Case Report
Case Report

Affiliation:
1Diagnostic Medical Sonography, Department of Radiology, Advocate Christ Medical Center, Oak Lawn, IL 60456, USA
†
ORCID: https://orcid.org/0009-0008-9571-3594
Affiliation:
2Department of Medicine, Division of Gastroenterology, Joint Base Elmendorf-Richardson Medical Center, Elmendorf AFB, AK 99506, USA
†
Email: ted.a.spiewak.mil@health.mil
ORCID: https://orcid.org/0000-0002-6990-7781
Affiliation:
3Department of Medicine, Division of Gastroenterology & Hepatology, Brooke Army Medical Center, Fort Sam Houston, TX 78234, USA
ORCID: https://orcid.org/0000-0002-1880-7991
Explor Dig Dis. 2025;4:1005103 DOI: https://doi.org/10.37349/edd.2025.1005103
Received: February 04, 2025 Accepted: October 10, 2025 Published: December 01, 2025
Academic Editor: Jochen Mattner, Friedrich-Alexander Universität Erlangen-Nürnberg (FAU), Universitätsklinikum Erlangen (UKER), Germany
Interleukin-17 inhibitors (IL-17i) are used for dermatologic and rheumatologic immune-mediated inflammatory diseases (IMIDs), yet paradoxical inflammatory bowel disease (IBD) can occur. Although trials report low incidence, recognition and management remain difficult outside tertiary care centers. A 54-year-old woman treated with ixekizumab (IXE) for presumptive psoriatic arthritis (PsA) without definitive confirmation developed anorexia, weight loss, abdominal pain, rectal urgency, and hematochezia 16 weeks after IXE initiation. Limited access to gastroenterology contributed to the delayed workup. Catastrophic complications, including bowel perforation, postoperative abscesses, and severe malnutrition, resulted from the cumulative effects of longstanding, inadequately treated disease; excessive immunosuppression with high-dose corticosteroids and infliximab; and concurrent use of opioids and antidiarrheals, among other factors. On transfer to a center skilled in IBD, care included withdrawal of excessive immunosuppression, targeted antimicrobials, and nutrition rehabilitation. Histopathology of the surgical specimen was most consistent with features of Crohn’s disease (CD). After recovery, she achieved clinical, endoscopic, and histologic remission. On rheumatologic reassessment at an independent practice, she did not meet classification criteria for PsA. With continued specialty follow-up, the patient has remained in sustained clinical, laboratory, and endoscopic remission for 16 months, underscoring that timely recognition and disciplined, evidence-based care grounded in the principles used for severe IBD and drug-induced colitis can deliver favorable long-term outcomes. This case highlights the need for structured, accessible clinical guidance, not only to support non-IBD specialists in managing IL-17i-associated complications but also to guide clinicians during the pre-therapy phase in selecting appropriate candidates for treatment and assessing potential gastrointestinal risks before initiating therapy. We present an evidence-informed framework for resource-limited settings that addresses screening, early recognition, diagnostic workup, and therapeutic decision-making to guide safer IL-17i use and improve outcomes.
The IL-23/IL-17 axis is central to several chronic immune-mediated inflammatory diseases (IMIDs). IL-17, a pro-inflammatory cytokine secreted by various immune cells, including T-helper 17 (Th17) cells, is released in response to IL-23 signaling from antigen-presenting cells, triggering an inflammatory cascade of cytokines and chemokines [1]. Monoclonal antibodies targeting IL-17 isoforms (IL-17A, IL-17E, IL-17AF) or the IL-17 receptor A have significantly impacted the treatment of certain dermatological and rheumatological disorders. Currently, four FDA-approved interleukin-17 inhibitors (IL-17i) exist: bimekizumab (BKZ), brodalumab (BRO), ixekizumab (IXE), and secukinumab (SEC). These biologics are individually labeled for conditions including ankylosing spondylitis (AS) (BKZ, IXE, SEC), hidradenitis suppurativa (BKZ, SEC), non-radiographic axial spondyloarthritis (BKZ, IXE, SEC), plaque psoriasis (PsO) (BKZ, BRO, IXE, SEC), and psoriatic arthritis (PsA) (BKZ, IXE, SEC), demonstrating strong therapeutic efficacy within their approved domains [1–3].
Basic science research has demonstrated increased levels of Th17 cells, IL-17, and other Th17-related cytokines in the serum and mucosa of patients with active ulcerative colitis (UC) and Crohn’s disease (CD) [4]. This prompted evaluation of IL-17 inhibition for the treatment of inflammatory bowel disease (IBD). However, clinical trials paradoxically suggested a lack of efficacy compared to placebo, and even indicated disease worsening, leading to early termination of the studies [5, 6]. The precise pathophysiological mechanisms underlying these paradoxical findings remain incompletely understood. Current research suggests that while IL-17 inhibition effectively reduces skin and joint inflammation, it simultaneously compromises intestinal barrier function, potentially triggering or worsening gut inflammation [1, 7]. All four approved IL-17i have been associated with exacerbation and new onset of IBD. While these events were reported as rare (less than 1%) in dermatological and rheumatological clinical trials, new cases continue to emerge in real-world clinical practice settings [7–13]. Given the higher baseline prevalence of IBD in patients with PsO, PsA, and other related IMIDs [14], it remains uncertain whether IL-17i truly induces IBD or merely unmask subclinical disease in susceptible individuals.
The limited guidance on managing this rare adverse event, derived primarily from systematic reviews [7–11], experiences with other drug-induced enterocolitis [15] and IBD [16–21], highlights the importance of each documented case in expanding our collective medical understanding. To our knowledge, this is the first reported case of a patient without a definitively diagnosed dermatological or rheumatological disease who developed new-onset CD following IL-17i use. Her severe clinical course, marked by diagnostic delays, bowel perforation, and multiple postoperative complications, underscores the need for a standardized approach to screening, management, and surveillance. Such guidance is particularly helpful for providers in rural or resource-limited settings, who may have limited experience managing these complex conditions. By presenting this unique case and proposing a structured framework for care, we aim to provide a practical, evidence-informed resource to support future best practices and ultimately improve patient outcomes.
A 54-year-old Caucasian female from an underserved rural community (roughly 375 miles from our institution) presented to her primary care physician with a 12-week history of progressive anorexia, 20-pound unintentional weight loss, lower quadrant abdominal pain, urgency, and up to 15 bloody bowel movements daily. She denied recent travel, sick contacts, antibiotic use, and any tobacco, alcohol, or illicit drug use. On examination, she was reportedly afebrile and nontoxic-appearing, but with signs of dehydration and mild lower abdominal quadrant tenderness without rebound or guarding. Her past medical history included Graves’ disease treated with radioactive iodine therapy and PsA diagnosed 2 years earlier. Family history revealed AS without a history of IBD or PsO, and the patient previously tested negative for HLA-B27.
Over the course of the 3 years preceding her acute presentation, the patient had numerous visits with her primary care physician for persistent joint pains involving her hands, feet, knees, and lower back, accompanied by nail ridging affecting both fingernails and toenails. Throughout this period, she consistently denied gastrointestinal symptoms, including abdominal pain, diarrhea, urgency, rectal bleeding, rectal pain, or perianal drainage. She also denied fever, fatigue, unintentional weight loss, ocular symptoms, or painful skin lesions. Vital signs during these visits were consistently within normal limits aside from chronic hypertension, and physical examinations revealed no evidence of ocular inflammation, oral (PO) ulcers, abdominal tenderness, or dermatological findings consistent with erythema nodosum or pyoderma gangrenosum. Baseline laboratory values obtained during this 3-year period were unremarkable, with hemoglobin (HGB) ranging from 13.3 to 14.1 g/dL (reference 12.0–16.0), albumin (ALB) from 4.1 to 4.4 g/dL (reference 3.5–5.2), white blood cell (WBC) count from 6.4 to 8.9 × 103/µL (reference 4.5–11), and platelet (PLT) count from 237 to 324 × 103/µL (reference 155–369). Vitamin B12, folate, and iron studies were consistently within normal limits. Her only medication was levothyroxine for secondary hypothyroidism. Additionally, a colonoscopy performed at age 52 for colorectal cancer screening reported a normal terminal ileum and colonic mucosa, without evidence of colorectal cancer, polyps, or inflammation.
