein expression strongly localizes to injured/ballooned hepatocytes, correlating with NASH severity. Also, mice lacking caspase-2 showed protection from western diet-induced obesity, dyslipidemia, and insulin resistance. Although there are no effective therapies for NASH and HCC, the evaluation of a pan-caspase inhibitor has reached a phase I/II in clinical trials for advanced liver disease. Nevertheless, a better understanding of caspase functions with the identification of specific proteolytic substrates is essential for future therapeutic developments. Bearing in mind the pressing need to identify new targets for NASH-HCC and its metabolic-related comorbidities, and the favorable effect of caspase-2 genetic inhibition in animal models, pharmacological caspase-2 inhibition arises as a promising strategy that should be further investigated.
Caspases comprise a family of cysteine-dependent aspartate-specific proteolytic enzymes, known best for their roles in cell death and immune responses. When caspases are activated, they cleave a wide variety of substrates involved in apoptosis and immune response. In humans, 11 genes were found encoding caspase-1 to caspase-10 and caspase-14. In mice, caspase-11 and -12 are orthologs of human caspase-4 and -5 and caspase-10 is absent. All other caspases in humans and mice are orthologs with the same function. The protein initially named caspase-13 is a homolog of caspase-4 in other mammals. On the basis of functional characteristics, caspases are grouped into pro-apoptotic (caspase-2, -3, -6, -7, -8, -9, and -10) and pro-inflammatory (caspase-1, -4, -5, and -14) caspases [1]. However, this classification would be an oversimplification due to the diverse roles of caspases in multiple cellular processes other than apoptosis and inflammation, and the fact that pro-inflammatory caspases could induce apoptosis. In this sense, a better classification is the grouping according to the length of their prodomain. Given this classification, caspases are divided into initiator with long prodomains (caspase-1, -2, -4, -5, -8, -9, -10, -11, and -12) and effector caspases with short prodomains (caspase-3, -6, and -7) [1–3]. To date, there are three well-described activation mechanisms for caspases in mammals that depend on their structural features: i) recruitment activation of the pro-enzyme into macromolecular complexes; ii) autocatalytic activation after proximity-induced dimerization; iii) trans-activation by an initiator caspase.
The first evidence on the cleavage specificities of caspases using the positional scanning combinational library approach revealed that the S1 and S3 subsites (first and third residue in the enzyme) are nearly identical among caspases, whereas the location of S2 and S4 subsites is conserved [4]. The S1 pocket is highly basic and accommodates an Asp at the P1 position of the substrate (the residue on the N-terminal of the cleavage site), while the S3 pocket has a preference for Glu. The S2 subsite can contribute to substrate differentiation in the sense that caspase-3 and -7, the executioner caspases, have an aromatic S2 subsite that preferentially accommodates small aliphatic residues (Ala, Val). By contrast, the S2 subsite is larger in initiator caspases—such as caspase-2—with long prodomains and therefore tolerates well residues with bulkier side chains [5], with the exception of caspase-9 which has a stringent specificity for His in P2 [4]. P4 is the most critical determinant of specificity. Some inflammatory caspases—caspase-1, -4, and -5—have an extended, shallow hydrophobic depression at the S4 subsite which can accommodate large aromatic/hydrophobic amino acids in the S4 position [5]. In contrast, caspase-2, -3, and -7 possess the narrowest pocket and prefer an Asp residue in the P4 position [3]. Finally, caspases-6, -8, -9, and -10 tolerate many different amino acids in P4 but prefer those with larger aliphatic side chains [4]. Moreover, caspase-2 requires an occupied S5 subsite, which binds the substrate residue N-terminal to the cleavage site (P5) with a preference for small hydrophobic residues at this position for efficient substrate cleavage [5]. Caspases are synthesized as inactive proenzymes containing a prodomain, a p20 large subunit and a p10 small subunit. Activation of the proenzymes by proteolytic cleavages divides the large and small subunits and removes the prodomain. The catalytic dyad (Cys-His) in the p20 subunit relies on the active site in the cysteine within the conserved pentapeptide sequence “QACXG”. Hence, current and future research studies must consider the particular features of each caspase member when studying their role, mechanisms, and pathophysiological implications. This review aims to provide an outline of the current scientific evidence showing the potential contribution of caspase-2 to the pathology of human diseases and to emphasize the challenges and future directions of research regarding this enigmatic enzyme.