Two years prior to the index presentation, the patient was referred to podiatry for evaluation of a left hallux nail abnormality and was diagnosed with onychocryptosis. During this podiatric evaluation, the patient described persistent finger and toe joint pains, alongside nail abnormalities characterized by ridging, brittleness, cuticle fissures, and erythematous periungual skin. Her podiatrist raised concerns for possible PsA, prompting referral to dermatology. Per review of the outside records, dermatologic assessment revealed irregular, moderately irritated, psoriasiform skin lesions primarily involving her fingers and fingernails, and the patient was diagnosed with psoriasiform dermatitis. Initial management was with topical clobetasol therapy. Due to ongoing joint symptoms despite several months of topical treatment, hand radiographs were obtained and showed “degenerative changes consistent with osteoarthritis, without erosions typical of PsA”. At follow-up, the patient continued to report joint discomfort, and examination revealed evolving psoriasiform lesions on her fingers, described as papules coalescing into plaques. Based on these persistent symptoms and cutaneous findings, her dermatologist initiated a “therapeutic trial” of adalimumab for what was described as PsA.
Following 7 months of adalimumab therapy without improvement in joint pain or nail changes, the patient was referred to rheumatology. Per review of the records, initial rheumatologic physical examination revealed Heberden’s and Bouchard’s nodes and persistent nail ridging, but no classic psoriatic skin lesions or signs of inflammatory joint swelling. The patient continued to report significant joint pain despite adalimumab treatment, and the rheumatologist elected to change therapy from adalimumab to ustekinumab based on the working diagnosis of PsA.
Prior to follow-up evaluation after 8 months of ustekinumab, lower body radiographs were obtained and demonstrated multilevel lumbar spondylosis, mild bilateral sacroiliac joint and femoroacetabular osteoarthritis, bilateral plantar fascia insertional enthesopathy, mild left-sided Achilles tendon insertional enthesopathy, and mild bilateral osteoarthritis of the feet. Laboratory studies prior to rheumatology follow-up revealed negative rheumatoid factor, WBC 6.14 × 103/µL, HGB 13.7 g/dL, PLT 248 × 103/µL, ALB 4.1 g/dL, vitamin B12 823 pg/mL, and 25-hydroxyvitamin D 71.2 ng/mL, all within normal limits. During the follow-up visit, review of systems from a gastrointestinal perspective was negative. Repeat physical examination again noted Heberden’s and Bouchard’s nodes and ongoing nail ridging, without evidence of inflammatory joint swelling, ocular inflammation, abdominal tenderness, or new cutaneous lesions. According to the medical records, due to ongoing patient-reported “morning stiffness, diffuse joint and muscle pain, and persistent nail changes” despite ustekinumab therapy, her rheumatologist elected to discontinue ustekinumab and initiate IXE to treat refractory joint pain. IXE was initiated with an induction dose of 160 mg subcutaneously, followed by 4 maintenance doses of 80 mg administered every 4 weeks. The patient developed her initial gastrointestinal symptoms approximately 16 weeks after the initial induction dose, marking the onset of the acute presentation described in this case report.
Initial laboratory studies obtained by her primary care physician at the time of her index presentation, 28 weeks after the initial IXE induction dose (and 12 weeks after the onset of gastrointestinal symptoms), revealed markedly elevated inflammatory markers, including C-reactive protein (CRP) 5.83 mg/dL (reference 0.00–0.50), erythrocyte sedimentation rate (ESR) 69 mm/hr (reference ≤ 30), and fecal calprotectin (FC) 3,650 mcg/g (reference 0–120). Based on the record review, basic serum labs were not obtained. Stool testing, including gastrointestinal PCR panel and ova and parasites, was negative. The patient was urgently referred to general surgery for a colonoscopy due to the lack of local gastroenterology subspecialty care, with the nearest major metropolitan city located more than 350 miles away. Unable to reach the patient’s rheumatologist, the primary care physician initiated PO prednisone 40 mg to manage symptoms of presumed colitis and serve as a bridge until further evaluation.
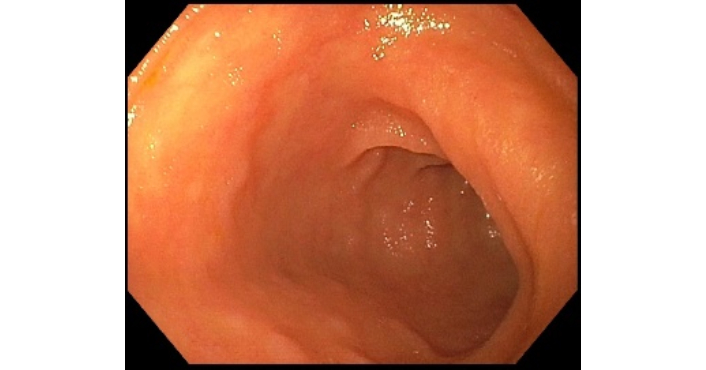
Due to limited access to general surgery care, the patient’s colonoscopy was delayed until 6.5 weeks after the initial presentation (34.5 weeks after IXE induction and 18.5 weeks after the onset of gastrointestinal symptoms), during which she experienced an additional 30-pound weight loss. Periprocedural labs revealed CRP 17.26 mg/dL and ESR 75 mm/hr. Once performed, the ileocolonoscopy report states the terminal ileum appeared normal, but the right colon displayed severe colitis, marked by pronounced erythema, friability, serpiginous ulcerations, and spontaneous bleeding. The sigmoid colon and rectum were reported as “relatively spared” (Figure 1). Histopathological examination of segmental biopsies obtained reported mild focal active ileitis and colitis without evidence of chronicity. Immunohistochemical staining for cytomegalovirus (CMV) was negative. IXE was discontinued, with the last dose administered 4 weeks prior to ileocolonoscopy.
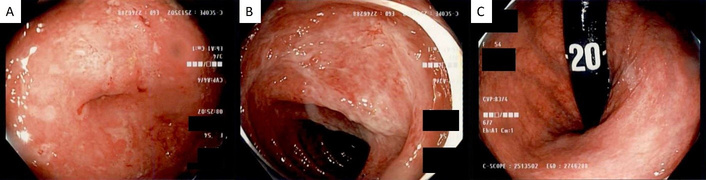
Ileocolonoscopy findings: (A) severe inflammation at the appendiceal orifice, and (B) hepatic flexure with (C) normal-appearing rectum visualized in retroflexion.
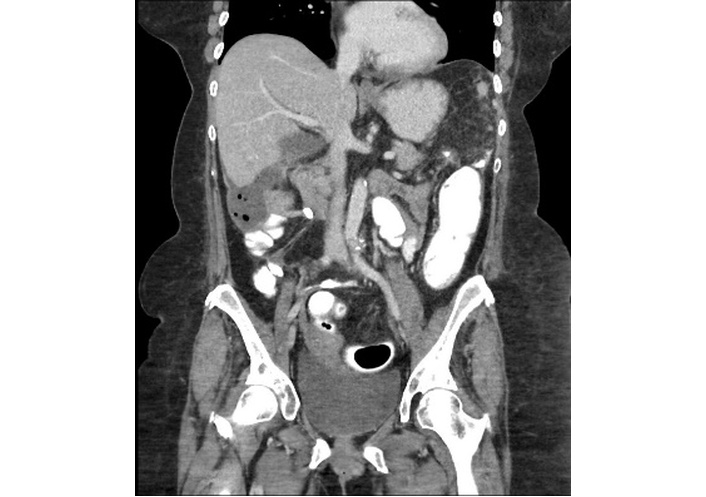
Seven days after the endoscopy, the patient presented to her local emergency department with progressively worsening symptoms. Upon presentation, the patient was tachycardic (heart rate 105 bpm), dehydrated, and exhibited mild lower abdominal tenderness. Laboratory tests revealed ALB 2.9 g/dL, HGB 9.8 g/dL, and a negative Clostridioides difficile PCR. Abdominal and pelvic computed tomography (CT) in the emergency department demonstrated fluid-filled small bowel loops with mild circumferential wall thickening involving the splenic flexure and descending colon (Figure 2). She was admitted to general surgery with internal medicine consultation and was started on intravenous (IV) methylprednisolone 125 mg daily, along with PO budesonide 9 mg daily and PO mesalamine 4 g daily. After a negative infectious workup, PO loperamide was started for diarrhea. The patient’s abdominal pain worsened, and IV morphine and PO ibuprofen were ordered.