Caspase-2, one of the first members of caspases to be characterized and one of the best evolutionary conserved caspase across species, has characteristics of both initiator and effector caspases making it unique among this type of proteases [6]. Caspase-2 has a long amino-terminal prodomain called caspase activation and recruitment domain (CARD) through which it can interact with adaptor proteins, which is typical for initiator caspases [1, 2]. The C-terminal catalytic domain of caspase-2 has a large subunit (p18) containing the active site and a small subunit (p12) (Figure 1A). Despite the biochemical characteristics pointing to an initiator role of caspase-2, its predicted cleavage specificity is closer to effector caspases (concretely caspase-3 and -7) [2]. In certain circumstances, full length procaspase-2 (p51) is recruited to a large oligomeric complex called the PIDDosome, consisting of the C-terminal domain of the p53-induced death domain protein 1 (PIDD1) and the death domain-containing protein [CRADD, also called receptor interacting protein-associated protein with a death domain (RAIDD)] (Figure 1B) [7]. In this complex, the CARD of caspase-2 dimerizes, promoting its own cleavage at Asp333, and dividing the protein into two large p32 and p33 subunits (others detected these as p37 and p38 subunits) and a small p14 subunit [8, 9]. Subsequently, the prodomain and the linker region are removed at Asp169 and Asp347, respectively, generating the heterotetrameric complex consisting of two large p18 or p19, and two small p12 subunits [8, 10]. However, both activation independent of large activation complexes [1, 11, 12] and trans-activation by caspase-3, -7, and -8 were also described for caspase-2 [8]. Although dimerization and proteolytic cleavage of caspase-2 enhance caspase activity—especially for apoptotic functions—partial activation of caspase-2 upon dimerization and recruitment at activation complexes could be sufficient for caspase-2 activity in non-apoptotic functions. In addition to these cleavage site residues, Cys320 is the catalytic site and Ser384 is essential for the interactions within the caspase-2 active site [13]. Moreover, there are evidences of caspase-2 phosphorylation at a total of 11 sites in mammals (PhosphoSitePlus® database [14]): Ser24 [15], Ser80 [15], Ser157 [15–20], Thr161 [15], Ser139 [15, 21], Ser164 [15, 21, 22], Thr180 [15, 23], Ser220 [15], Ser340 [13, 15, 17, 18, 24–30], Ser346 [15], and Ser384 [15]. While specific protein kinase CK2 (PKCK2) has been reported to phosphorylate caspase-2 at Ser157 [20], calcium/calmodulin-dependent protein kinase II (CaMKII) at Ser164 [21, 22], and cyclin dependent kinase 1 (CDK1) at Ser340 [31], Aurora B kinase (AURKB) appears to be able to phosphorylate each and every one of the identified caspase-2 sites [15, 17]. Lastly, the kinase that phosphorylates Ser139 is still unknown [21]. Some of these caspase-2 post-translational modifications have been reported to suppress its activation in certain metabolic states to regulate cell survival [20, 22, 31, 32].
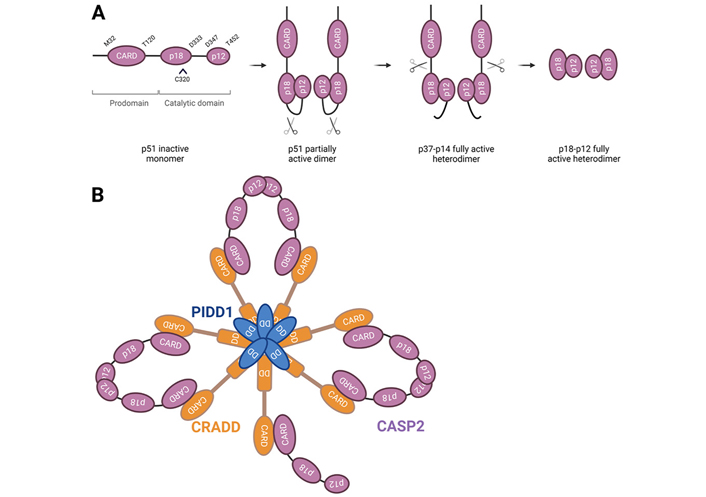
Caspase-2 processing and activation. A. Caspase-2 is synthesized as a proenzyme and is activated via a dimerization-dependent autocatalytic cleavage. Procaspase-2 monomers are recruited into close proximity and the catalytic domains can promote the cleavage between the large and small subunits of each monomer at D333. Intramolecular cleavage leads to the formation of an active stable tetrameric complex, which contains full enzymatic capacity and is completely processed upon removal of the prodomain and linker regions of the protein. C320 represents the cysteine of the active site; B. full length PIDD1 undergoes autoprocessing to generate an N-terminal PIDD-N fragment and two C-terminal fragments with distinct functions, PIDD-C and PIDD-CC. The latter interacts by its death domain (DD) with the DD of CRADD to form the PIDDosome. The CARD of CRADD interacts with procaspase-2-CARD domain. The core of the PIDDosome complex is formed by 5:5 PIDD-CC:CRADD molecules interacting via the DD, and two additional CRADD entities are placed on top protruding in diametric orientation. This structure would potentially allow up to seven caspase-2 monomers to be recruited via interaction with CRADD. The asymmetric assembly in which only six monomers could dimerize at one time may allow for enhanced stabilization of the complex, resulting in three active caspase-2 dimers. The binding CRADD to procaspase-2 promotes proximity-induced dimerization and auto-cleavage of caspase-2, yielding a fully active enzyme. Further processing results in the removal of the N-terminal CARD, generating a fully mature tetramer. CASP2: caspase-2