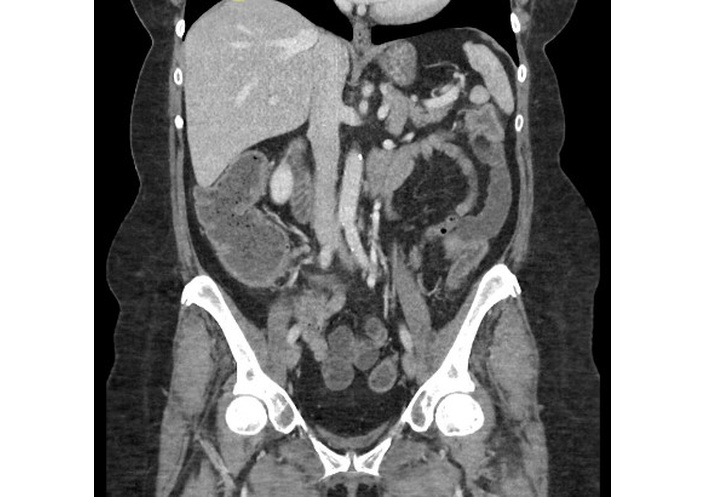
Coronal contrast-enhanced CT of the abdomen and pelvis demonstrates mild mucosal hyperenhancement with adjacent inflammatory stranding and edema involving the splenic flexure and descending colon. CT: computed tomography.
By hospital day 4, her condition had not improved. According to a review of the medical records, the physical examination performed that day did not show any signs of systemic toxicity and only revealed mild abdominal tenderness without significant distension. Additionally, the patient exhibited no altered mental status, and no objective signs of dehydration were reported. Vital signs were stable (temperature 36.6°C, heart rate 61 bpm, respiratory rate 16 breaths per minute, blood pressure 128/66 mmHg, oxygen saturation 95% on room air). Laboratory results included WBC 6.72 × 103/µL, HGB 9.4 g/dL, sodium 133 mmol/L, potassium 3.78 mmol/L, chloride 93 mmol/L, and corrected calcium 9.8 mg/dL. The record review indicates that the surgical team had low concern for toxic megacolon or the need for any urgent surgical intervention. Due to a persistent lack of improvement, the team contacted the nearest gastroenterology team, located approximately 375 miles away. Based on the clinical information provided, the specialist recommended discontinuing antidiarrheals, opioids, and nonsteroidal anti-inflammatory drugs (NSAIDs). With no immediate surgical indication identified by the surgical team, intensive infliximab therapy was initiated to manage presumed severe colitis medically.
An initial loading dose of IV infliximab 10 mg/kg was administered without any immediate issues. Later that evening, while ambulating to the restroom, the patient experienced severe left upper quadrant pain radiating to the left arm, accompanied by shortness of breath. A STAT abdominal radiograph showed significant intraperitoneal free air and marked colonic dilation with large air-fluid levels (Figure 3). She was immediately started on broad-spectrum antibiotics (IV piperacillin-tazobactam 3.375 g every 6 hours) and taken emergently to the operating room. Exploratory laparotomy revealed gross perforations at the splenic flexure, transverse colon, and ascending colon near the cecum, with pronounced inflammation in the left colon. An extended right hemicolectomy with end ileostomy was performed. Pathologic evaluation of the resected specimen showed severe acute colitis with ulceration, submucosal and subserosal abscesses, transmural inflammation, and non-caseating granuloma formation characterized by the presence of epithelioid cells, multinucleated giant cells, and lymphocytes, without necrosis (Figure 4).
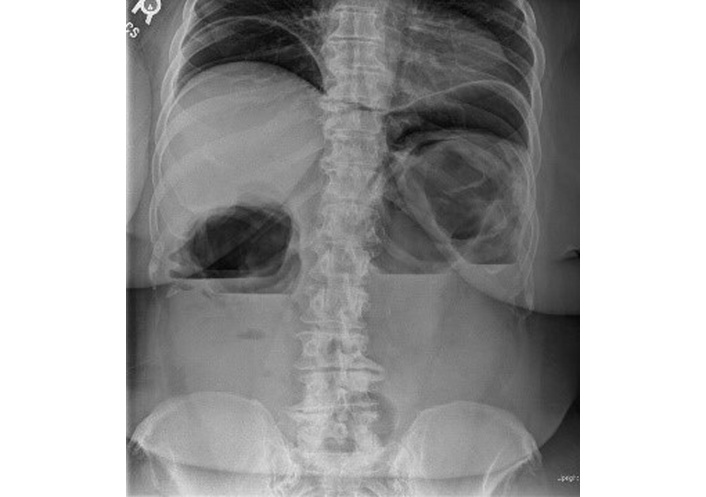
Upright abdominal radiograph demonstrating significant intraperitoneal free air, marked colonic dilation, and large air-fluid levels, consistent with bowel perforation.
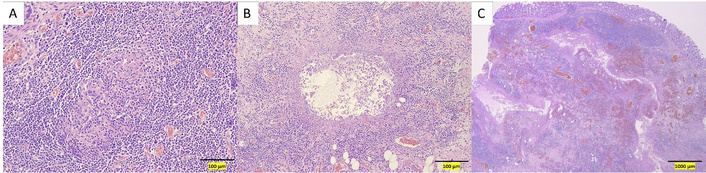
Hematoxylin and eosin-stained sections of the resected colectomy specimen. (A) High-power view showing a non-caseating granuloma. (B) High-power view showing a submucosal abscess. (C) Low-power view demonstrating acute, severe transmural inflammation.
Postoperatively, broad-spectrum antibiotics were continued, and IV methylprednisolone 50 mg daily was started due to intraoperative hypotension. Over several days, bowel function returned, abdominal drain output decreased, appetite improved, and abdominal pain lessened. At the outside hospital, enteral nutrition via tube feeding was not initiated, in part due to the patient’s strong aversion following a prior traumatic nasogastric tube placement earlier in her hospitalization. Additionally, the clinical team noted the absence of a prolonged postoperative ileus and opted to rely on PO intake as her gastrointestinal function appeared to recover, ultimately deferring tube feeding based on patient preference and observed clinical progress. Given the lack of severe malabsorption or sustained intolerance and the medical team’s desire to reduce risks associated with central venous access during the postoperative recovery period, total parenteral nutrition (TPN) was not initiated.
On postoperative day 6, she was transitioned to PO prednisone 40 mg daily. She completed 8 days of IV piperacillin-tazobactam 3.375 g every 6 hours, followed by PO ciprofloxacin 500 mg twice daily and PO metronidazole 500 mg four times daily for a total 14-day antibiotic course. During her 16-day hospitalization, she remained afebrile, and CRP decreased to 6.76 mg/dL. Other notable labs at the time of discharge included WBC 8.51 × 103/µL, HGB 7.10 g/dL, and ALB 2.8 g/dL. She was discharged on postoperative day 10 with prescriptions for rectal mesalamine 1,000 mg, PO oxycodone 5 mg as needed, and a PO prednisone taper starting at 40 mg and decreasing by 10 mg weekly to 5 mg. Infliximab 10 mg/kg was resumed after discharge, with the second induction dose given 3 days post-discharge without issue.
Five days post-discharge, the patient returned to the general surgery clinic with purulent drainage from a right pelvic Jackson-Pratt drain and right flank pain and was sent directly to her local emergency department for further evaluation. Vitals revealed (temperature 37.3°C, heart rate 115 bpm, respiratory rate 18 breaths per minute, blood pressure 126/76 mmHg, oxygen saturation 98% on room air). Laboratory results included WBC 18.9 × 103/µL, HGB 9.4 g/dL, PLT 490 × 103/µL, ALB 3.0 g/dL, and CRP 13.98 mg/dL. A contrast-enhanced (IV, PO, and rectal) abdominal CT showed post-surgical changes but no small bowel obstruction or contrast extravasation. A 4.0 cm × 7.4 cm × 7.2 cm rim-enhancing fluid collection with multiple air foci was identified in the right hemiabdomen (Figure 5). Physical examination, once admitted to the general surgery service, revealed greenish discoloration on the surgical dressing. Upon removal, a wound gap with fascial separation was identified, along with fecal leakage into the wound from a separated stoma appliance. Exploratory laparotomy confirmed mid-abdominal fascial dehiscence and a peri-hepatic abscess. The abscess was evacuated, the right Jackson-Pratt drain was repositioned, and a new left upper quadrant drain was placed. Wound cultures were polymicrobial, including 1+ Candida albicans. The treating teams initiated IV ciprofloxacin, IV metronidazole, and IV fluconazole, while low-dose PO prednisone 20 mg was continued due to concerns for secondary adrenal insufficiency from the prior hospitalization. By hospital day 3 of readmission, she had remained afebrile and without leukocytosis for 24 hours. She was discharged on PO ciprofloxacin, PO metronidazole, and PO fluconazole to complete a 3-week course.
Coronal contrast-enhanced CT of the abdomen demonstrating a 4.0 cm × 7.4 cm × 7.2 cm rim-enhancing fluid collection with multiple air foci located in the right hemiabdomen, consistent with an intra-abdominal abscess.
Initially, the patient’s abdominal pain improved with the PO antimicrobial regimen. However, 9 days post-discharge, she developed new left-sided abdominal pain, intermittent nausea, malaise, and subjective fevers, prompting her return to her local emergency department. Initial vital signs were within normal limits, and the patient was afebrile. Labs were notable for WBC 14.0 × 103/µL, HGB 7.6 g/dL, PLT 584 × 103/µL, ESR > 140 mm/hr, CRP 11.60 mg/dL, ALB 1.6 g/dL, and lactate 1.7 mmol/L (reference 0.5–2.0). Repeat contrast-enhanced (IV, PO, and rectal) CT imaging of the abdomen revealed no evidence of contrast extravasation from the gastrointestinal tract and resolution of right hemiabdomen fluid collection. However, findings now included long-segment small bowel wall thickening with surrounding fat stranding, a 2.0 cm × 1.3 cm superficial fluid collection located midline above the umbilicus, and a 9.0 cm × 1.2 cm left subdiaphragmatic fluid collection with peripheral enhancement (Figure 6). The patient was readmitted and started on IV ciprofloxacin and IV metronidazole. The surgical and internal medicine teams contacted our Gastroenterology service for transfer to our center, as they solely attributed the postsurgical complications, including recurrent abscess formation, to active severe penetrating CD.
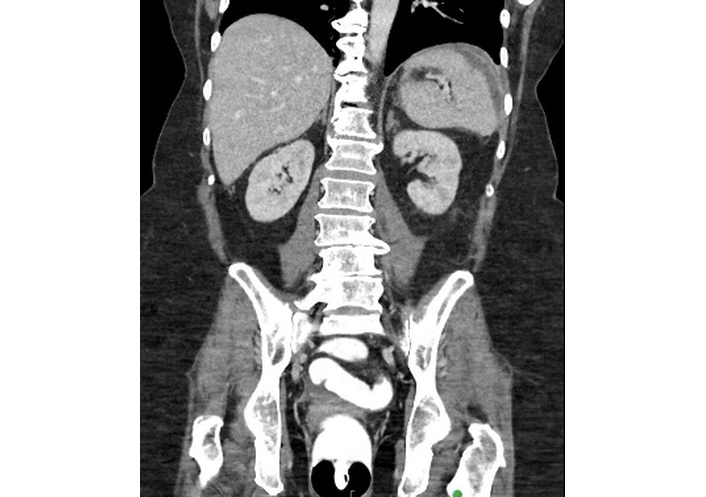
Coronal contrast-enhanced CT of the abdomen demonstrating a 9.0 cm × 1.2 cm left subdiaphragmatic fluid collection with peripheral enhancement. CT: computed tomography.
Upon transfer, the patient was afebrile, with normal ileostomy output and appearance, and no evidence of stomal ulcerations. She denied rectal bleeding and discharge. The patient had lost more than 70 pounds since the onset of her initial symptoms, there was evidence of significant muscle wasting, and ALB 1.9 g/dL, consistent with severe protein-calorie malnutrition. Vitals revealed (temperature 37.3°C, heart rate 115 bpm, respiratory rate 18 breaths per minute, blood pressure 126/76 mmHg, oxygen saturation 98% on room air). Laboratory results were significant for WBC 15.6 × 103/µL, HGB 9.2 g/dL, PLT 524 × 103/µL, ESR > 140 mm/hr, CRP 14.80 mg/dL, ileostomy FC 61 mcg/g, and negative blood cultures.
In light of persistent intra-abdominal infection without source control, most consistent with bacterial seeding from the prior colonic perforation and compounded by profound iatrogenic immunosuppression, severe protein-calorie malnutrition, and postoperative wound dehiscence with contamination of the abdomen by stoma effluent, all immunosuppressive therapies were discontinued, and antimicrobial therapy was continued. A multidisciplinary team was assembled, including specialists from gastroenterology, general surgery, infectious disease, interventional radiology, nutrition, and physical therapy.
Interventional radiology determined that the left subdiaphragmatic fluid collection was not amendable to percutaneous drainage. Infectious disease recommended continuing antimicrobial therapy with short-interval repeat imaging. Follow-up CT imaging performed 7 days later revealed a left pleural effusion with a split pleura sign, raising concern for empyema. Additionally, midline rim-enhancing fluid collections were observed within the postsurgical midline incision, along with a reduction in the size of the perisplenic fluid collection (Figure 7). Antimicrobials were broadened to IV piperacillin-tazobactam and IV micafungin, guided by infectious disease specialist input.
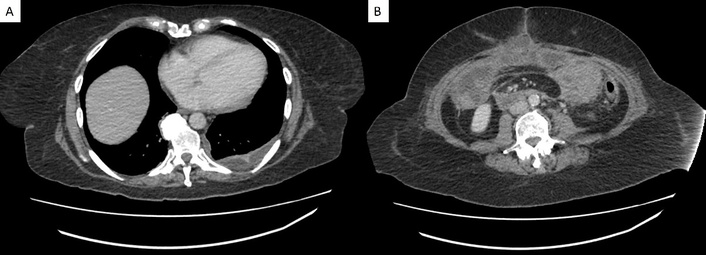
Follow-up transverse CT imaging findings. (A) Left pleural effusion with a split pleura sign, indicative of empyema. (B) Midline rim-enhancing fluid collections within the postsurgical midline incision. CT: computed tomography.
Subsequently, appetite improved significantly, and the patient began consistently meeting her nutritional goals through PO intake, verified by daily inpatient assessments conducted by our registered dietitian. Enteral nutrition via tube feeding was considered but ultimately not initiated due to the patient’s demonstrated clinical improvement, absence of ileus or gastrointestinal dysfunction impairing PO intake, and a documented history of intolerance to nasogastric tube placement during an earlier hospitalization. After counseling, she also declined tube feeding. Given these factors, PO nutrition was favored due to its lower risk profile compared to tube feeding, including a reduced risk of aspiration, diarrhea, and device-related complications.
Over several days, she showed marked clinical improvement, with decreasing leukocytosis and thrombocytosis. She remained afebrile, and antimicrobials were transitioned to PO amoxicillin-clavulanate 875 mg/125 mg twice daily and PO fluconazole 400 mg once daily, with a planned 4-week outpatient course and follow-up imaging. She tolerated this regimen while admitted and was discharged home after a 12-day hospital course.
After completing 17 days of PO amoxicillin-clavulanate, the patient’s antibiotic regimen was adjusted by her primary care team due to reported antibiotic-related gastrointestinal upset. The new regimen included PO ciprofloxacin 500 mg twice daily and PO metronidazole 500 mg three times daily, while fluconazole 400 mg once daily was continued. Twenty-four days post-discharge, inflammatory markers improved significantly. Repeat labs revealed WBC 9.27 × 103/µL, HGB 11.3 g/dL, PLT 469 × 103/µL, ESR 56 mm/hr, ALB 3.5 g/dL, and CRP 0.49 mg/dL. To assess small-bowel involvement, CT enterography (CTE) was performed as magnetic resonance enterography was unavailable locally. Imaging demonstrated persistent mesenteric stranding near the colonic stump in the left upper quadrant but revealed otherwise normal stomach and bowel caliber, without evidence of obstruction, strictures, small bowel wall thickening, masses, or fistulas.