As stated earlier, caspases recognize at least four contiguous amino acids in their substrates, P4–P3–P2–P1, and cleave after the C-terminal residue (P1)—usually an Asp residue [5]. The cleavage pattern of caspase-2 was defined as exceptional in its requirement for an occupied pocket 5 subsite [5]. Thus, caspase-2 is the only caspase that hydrolyzes pentapeptides much more efficiently than tetrapeptides, as required for efficient cleavage by other caspases [6, 33]. These structural characteristics determine the preference for their substrates, which are believed to be nearly 300 (MEROPS database [34]). The subcellular localization of caspase-2 is associated with the activity on its substrates to mediate several functions in different cellular compartments. Specifically, caspase-2 has been found in Golgi, endoplasmic reticulum (ER), mitochondria, nucleus, nucleolus, and cytoplasm [35–38]. Caspase-2 has a nuclear localization signal (NLS) in the C-terminal linker of the prodomain and is imported by the classical nuclear import machinery through the interaction of this NLS with the a/b-importin heterodimer [39]. However, it has been described that the subcellular distribution of caspase-2 is independent of its state of processing [40]. Caspase-2 is retained in the nucleus until the late stage of apoptosis [39, 41]. Moreover, PIDD1, the member of the caspase-2 activation complex shuttles from the cytoplasm to the nucleus in response to various genotoxic stimuli [42]. In the cytoplasm, caspase-2 is activated by a CRADD-dependent but PIDD1-independent mechanism in response to various stressors [38, 43]. The localization of caspase-2 in the Golgi is involved in the cleavage on a unique site at Golgin 160 and the subsequent fragmentation of the Golgi complex during apoptosis [36]. Also in response to ER stress, caspase-2 trafficking occurs between ER and Golgi to execute the mitochondrial apoptotic pathway [37, 44, 45]. Caspase-2 activity was also detected in the nucleolus and its activation is dependent on the PIDDosome, assembled by the nucleolar phosphoprotein nucleophosmin 1 (NPM1) after DNA damage [38].
Several studies have shown that caspase-2 has both positive and negative regulatory functions in apoptosis depending on the cell type, state of growth and death stimuli, which have been related to the caspase-2 isoforms. Alternative splicing of caspase-2 messenger RNA (mRNA) induces exon 9 skipping and generates a long isoform of 435 amino acids, procaspase-2L, which has been described as a cell death inductor, whereas the insertion of the alternative exon 9 induces a premature stop codon which leads to the formation of a truncated isoform of 312 amino acids, procaspase-2S, which is devoid of the small subunit and was reported to antagonize cell death [46–49]. Although many tissues express both variants, some degree of tissue-specificity has been shown. While the long isoform is the dominant form expressed in a wide variety of tissues, the short isoform is mainly expressed in the heart, brain, and skeletal muscle [48, 49]. Moreover, the expression of the anti-apoptotic isoform caspase-2S is enhanced as a result of the treatment with anti-cancer drugs causing DNA damage, to delay apoptosis and enable DNA repair [46, 47].
The activity of caspase-2 on its substrates and its subcellular localization indicates a role of caspase-2 in apoptosis, cell cycle control, control of genomic stability, response to ER stress, metabolism, oxidative stress, and cancer [2, 10, 50–54]. To obtain a general overview of caspase-2 functions, a gene ontology (GO) enrichment analysis was performed with the substrates available in the MEROPS database. The enriched biological processes within proteins described as caspase-2 substrates (MEROPS database) pinpointed a role of caspase-2 in chromosome segregation, metabolic and biosynthetic processes, splicing, unfolded protein response (UPR), response to DNA damage, cell cycle control and apoptosis, among others (Table 1). The biological locations and most representative functions of caspase-2 substrates are depicted in Figure 2, obtained from the GO analysis of cellular components and biological processes enriched within proteins described as caspase-2 substrates (compiled by MEROPS database). Considering the analysis of cellular components, caspase-2 substrates are mainly found in the following subcellular structures and macromolecular complexes: cytoplasm, nucleus, spliceosome, cytoskeleton, chromosomes, centromeres, and microtubules (Figure 2A).