Approximately 4 months after discontinuing IXE and 3 months post-extended right hemicolectomy with end ileostomy, the patient underwent ileoscopy and flexible sigmoidoscopy with our team for disease surveillance. Ileoscopy revealed a normal-appearing ileum, with findings confirmed on histology (Figure 8). Flexible sigmoidoscopy showed normal descending and sigmoid colon and mild rectal changes, including faint erythema and punctate hemorrhages with air insufflation, endoscopically suggestive of mild diversion colitis (Figure 9). Sigmoid biopsies revealed focal active colitis without evidence of chronic changes, whereas rectal biopsies demonstrated mild chronic active colitis with non-necrotizing granulomata. Our team recommended a 3-month course of short-chain fatty acid enemas, followed by repeat flexible sigmoidoscopy and routine IBD surveillance labs to evaluate for resolution of mild colitis.
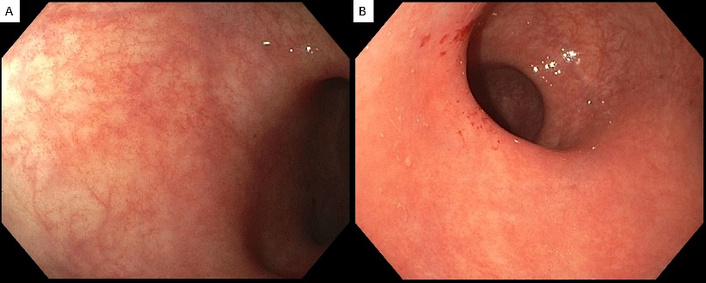
Flexible sigmoidoscopy findings. (A) Normal-appearing sigmoid colon. (B) Rectal changes are characterized by faint erythema and punctate hemorrhages caused by air insufflation.
Repeat flexible sigmoidoscopy performed 3 months after short-chain fatty acid enemas demonstrated an overall normal-appearing left colon (Figure 10). Histopathological examination of biopsies from the left colon and rectum showed no acute or chronic colitis, no granulomas, and minimal crypt architectural distortion (Figure 11). Overall, this reflected the complete resolution of disease activity, consistent with clinical, endoscopic, and histologic remission. There were notable improvements in HGB 13.1 g/dL, ESR 21 mm/hr, and CRP < 0.30 mg/dL. Because her PsA diagnosis was uncertain and the presentation favored CD induced by an IL-17i rather than primary severe, penetrating Crohn’s, we recommended conservative management without advanced therapy. We also advised referral to a new rheumatologist to re-evaluate the PsA diagnosis.
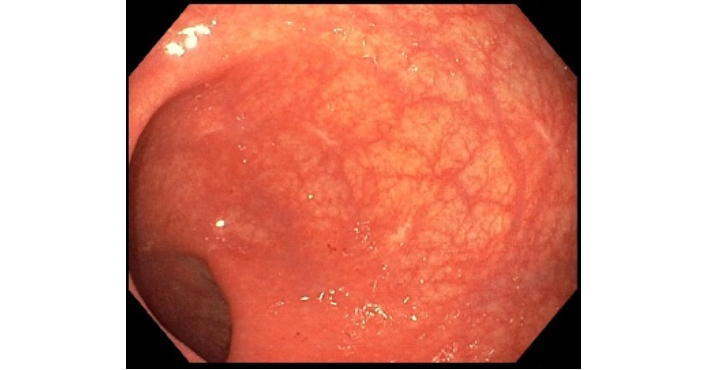
Flexible sigmoidoscopy showing a normal-appearing rectum with several (4) post-biopsy scars from prior endoscopic evaluation.
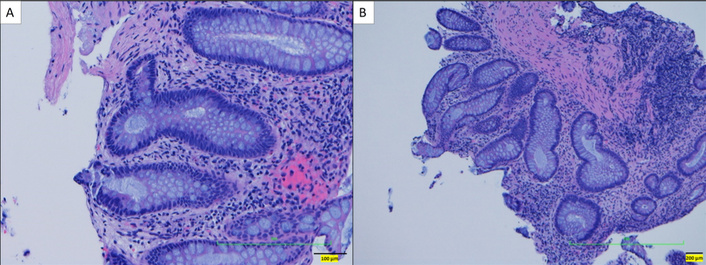
Hematoxylin and eosin-stained sections of the left colon and rectum showing mild crypt architectural distortion without evidence of acute or chronic inflammation at both high-power (A) and low-power (B) views.
At her second-opinion visit, the patient reported that PsA had never been firmly established; the earlier label rested mainly on longstanding foot pain and nail ridging without documented synovitis. Prior dermatology notes described “psoriasiform lesions” on the fingers, but no biopsy was performed, and she denied any personal history of cutaneous PsO. A comprehensive assessment, including expert review of peripheral and axial imaging, showed no erosive or inflammatory changes, and inflammatory markers were normal; the joint exam again revealed no synovitis. Mild entheseal tenderness was present but judged non-specific and more compatible with degenerative or mechanical causes. Applying the CASPAR framework, the entry criterion of inflammatory articular disease was considered met by enthesitis, yet the score totaled only 2 points: nail pitting (+1) and rheumatoid factor negativity (+1), with no current, past, or family history of PsO, no dactylitis, and no juxta-articular new bone formation on hand or foot radiographs. Because at least 3 points are required, classification as PsA was not supported, and she was transitioned to conservative symptom management with acetaminophen and localized corticosteroid injections, which have provided effective relief.
Since her acute hospitalization, the patient has not required any pharmacologic treatment for IBD during the recovery period, including corticosteroids. Eight months after her initial presentation, she underwent ileostomy takedown with ileocolonic anastomosis. Four months following surgical reversal, repeat disease-staging colonoscopy demonstrated a normal-appearing neoterminal ileum and colonic mucosa. A small, isolated 1 mm ulceration was noted at the ileocolonic anastomosis, with no surrounding inflammation. Biopsies from the neoterminal ileum and colonic segments revealed no evidence of active inflammation, chronicity, or granulomas. This focal ulcer was interpreted as a nonspecific postoperative finding, commonly observed at anastomotic sites. The patient remained entirely asymptomatic, with normal laboratory markers, including CRP < 0.30 mg/dL and FC 18 mcg/g. At present, 18 months after initial acute hospitalization and 16 months following endoscopic confirmation of remission, the patient remains clinically stable, with recovered nutritional status, and sustained remission across clinical, laboratory, and endoscopic parameters (Figure 12).