Biological functions of caspase-2 substrates. GO enrichment analysis of significantly enriched biological processes within proteins described as caspase-2 substrates (compiled by MEROPS database)
| GO biological process complete | FE | FDR |
|---|---|---|
| Female meiosis chromosome segregation | 42.02 | 1.40 × 10–2 |
| Positive regulation of chromosome segregation | 16.16 | 4.11 × 10–3 |
| Phosphatidylinositol-3-phosphate biosynthetic process | 16.01 | 1.93 × 10–2 |
| Negative regulation of mRNA splicing, via spliceosome | 16.01 | 1.92 × 10–2 |
| Cellular response to misfolded protein | 15.28 | 2.12 × 10–2 |
| Protein quality control for misfolded or incompletely synthesized proteins | 12.45 | 3.58 × 10–2 |
| Regulation of heterochromatin assembly | 12.01 | 3.98 × 10–2 |
| Regulation of androgen receptor signaling pathway | 12.01 | 3.96 × 10–2 |
| Positive regulation of chromosome separation | 11.59 | 4.34 × 10–2 |
| Establishment of spindle orientation | 10.77 | 1.66 × 10–2 |
| Regulation of mitotic sister chromatid segregation | 10.70 | 1.44 × 10–3 |
| mRNA splicing, via spliceosome | 8.79 | 5.12 × 10–12 |
| Histone deacetylation | 8.69 | 1.22 × 10–2 |
| Neuron apoptotic process | 7.64 | 5.00 × 10–2 |
| Regulation of mitotic metaphase/anaphase transition | 7.39 | 3.29 × 10–3 |
| Positive regulation of chromosome organization | 7.20 | 1.45 × 10–3 |
| Positive regulation of DNA recombination | 7.10 | 2.60 × 10–2 |
| Regulation of translational initiation | 6.15 | 4.46 × 10–2 |
| Positive regulation of translation | 5.92 | 2.16 × 10–3 |
| Positive regulation of apoptotic signaling pathway | 5.86 | 5.34 × 10–3 |
| Nuclear export | 5.82 | 5.50 × 10–3 |
| Ribonucleoprotein complex assembly | 5.81 | 2.02 × 10–4 |
| Regulation of mRNA stability | 5.57 | 6.23 × 10–4 |
| Negative regulation of translation | 5.03 | 6.51 × 10–3 |
| Positive regulation of proteasomal protein catabolic process | 5.03 | 4.86 × 10–2 |
| mRNA transport | 4.84 | 3.01 × 10–2 |
| Positive regulation of response to DNA damage stimulus | 4.61 | 2.06 × 10–2 |
| Mitotic nuclear division | 4.48 | 2.43 × 10–2 |
| Positive regulation of leukocyte differentiation | 4.30 | 3.02 × 10–2 |
| Negative regulation of organelle organization | 4.18 | 2.90 × 10–4 |
| Chromatin remodeling | 3.97 | 5.46 × 10–3 |
| Negative regulation of catabolic process | 3.36 | 1.92 × 10–2 |
| Translation | 3.34 | 8.75 × 10–3 |
| Ubiquitin-dependent protein catabolic process | 2.97 | 3.21 × 10–3 |
| Cell division | 2.81 | 1.75 × 10–2 |
| DNA repair | 2.74 | 2.96 × 10–2 |
| ncRNA metabolic process | 2.73 | 2.24 × 10–2 |
| Regulation of plasma membrane bounded cell projection organization | 2.70 | 1.00 × 10–2 |
| Intracellular signal transduction | 2.17 | 1.57 × 10–3 |
| Cellular protein localization | 2.06 | 1.40 × 10–2 |
| Protein transport | 2.03 | 3.42 × 10–2 |
The most specific GO term is shown (hierarchically sorted terms). Fold enrichment (FE) ≥ 2 and Fisher’s exact test with false discovery rate (FDR) less than 0.05 as calculated by the Benjamini-Hochberg procedure were used as thresholds in the enrichment analysis. ncRNA: noncoding RNA
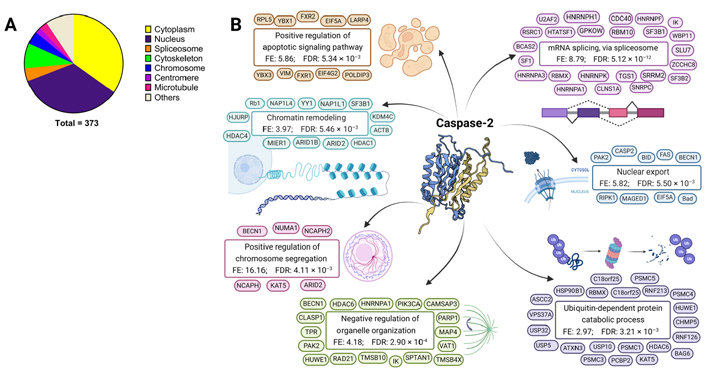
Biological locations and functions of caspase-2 substrates. GO cellular components and biological processes enriched within proteins described as caspase-2 substrates (compiled by MEROPS database). A. GO enrichment analysis of significantly enriched cellular components for caspase-2 substrates; B. GO analysis of biological processes enriched within caspase-2 protein substrates. Caspase-2 structure from Protein Database (3R7S) representing the crystal structure of apo caspase-2 (conformational states corresponding to ligand-unbound) [6]. Representative substrates are shown for each GO term. BID: Bcl-2 Homology 3-interacting domain death agonist
Caspase-2 has been implicated in programmed cell death in both mitochondria-dependent and death-receptor-mediated apoptosis. Caspase-2 is implicated in mitochondrial-mediated apoptosis in response to a variety of death stimuli including heat shock, DNA damage, aneuploidy, and microtubule disruption [55]. Upstream of mitochondria, caspase-2 induces apoptosis by the permeabilization of the outer mitochondrial membrane and the induction of cytochrome c and the second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO) release from these organelles [56, 57]. Also, caspase-2 initiates the mitochondrial apoptosis pathway indirectly through the proteolytic cleavage of the BID [45]. Regarding the death-receptor-mediated apoptosis, caspase-2 has been implicated in the tumor necrosis factor (TNF)-related apoptosis inducing ligand (TRAIL) pathway in a cell- or tissue-specific manner [1, 55]. Moreover, long-chain fatty acids such as palmitate induce caspase-2 activation and the subsequent initiation of apoptosis or lipoapoptosis [58]. On the other hand, caspase-2 is an important mediator in developmental apoptotic machinery in mammals, a crucial physiological phenomenon allowing embryonic development [59, 60]. Although caspase-2 shares a great number of substrates with the apoptotic executioner caspases-3 and -7, suggesting a role of caspase-2 in apoptosis, its function could depend on the context in which caspase-2 is activated in response to stimuli.
Several studies showed that caspase-2 is involved in genomic instability—changes in nucleic acid sequences and alterations to the karyotype—a hallmark of cancer. Caspase-2 knock-out (Casp2 KO) mouse embryonic fibroblasts treated with cytoskeleton disruptors that prevent normal segregation of chromosomes during mitosis—the microtubule depolymerizing agent vincristine, the microtubule disruptor paclitaxel, or the actin polymerization inhibitor cytochalasin D—were resistant to death, emphasizing that cell death due to cytoskeletal disruption requires caspase-2 [61, 62]. Livers from Casp2 KO mice displayed karyomegaly, a feature commonly associated with aging and aneuploidy [63]. Also, splenocytes appear to require catalytically active caspase-2 to limit aneuploid cells when exposed to anti-mitotic compounds [64]. Caspase-2 has been shown to be essential for mitotic catastrophe—delayed mitosis-linked cell death—upstream of cytochrome c release, followed by caspase-3 activation and chromatin condensation in a p53-independent manner [65]. In this study, when caspase-2 is suppressed, the mitotic catastrophe is inhibited and cells continue the cell cycle beyond the metaphase, leading to asymmetric distribution of the cytoplasm, DNA, and chromosomes. Of note, it has been shown that the PIDDosome limits hepatocyte ploidy and is instructed by the E2F network to control p53 activation in the developing as well as regenerating liver [66], suggesting that caspase-2 is at least indirectly involved in liver development and regeneration.
Caspase-2 has been associated with combating oxidative stress, as well as delaying the aging process in mice [67–69]. Casp2 KO mice showed hair loss, early hair graying, increased bone loss, reduced body weight and higher levels of oxidized proteins [67, 68]. Moreover, Casp2 KO mice showed impaired antioxidant defense, increased inflammation, and increased cellular stress [63]. Also, these mice exhibited accelerated aging and a decreased lifespan [67, 68]. In hepatocytes derived from Casp2 KO mice, the absence of caspase-2 accelerates age-dependent changes in terms of mitochondrial reactive oxygen species (ROS) neutralization [70] as well as in the remodeling of the metabolic and proteomic profile during aging [69]. Given these findings, caspase-2 could regulate processes in order to neutralize oxidative stress and could be counteracting premature aging. Despite some of these age-related differences that could be associated with the metabolic functions of caspase-2, young Casp2 KO mice showed lower expression of mitochondrial components [69], in agreement with the age-related characteristics associated with increased ROS and mitochondrial dysfunction [70]. Furthermore, it has been observed that caspase-2 is involved in the regulation of the inhibition of autophagy both after oxidative damage and in the absence of any external stressor [62].
An unexpected role of caspase-2 in metabolism has been identified, which was unpredictable for a member of the caspase family. Recent evidence indicates that caspase-2 has a role in metabolic homeostasis through the regulation of lipid and free cholesterol accumulation. Moreover, caspase-2 is required for lipotoxicity-induced apoptosis in hepatocytes, contributing to NASH progression [71, 72]. Caspase-2 expression is upregulated in humans and rodents with NASH and strongly localizes to injured/ballooned hepatocytes, correlating with NASH severity [71–73]. Above and beyond this, Casp2 KO mice are protected from hepatocyte apoptosis triggered during diet- and genetically-induced NASH, as a result of reduced ductular reaction and decreased liver fibrosis, lower hepatic cholesterol and triglyceride levels, less hepatocyte ballooning, and macrophage infiltration [71–73]. Mechanistically, ER stress-mediated upregulation of caspase-2 activates sterol regulatory element-binding proteins (SREBPs) leading to the buildup of cholesterol and triglyceride, thus promoting NASH development [72]. In this study performed by Kim and colleagues [72], the activation of SREBP was attributed to a caspase-2-dependent cleavage of site-1 protease (S1P). Moreover, caspase-2 translation depends on the inositol-requiring enzyme (IRE)-1α branch of the UPR [74] and upon inhibition of this branch of ER stress, and caspase-2 translation is blocked and SREBP activation is prevented [72]. Remarkably, caspase-2 is under the transcriptional regulation of SREBP2, which might boost further SREBP activation through a positive feedback loop [74]. Moreover, under lipotoxic conditions, caspase-2 is involved in the hedgehog pathway-induced lipoapoptosis with consequent production of hedgehog ligands that promote liver fibrosis [71]. Casp2 KO mice are also protected from western diet-induced obesity, dyslipidemia, and insulin resistance, contributing to the amelioration of metabolic syndrome (MetS). For example, caspase-2 has a role in basal energy metabolism by regulating adipocyte biology and fat expansion, shifting the balance in fuel choice towards increased carbohydrate utilization [73, 75]. Obesity-induced whitening of brown adipose tissue was prevented in Casp2 KO mice, which had also increased levels of thermogenesis markers in both white and brown adipose tissues [72]. Besides, Casp2 KO mice prevented western diet-induced hyperplasia of pancreatic Langerhans islands, hyperinsulinemia, and hyperglycemia, improving overall glucose homeostasis and pancreatic function [73, 75, 76]. Casp2 KO mice were protected from western diet-induced dyslipidemia through a reduction in total cholesterol, triglycerides and non-esterified fatty acids [73, 75, 76].