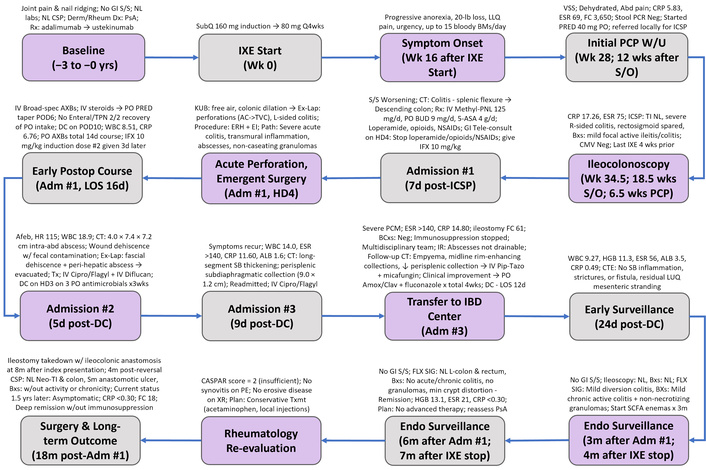
Timeline of presentation, treatment, complications, and recovery in a 54-year-old woman with severe IL-17 inhibitor-associated Crohn’s disease. 2/2: secondary to; 5-ASA: 5-aminosalicylic acid; ABD: abdominal; AC: ascending colon; ADM: admission; AFEB: afebrile; ALB: albumin; AMOX/CLAV: amoxicillin–clavulanate; AXBS: antibiotics; BCXS: blood cultures; BMS: bowel movements; BROAD-SPEC: broad-spectrum; BUD: budesonide; BXS: biopsies; CASPAR: Classification Criteria for Psoriatic Arthritis; CIPRO: ciprofloxacin; CMV: cytomegalovirus; CRP: C-reactive protein; CSP: colonoscopy; CT: computed tomography; CTE: computed tomography enterography; DC: discharge; DX: diagnosis; EI: end ileostomy; ENDO: endoscopic; ERH: extended right hemicolectomy; ESR: erythrocyte sedimentation rate; EX-LAP: exploratory laparotomy; FC: fecal calprotectin; FLX SIG: flexible sigmoidoscopy; GI: gastrointestinal; HD: hospital day; HGB: hemoglobin; HR: heart rate; IBD: inflammatory bowel disease; ICSP: ileocolonoscopy; IFX: infliximab; IR: interventional radiology; IV: intravenous; IXE: ixekizumab; KUB: kidneys-ureters-bladder radiograph; LLQ: left lower quadrant; LOS: length of stay; LUQ: left upper quadrant; METHYL-PNL: methylprednisolone; NEG: negative; NL: normal; NSAIDs: nonsteroidal anti-inflammatory drugs; PATH: pathology; PCM: protein-calorie malnutrition; PCP: primary care provider; PE: physical examination; PIP-TAZO: piperacillin–tazobactam; PO: oral; POD: postoperative day; PRED: prednisone; PSA: psoriatic arthritis; RX: treatment; S/O: symptom onset; S/S: signs and symptoms; SB: small bowel; SCFA: short-chain fatty acid; SM: small; SUBQ: subcutaneous; TI: terminal ileum; TPN: total parenteral nutrition; TVC: transverse colon; TX: treatment; TXMT: treatment; VSS: vital signs stable; W/: with; W/U: workup; WBC: white blood cell; XR: X-ray.
IL-17i are pivotal therapies for PsO, PsA, and AS, with demonstrated efficacy, and are likely to remain a cornerstone given limited alternatives [1–3, 22, 23]. However, these agents carry a small but clinically meaningful risk of inducing de novo IBD or unmasking subclinical disease [7–12], creating a paradoxical safety signal that can complicate timely diagnosis and management. In the absence of prospective, dedicated studies, our current understanding and approaches rely on retrospective case analyses and extrapolations from trials not designed to specifically evaluate management of this complication [7–11].
This case emphasizes the significant clinical challenges and healthcare disparities inherent in diagnosing and managing uncommon disease presentations, especially in geographically isolated and resource-limited settings. Our patient lived about 375 miles from gastroenterology specialty care, which delayed recognition of severe CD and complicated initial management. While caring for this patient, we encountered a notable lack of awareness of IL-17i-associated IBD among non-academic, community-based providers; a knowledge gap with tangible clinical consequences, as delayed recognition and empiric management in the absence of structured guidance can contribute to poorer outcomes.
The patient’s perioperative course illustrates how deviations from best practice in severe IBD increase risk in resource-limited settings. At the referring rural hospital, opioids, antidiarrheals, and aminosalicylates were used despite acute, severe disease, and budesonide was added while the patient was already receiving high-dose systemic corticosteroids. In this context, opioids and antimotility agents slow transit, blunt abdominal findings, and raise intraluminal pressure, while overlapping immunosuppression impairs mucosal healing and host defense, together masking deterioration and increasing the risk of perforation and postoperative infection. Surgical urgency was further underestimated because toxic megacolon criteria were never met, and the initial admission CT did not show overt severe findings, which likely fostered false reassurance. Repeat cross-sectional imaging was not performed despite clinical worsening after corticosteroids and antimotility agents. Although vital signs and basic laboratories were relatively reassuring, the lack of improvement warranted consideration of re-imaging and early surgical involvement given prolonged severe clinical course, failure of an extended outpatient prednisone course, nonresponse to high-dose IV corticosteroids, markedly elevated CRP and FC, progressive malnutrition, and right-sided colitis on endoscopy, features that signal high risk for transmural complications and perforation if operative planning is delayed [24]. Ongoing postoperative complications were likely driven by bacterial seeding from the prior perforation, compounded by profound iatrogenic immunosuppression, severe protein-calorie malnutrition, and wound dehiscence with stoma effluent contamination, all of which predispose to recurrent abscess formation [20].
Further complicating this scenario was the inconclusive initial diagnosis of PsA, established by a community-based dermatologist and subsequently adopted by the patient’s rheumatologist without objective confirmation or fulfillment of standard classification criteria. Specifically, retrospective evaluation of the patient’s outside records demonstrated that the patient did not satisfy the CASPAR classification criteria, scoring only two points based on mild enthesitis, nail pitting, and negative rheumatoid factor. The patient lacked classic clinical hallmarks of PsA, including psoriatic skin plaques, scalp PsO, or dactylitis [25]. Moreover, dermatology clinically diagnosed psoriasiform dermatitis without biopsy, using the term to describe PsO-like plaques. Psoriasiform is a morphologic label that can reflect many conditions (drug eruption, tinea, eczema, secondary syphilis) and is not equivalent to clinical PsO [26]. Additionally, serial imaging over 5 years failed to identify characteristic radiographic abnormalities such as “pencil-in-cup” deformities, phalangeal erosions, periostitis, juxta-articular new bone formation, or evidence of axial involvement [25]. We believe that because the patient continued to report joint pains and the presence of the word “psoriasiform” in the medical record, diagnostic momentum likely shifted clinicians toward a PsA label in an Occam’s razor type scenario. Consequently, escalation to IL-17i therapy underscores the essential importance of adhering to rigorous, evidence-based diagnostic standards prior to initiating biologic therapies with the potential for severe adverse outcomes.
An alternative explanation, pre-existing subclinical IBD with extraintestinal manifestations, is less likely. IBD is associated with other IMIDs [27]; however, our patient’s only confirmed autoimmune condition was Graves’ disease. A recent Mendelian randomization study suggested a potential, though statistically insignificant, association between genetically predicted Graves' disease and CD, with no evidence linking Graves’ disease to an increased risk of UC [28]. Across multi-institutional records, there were no prior gastrointestinal symptoms or extraintestinal manifestations, repeatedly normal laboratory markers, and a normal colonoscopy within 2 years of IL-17i initiation. Symptoms began approximately 3 months after starting IXE, aligning with the reported median onset of 2.9 months (range 0.47–48) for IL-17i-associated IBD [7–11]. The clinical course was unusually fulminant, with rapid escalation, profound weight loss, markedly elevated inflammatory markers, bowel perforation, and serious postoperative complications. After IXE withdrawal and short-term acute management, she achieved sustained remission for more than 1 year without maintenance immunosuppression, an outcome our team rarely observes in severe CD. Histopathology from the right hemicolectomy was most consistent with CD (acute-on-chronic transmural inflammation with ulceration, non-caseating granuloma, and submucosal/subserosal abscesses) [29]. Lastly, prior exposure to adalimumab and ustekinumab, agents that treat rather than induce intestinal inflammation, makes them unlikely culprits. Taken together, neither preexisting IBD, coexisting autoimmunity, nor alternative biologic exposures strongly explain this patient’s severe CD, suggesting a causal role of IL-17 inhibition in triggering de novo CD.
Although addressing specific systemic healthcare disparities was not the central objective of this manuscript, our experience highlights the need to consolidate fragmented literature on IL-17i-associated IBD into a single, accessible, clinician-focused resource. Given this condition’s rarity, formal consensus guidelines from major gastroenterology societies are unlikely; as a result, clinicians are often left without clear, evidence-informed protocols when encountering IL-17i-associated IBD in the acute settings. To address this gap, we present an “extended case report” with practical guidance on screening, diagnosis, and management of IL-17i-associated IBD. The framework is designed for clinicians without formal subspecialty training who often manage complex cases in resource-limited settings without direct gastroenterology support. In these situations, clinicians may not have the time or capacity to synthesize scattered case reports and retrospective reviews, increasing the risk of suboptimal care.
Before initiating an IL-17i, dermatologists and rheumatologists should screen for IBD. Pre-therapy evaluation should include focused history, review of prior GI workups, and personal/family history of autoimmune disease. We recommend that a confirmed history of IBD be considered a contraindication, unlike immune checkpoint inhibitors (ICIs), whose life-saving benefit can justify gastrointestinal risk with close surveillance [15]. When IL-17i use is unavoidable (e.g., refractory disease or lack of alternatives), initiate treatment only after shared decision-making and in consultation with a gastroenterologist, with plans for close clinical monitoring. We recommend obtaining pre-therapy FC for all candidates; obtain CRP and stool GI pathogen testing in symptomatic patients (e.g., abdominal pain, diarrhea, etc.) [8, 30–32]. FC is more sensitive for colonic than small-bowel inflammation (83–100% vs. 60–70% at > 50 µg/g) [33]; manufacturer cutoffs of 50 µg/g are seldom used in practice [34], and IMID cohorts suggest improved performance at > 132 µg/g (SN 78.7%, SP 76.9%) and > 266 µg/g (SN 100%, SP 66.7%) [35, 36]. Because CRP may reflect systemic inflammation rather than luminal activity, interpret it cautiously, ideally when the primary IMID is quiescent or a baseline is available for comparison. Given the potential for severe gastrointestinal complications with IL-17 blockade, we recommend gastroenterology referral for endoscopy (with consideration of small-bowel imaging) for FC > 150 µg/g or if CRP > 0.5 mg/dL when there is a high clinical suspicion for underlying IBD, thresholds that balance test performance with clinical risk in this context [31, 32].
Informed consent should explicitly address the risk of de novo IBD or unmasking of subclinical disease, and this discussion should be documented, ideally with a medication-specific consent form. Patients should be counseled to report new gastrointestinal or extraintestinal symptoms promptly. Because early adverse effects are often managed in dermatology or rheumatology, clinicians should maintain a high index of suspicion for clinically meaningful gastrointestinal manifestations. The side effect of mild to moderate diarrhea occurs in ~5–10% of patients on IL-17i, underscoring the need for clinical vigilance [37]. FC testing can be considered every 3–6 months during therapy, especially in the first year, when most IL-17i-associated IBD is diagnosed [7–11, 38], with a low threshold for gastroenterology referral for sustained FC elevation or new symptoms. Again, recognizing that FC is less sensitive for isolated small-bowel disease, persistent symptoms with normal FC warrant further evaluation despite reassuring biomarkers.
If a patient receiving IL-17i therapy develops diarrhea, abdominal pain, rectal bleeding, fever, or weight loss, IL-17i therapy should be held, and an initial workup performed. We recommend obtaining FC, CRP, and stool studies for C. difficile or a GI PCR to exclude enteric pathogens. Although IL-17i therapies rarely lead to infectious diarrhea [37], their impact on mucosal immunity can increase susceptibility to bacterial or fungal gastrointestinal infections [1, 39], emphasizing the importance of investigating infectious causes. Routine stool ova and parasite testing is not recommended unless specific risk factors exist [40]. If C. difficile infection is highly suggested (≥ 3 unformed stools in 24 hours), a 2-step testing approach is recommended: first, a highly SN test (e.g., nucleic acid amplification test or glutamate dehydrogenase), followed by a toxin enzyme immunoassay if positive, to distinguish true infection from colonization. Up to 5–10% of IBD patients may be colonized with C. difficile [41], stressing the need for careful interpretation of test results. Although laboratory tests can help gauge disease severity, elevated WBCs, PLTs, and inflammatory markers (ESR, CRP) may be nonspecific if coexisting dermatological or rheumatological disease is active [19, 21, 31, 32].
Because clinical presentation alone often fails to distinguish UC from CD, further workup is necessary. Ideally, the gastroenterologist plays a pivotal role in suspected IL-17i-associated IBD, which includes endoscopic evaluation, disease severity assessment, and histologic confirmation. The differential diagnosis is broad. In addition to IBD and infectious etiologies, other considerations include other drug-induced colitides (e.g., NSAIDs, ICIs), malignancy, ischemic colitis, diverticulitis, and microscopic colitis, among others. In severe cases, hospital admission should be strongly considered to facilitate timely, comprehensive evaluation and management, including prompt consultation with gastroenterology [16–21]. Given the likelihood of requiring advanced therapy to control confirmed IBD, comprehensive pre-therapy labs, if not previously performed before IL-17i initiation, should include qualitative human chorionic gonadotropin (biologic females), human immunodeficiency virus, hepatitis C, hepatitis B (surface antigen, surface antibody, and core antibody), and latent tuberculosis testing (interferon-gamma release assay or Mantoux test) [42, 43].
If the initial workup does not indicate an infectious etiology or IBD, IL-17i can be cautiously resumed with close monitoring. Concurrently, alternative causes for symptoms (e.g., other medication-induced diarrhea, irritable bowel syndrome, celiac disease, or food intolerances, etc.) should be evaluated. In patients with mild to moderate gastrointestinal symptoms, a negative infectious workup, and FC > 150 mcg/g and/or CRP > 0.5 mg/dL, we recommend outpatient referral to gastroenterology for ileocolonoscopy. A proposed pre-IL-17i therapy algorithm is presented in Figure 13.
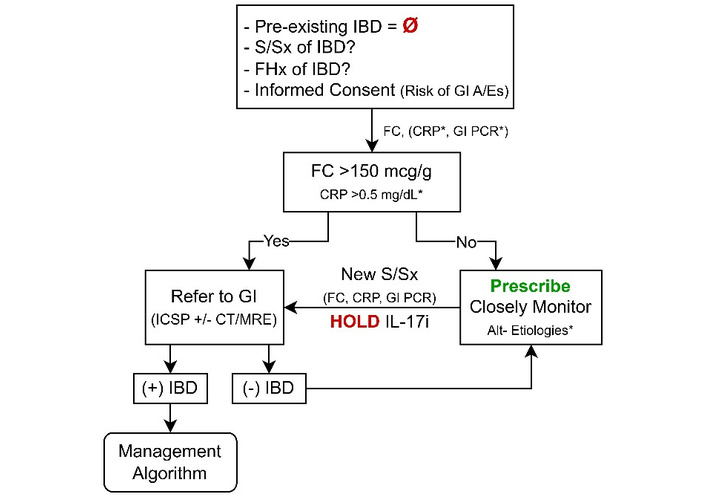
Recommendations for pre-treatment screening and monitoring of IL-17i therapies. A/Es: adverse events; Alt: alternative; CRP: C-reactive protein; CT: computed tomography; FC: fecal calprotectin; FHx: family history; GI: gastrointestinal; IBD: inflammatory bowel disease; ICSP: ileocolonoscopy; IL-17i: interleukin-17 inhibitor; MRI: magnetic resonance imaging; S/Sx: signs and symptoms. *: Items marked with an asterisk are discussed in detail in the main text, which outlines specific caveats and clinical contexts in which they should or should not be considered.
Cross-sectional imaging with CT can be useful in severe IBD flares, but it cannot definitively distinguish Crohn’s colitis from UC, nor from infectious or ischemic colitis. Consequently, imaging should be reserved for cases where alternative etiologies (e.g., diverticulitis) or complications (e.g., toxic megacolon, perforation, intra-abdominal abscess, or obstruction) are suspected [15, 19, 21]. Cases of toxic megacolon have been reported in the literature as a complication of IL-17i-associated IBD [8–10, 44]. Toxic megacolon is classically diagnosed by concerning abdominal exam findings and colonic distension > 5.5 cm on abdominal X-ray, plus at least 3 of the following: fever > 38°C, heart rate > 120 beats/min, neutrophilic leukocytosis > 10.5 × 103/µL, anemia, dehydration, altered sensorium, electrolyte disturbances, or hypotension [24, 45]. If small bowel CD is suspected [10, 38, 46, 47], CTE or magnetic resonance enterography [47] can be considered after initial acute workup and management, as these tests are not usually performed urgently but can be critical for accurate disease phenotyping [21].