Reduced levels of caspase-2 mRNA and protein expression have been observed in cancer cells compared to normal tissues [77]. Moreover, caspase-2 expression within cancer samples has been correlated both positively [78] and negatively [79, 80] with survival outcomes and/or therapeutic responses. For instance, procaspase-2 protein levels were decreased in children with drug-resistant acute lymphoblastic leukemia [78]. Also, mouse embryonic fibroblasts lacking caspase-2 showed enhanced proliferation, elevated clonogenicity, accelerated anchorage-independent growth, and transformation and were highly tumorigenic, rapidly producing large tumors in athymic nude mice [77]. In this line, caspase-2 plays a role in the prevention and limitation of breast and lung cancer progression [81, 82], since mice with mutated Casp2 combined with cancer-driven mutations accelerated carcinogenesis. Despite these findings suggesting a role of caspase-2 as a tumor suppressor, the opposite situation has been observed in neuroblastoma, where Casp2 KO delayed tumorigenesis in a mouse model of this type of tumor [79]. In gastric cancer, the therapeutic response could be dependent on the activation of caspase-2-induced apoptosis [80]. Caspase-2 is required for sphingosine kinase 1 (SK1) degradation—SK1 is a lipid kinase whose activity produces sphingosine-1 phosphate, a pro-survival lipid associated with proliferation, angiogenesis, and invasion. SK1 has been described as a caspase-2 substrate in human breast cancer cells, and loss of caspase-2 significantly affects sphingolipid metabolism in response to DNA damage [83]. In aneuploid colorectal cancer, a mutation in the B-cell CLL/lymphoma 9-like (BCL9L) gene led to caspase-2 dysfunction and tolerance to chromosome missegregation independently of p53 status [84]. On the other hand, the role of this caspase in hepatocellular carcinoma (HCC) remains controversial, with some works describing anti-tumorigenic properties for caspase-2 [85], while others revealed pro-tumorigenic properties in a chemically-induced HCC mouse model [86]. Given the relationship between NASH and HCC, it has been suggested that caspase-2 could be involved in the risk of liver cancer development via the previously mentioned S1P/SREBP pathway [54, 55]. Despite the reasons for these discrepancies being unclear, the aforementioned studies with Casp2 KO mice [54, 55, 77–86] clearly prove that caspase-2 is not a primary driver of tumorigenesis and suggest a tissue and context-specific role for caspase-2 in cancer development. One should be cautious with the potential role of caspase-2 as a tumor suppressor since this role is only manifested in animals engineered to express potent oncogenes or lacking crucial tumor suppressor genes [87]. Thus, it seems unlikely that caspase-2 was kept throughout mammalian evolution for its cancer-promoting function.
Apoptosis and inflammation are known to play central roles in the pathogenesis of metabolic diseases such as obesity and the MetS, and the progression of non-alcoholic fatty liver disease (NAFLD) to non-alcoholic steatohepatitis (NASH) and more severe disease stages. Despite NAFLD is the most prevalent liver disease in the world, current treatment strategies—including lifestyle and dietary changes, off-label use of pharmacotherapy, and surgery—are limited and there are no approved pharmacological treatments [88, 89]. The off-label use of drugs for the treatment of NAFLD/NASH includes the prescription of vitamin E, insulin sensitizers, and statins, mainly aimed at treating NAFLD comorbidities—such as diabetes and cardiovascular disease [89]. Likewise, in patients with severe obesity, bariatric surgery could be an option, but it is only considered for a small proportion of patients in early stages and without irreversible end-stage liver disease [90]. Liver transplantation could be indicated for NASH patients with end-stage liver disease, although it is a most invasive approach and does not guarantee the complete recovery of liver function [91].
Potential drug therapy with the pan-caspase inhibitor Emricasan—formerly named IDN-6556 or PF-03491390, which acts by blocking apoptosis and inflammation—has reached phase I/II clinical trials [92, 93]. Up to date, eleven phase II trials have been carried out in patients with severe liver disease, and several are under investigation in the context of NAFLD/NASH [93–98]. In this sense, the treatment with Emricasan for 28 days showed significantly reduced alanine aminotransferase (ALT) serum levels and liver disease biomarkers in subjects with NAFLD [98]. However, another study showed that the treatment with this pan-caspase inhibitor over 72 weeks did not improve liver histology in patients with NASH fibrosis, and might even worsen fibrosis and ballooning [96]. Moreover, the treatment with Emricasan for 48 weeks was unable to reduce portal hypertension in patients with NASH-related cirrhosis and severe portal hypertension [95], and was insufficient to decrease clinical events in patients with decompensated NASH-cirrhosis [97]. Emricasan has been also investigated in other etiologies of liver disease. Chronic hepatitis C patients with residual fibrosis or cirrhosis treated with Emricasan for 24 months achieved an improvement and/or stabilization in the fibrosis stage [99]. Emricasan administration over 3 months improved hepatic function in patients with decompensated cirrhosis and more severe end-stage liver disease (including NASH patients) [100]. In shorter periods of time, this pan-caspase inhibitor produced an improvement in liver disease biomarkers [94, 101–103]. For example, patients with chronic hepatitis C who were given 12 days of Emricasan treatment improved their ALT and aspartate aminotransferase (AST) serum levels, which returned to baseline levels [101]. Also, Emricassan treatment for 14 days resulted in an amelioration of ALT and AST in patients with hepatitis C but not in other causes of liver disease [102]. Another study showed that treatment with Emricasan for 28 days improved several liver disease biomarkers in subjects with acutely decompensated cirrhosis but failed to do so in those with advanced cirrhosis [103]. Patients with compensated cirrhosis and severe portal hypertension showed reduced serum ALT and AST levels and liver disease biomarkers, along with decreased portal hypertension after 28 days of treatment with Emricasan [94]. Moreover, in animal models, Emricasan had a clear anti-apoptotic effect in liver cells, with consequent downregulation of circulating pro-inflammatory cytokines and a favorable impact on liver fibrogenesis and portal pressure [93]. Apoptosis was substantially attenuated, and liver injury, fibrosis, and inflammation were reduced in diet-induced obese and NASH mice treated with this pan-caspase inhibitor for 20 weeks [104]. Both a short-term and long-term treatment with Emricasan (10 days and 20 days, respectively) in mice following common bile-duct ligation (BDL) showed decreased portal pressure and increased survival [105]. In the anti-Fas mouse model of acute liver injury, Emricasan administration both 2 h before and 4 h after Fas stimulation resulted in reduced ALT serum levels, apoptosis, and caspase-3 and -7 activity [106, 107]. Additionally, there is no evidence of Emricasan-related tumor formation in any tissue, albeit non-neoplastic lesions (not pre-neoplastic) were observed in Tg.rasH2 mice used for short-term carcinogenicity studies [108]. Thus, Emricasan was not considered carcinogenic.
The above-mentioned clinical trials were carried out because preclinical studies suggested that Emricasan could be effective to decrease the excessive caspase-mediated apoptosis and inflammation, crucial drivers of liver injury in NAFLD/NASH. However, this pan-caspase inhibitor has shown conflicting results regarding fibrosis and cirrhosis amelioration, and its effects on portal hypertension. The lack of effect or even worsening of fibrosis might be explained by the fact that apoptosis is also the main mechanism promoting the resolution of fibrosis [109, 110]. Thus, by inhibiting the apoptosis-related caspases, Emricasan could be impairing the reversion of fibrosis in liver disease. These highly heterogeneous results might be due to their pharmacological activity. Emricasan inhibits all caspases with different affinities, with the highest affinity for anti-inflammatory caspases-4 and -5 [the half maximal inhibitory concentration (IC50) of 0.06 nmol/L and 0.01 nmol/L, respectively], and showing the lowest affinity for caspase-2 (IC50 of 20 nmol/L) [111]. Therefore, the affinity of Emricasan for caspase-2—measured by the IC50, as reported by the owner companies Conatus and Novartis—is 100 times lower than that for caspase-1, -7, -8, and -9, and 1,000 times lower than that for caspase-4 and -5. The lack of selectivity for caspase-2, together with the irreversible inhibition of other caspases by Emricasan, could contribute to its reduced inhibitory activity of caspase-2. This would explain the limited clinical efficacy on steatosis-related parameters, as caspase-2 has been involved in intracellular lipid accumulation and lipoapoptosis. These data contribute to understanding the poor performance of Emricasan observed in preclinical and clinical trials in liver disease. Interestingly, new caspase-2 inhibitors have been recently developed with the aim of improving colon cancer therapy [33], as well as for the treatment of neurodegenerative diseases [112, 113].
Despite an active research activity in the field, the specific function of each caspase member is still unclear, and evidence of crosstalk between alternative cell death pathways may complicate the therapeutic blocking of caspases to treat metabolic diseases [92]. Due to the high overlap in substrate selectivity among caspase family members, there is currently a lack of available specific small-molecule inhibitors—although several compounds have been patented [114]. Firstly, however, there is a need to better elucidate the mechanism of caspase functions, with the identification of their specific proteolytic substrates, which is essential for future therapeutic developments. In light of the multiple pathways in which caspase-2 is involved, one such promising therapeutic strategy could be targeting this caspase. In view of the potential roles of caspase-2, this could be an appealing therapeutic approach for NAFLD/NASH, their progression towards HCC, and comorbidities such as obesity and MetS-related disorders.
Caspase-2 is a protease at odds with current dogma due to its particular properties from its processing and activation to its substrates and functions. However, it is still unclear whether it requires an activation platform or if it is capable of dimerizing and cleaving autonomously, and thus self-activating. Moreover, caspase-2 processing and activation modes could be stimulus and cell-type dependent. Although there are many caspase-2-specific substrates already identified, the relevance of some of these to caspase-2- dependent processes needs further investigation. Several reports indicate that cleavage might not be a requisite for caspase-2 enzymatic activity, which adds another difficulty in the characterization of its activity and functions. Estimating the specific enzymatic activity of a single caspase member is quite difficult due to their overlap in substrate preferences. Therefore, current approaches to better describe the unknowns of this protease usually involve in vitro models, purified protein, and artificial inhibitors, and may not resemble the actual cellular and organ milieu. On the other hand, the use of genetically modified models, especially caspase-2 deficient mice, has shown promising results regarding the potential benefits of caspase-2 downregulation. Yet, this model could be compensated by increased activity of other caspases, or even several other proteins involved in the pathways in which caspase-2 is acting, masking some caspase-2 functions.
In contrast to other caspases with a clear direct apoptotic function, caspase-2 could be involved in tumor suppression indirectly, by acting on other multiple pathways such as cytoskeletal disruption, aneuploidy and oxidative stress, and lipotoxicity. Interestingly, several reports suggest that caspase-2 is an attractive therapeutic target for blocking apoptosis, inhibiting lipogenesis and reducing inflammation, thus preventing fibrosis and slowing the progression of NAFLD and NASH to end-stage liver disease—i.e. cirrhosis and HCC. This therapeutic approach is an appealing and promising therapeutic option since the inhibition of caspase-2 activity could be beneficial for other metabolic organs affected by metabolic disorders, such as the heart, adipose tissue, and pancreas, especially when considering caspase-2 involvement in lipoapoptosis and metabolism. Taken together, further studies are needed to better understand the role played by caspase-2 under physiological conditions in different cell type-dependent scenarios.
ALT: alanine aminotransferase
AST: aspartate aminotransferase
BID: Bcl-2 Homology 3-interacting domain death agonist
CARD: caspase activation and recruitment domain
Casp2 KO: Caspase-2 knock-out
CRADD: death domain-containing protein
ER: endoplasmic reticulum
GO: gene ontology
HCC: hepatocellular carcinoma
IC50: the half maximal inhibitory concentration
MetS: metabolic syndrome
mRNA: messenger RNA
NAFLD: non-alcoholic fatty liver disease
NASH: non-alcoholic steatohepatitis
PIDD1: p53-induced death domain protein 1
SK1: sphingosine kinase 1
SREBPs: sterol regulatory element-binding proteins
All the figures have been created with the BioRender program (BioRender.com).
ALP: conceptualization, formal analysis, visualization, writing original draft, review and editing. MC: critical review and editing. MA: funding acquisition, critical review and editing. CB: funding acquisition, critical review and editing. CH: critical review and editing. MAA: conceptualization, funding acquisition, critical review and editing. MGFB: conceptualization, funding acquisition, critical review and editing. All authors have read and agreed to the published version of the manuscript.
ALP is employed by Kintsugi Therapeutics S.L. CH is Co-founder and CEO of Kintsugi Therapeutics S.L. MC is Co-founder and COO of Kintsugi Therapeutics S.L., a start-up developing capase-2 inhibitors as therapeutic agents for liver disease. MA, CB, MAA, MGFB declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Work in the authors’ laboratory is supported by: CIBERehd; grant PI19/00613 from Instituto de Salud Carlos III (ISCIII) co-financed by “Fondo Europeo de Desarrollo Regional” (FEDER) “Una manera de hacer Europa” grant 2018-055 from Gobierno de Navarra; grants SAF2014-54191-R, SAF2017-88933-R, PID2019-104878R-100/AEI/10.13039/501100011033 and PID2019-104265RB-100/AEI/10.13039/501100011033 from FEDER/Ministerio de Ciencia, Innovación y Universidades-Agencia Estatal de Investigación; AECC LAB Grant 2020; AECC post-doctoral fellowship POSTD18014AREC to MA and Ramón y Cajal Program contract RYC2018-024475-1 to MGFB; grant CPP2021-008411 “Proyectos de colaboración público-privada” co-founded by the Ministerio de Ciencia e Innovación and European Union NextGenerationEU/PRTR. The generous support of Mr. Eduardo Avila is acknowledged. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