Endoscopy with biopsy permits direct visualization, histology, and immediate severity triage [19, 21]. For non-GI/IBD specialist endoscopists, we propose a practical, phenotype-agnostic rubric to guide endoscopic grading. Mild disease should be classified in the setting of erythema, granularity, mild friability, decreased vascular pattern, or scattered aphthous ulcers ≤ 5 mm without confluence, no spontaneous bleeding, no strictures, and limited extent. Classify moderate disease when there is a significant number of erosions, ulcers are larger or confluent (5–10 mm) but shallow, involve roughly < 10–20% of the surface area, and there is marked erythema, friability with contact bleeding, absent vascular pattern, or exudate, yet no deep/penetrating ulcers, strictures, fistulas, or toxic dilation. Classify severe disease when there are deep linear/serpiginous ulcers, broad ulcerated surfaces (> 10–20% or diffuse), spontaneous bleeding, cobblestoning, strictures, fistulas, active hemorrhage requiring hemostasis, or endoscopic concern for toxic megacolon or impending perforation. Document distribution to suggest phenotype, continuous distal-to-proximal involvement supports UC, whereas skip areas, serpiginous ulcers, and terminal-ileal disease support CD, but manage urgency according to severity rather than phenotype alone [48–50]. Obtain segmental biopsies from rectum through terminal ileum, sample both inflamed and uninvolved mucosa, and request CMV staining in steroid-exposed or severe colitis. Gastroenterologists should use established scores (Mayo endoscopic subscore for UC; SES-CD for CD) [49–51]. If upper-GI symptoms are present or ileocolonoscopy is unrevealing, we recommend performing esophagogastroduodenoscopy.
No prospective trials specifically address treatment of IL-17i-associated IBD; management therefore relies on retrospective reports [7–11, 38] and extrapolation from IBD [16–21] and ICI-colitis guidance [15]. Care should be coordinated with the prescribing dermatologist/rheumatologist. We recommend an advanced-therapy-first strategy. Advanced therapy should even be considered for mild presentations. Emerging data supporting early advanced therapy use in UC [18] and CD [52] further strengthen this strategy. This approach is preferred over 5-ASA or budesonide monotherapy, and over prolonged corticosteroids, which are frequently ineffective in this setting and often provide only transient benefit [9, 38, 44, 53, 54]. We recommend that agent selection should consider infection risk, expected treatment duration, comorbidities, the primary indication for which the IL-17i was prescribed, and the IBD phenotype (Table 1). When disease is recognized early, steroid-free induction can often be achieved with guselkumab, risankizumab (IL-23i), ustekinumab (IL-12/23i), infliximab (TNF-α inhibitor), or tofacitinib, upadacitinib [JAK inhibitors (JAKi)] [8–11, 38, 46, 55]; when feasible, we generally favor IL-23i or ustekinumab due to their proven efficacy and safety in IMIDs and IBD.
FDA-labeled indications for proposed treatments in IL-17-associated IBD.
| Condition | Risankizumab | Guselkumab | Ustekinumab | Infliximab | Upadacitinib | Tofacitinib |
|---|---|---|---|---|---|---|
| Plaque psoriasis | Yes | Yes | Yes | Yes | No | No* |
| Psoriatic arthritis | Yes | Yes | Yes | Yes | Yes* | Yes* |
| Ankylosing spondylitis | No | No | No | Yes | Yes* | Yes* |
| Nonradiographic axial spondyloarthritis | No | No | No | No* | Yes* | No |
| Crohn’s disease | Yes | No* | Yes | Yes | Yes* | No |
| Ulcerative colitis | Yes | Yes | Yes | Yes | Yes* | Yes* |
*May be used off-label, is under FDA applicational review, or additional requirements (e.g., disease severity criteria, prior therapy failure, or in combination with other medications) may apply. Refer to each drug’s prescribing information for detailed guidance.
Corticosteroids should be used as a short bridge when symptoms significantly impair quality of life, the disease is severe, or initiation of advanced therapy is delayed or contraindicated. For mild to moderate disease, initiate prednisone (or equivalent) 40 mg daily; higher starting doses do not improve efficacy. Maintain for 1–2 weeks based on clinical response, then taper by 10 mg per week if 40 mg was used for 1 week, or by 5 mg per week if used for greater than 2 weeks, until discontinuation. Avoid prolonged courses, and do not use corticosteroids for maintenance [16–21, 56]. Brief bridging may be appropriate when resources or insurance limit access, baseline labs/vaccinations are pending, IBD phenotype is uncertain, or the patient is hesitant to start new advanced therapy after an IL-17i adverse event. Reassess within 1 week; if there is no meaningful improvement, or if symptoms recur during the taper, consider obtaining cross-sectional imaging to exclude complications (e.g., abscess) and transition to advanced therapy. For patients who fail initial therapy or present with severe disease, admit to the hospital and initiate IV methylprednisolone 60 mg/day (or equivalent), avoiding higher doses [17, 19–21, 24]. Start advanced therapy, preferably within 72 hours, if there is no clinical improvement, in coordination with dermatology and rheumatology. In steroid-refractory cases, obtain imaging to evaluate for complications and consider performing endoscopy with colonic biopsy to exclude CMV (reported in IL-17i-associated UC); note that low-level positive CMV PCR may reflect disease severity in a colonized patient rather than active infection, underscoring the value of tissue diagnosis [17, 19–21, 24, 53].
When advanced therapy is needed, we favor IL-23i (guselkumab or risankizumab) as first line, with ustekinumab a close alternative when concomitant PsO or PsA is present. This preference reflects cross-indication efficacy and a favorable safety profile supported by head-to-head trials, comparative effectiveness studies, and meta-analyses in IBD and dermatology/rheumatology [18, 57, 58]. In PsO, risankizumab outperformed ustekinumab for PASI-90 at week 16 in ultIMMa-1/-2 (75% vs. 42%) and surpassed infliximab in a meta-analysis (greater efficacy and safety) [59, 60]. In CD, SEQUENCE showed higher week-48 clinical and endoscopic remission with risankizumab vs ustekinumab after tumor necrosis factor inhibitor (TNFi) failure (60.8% vs. 40.8%; 31.8% vs. 16.2%) [61], while SEAVUE demonstrated ustekinumab efficacy comparable to adalimumab in biologic-naïve CD with a slightly better safety profile [62]. Although direct comparisons across all IMIDs remain limited, the aggregate evidence and the absence of boxed warnings for IL-23-pathway agents position them as a preferred starting point for patients who develop IL-17i-associated IBD.
Infliximab (TNF-α inhibitor) is approved for moderate-severe PsO, PsA, and AS [58] and has a long, well-characterized track record in IBD, which supports clinician comfort with dosing, monitoring, and adverse events [17–21, 42, 43]. It is particularly attractive when AS coexists with IL-17i-associated IBD [63]. For mild-moderate presentations, standard induction (5 mg/kg IV at weeks 0, 2, and 6) is appropriate, with usual safety precautions, avoiding use in advanced heart failure, demyelinating disease, and active/recent hematologic malignancy [64]. Clinicians should be aware that while infliximab often controls gastrointestinal inflammation, PsO may worsen in some patients, prompting a switch to an IL-23i or ustekinumab, or addition of apremilast to optimize the primary IMID [8–11, 55, 65]. Overall, infliximab remains a cornerstone option across IBD and select IMIDs given its robust evidence base and extensive real-world use [17–21, 42, 43, 58].
For severe or steroid-refractory disease, escalation to inpatient infliximab should be considered. Although no prospective studies define optimal dosing in IL-17i-associated IBD, pathophysiology favors intensive strategies, shortened intervals (“dose stacking” < 2 weeks) and/or higher doses (10 mg/kg), over the standard regimen in patients with high inflammatory burden, hypoalbuminemia, and malnutrition, which increase drug clearance and fecal loss [17, 24]. Observational data support the safety and effectiveness of inpatient intensive dosing for acute severe CD flares, with avoidance of surgery and low complication rates [66], and case reports document successful rescue of IL-17i-associated IBD using high-dose schedules (e.g., 10 mg/kg at weeks 0, 1, and 5, then every 4 weeks) with endoscopic and histologic remission [38, 65]. Accordingly, when treating hospitalized, steroid-refractory patients in this setting, we recommend considering a higher-dose, dose-stacked induction consistent with the University of Michigan Severe UC Protocol, in collaboration with dermatology and rheumatology [17, 24].
Tofacitinib and upadacitinib are rapidly acting, non-immunogenic PO options that can bridge IMID and IBD needs, particularly with concomitant nonradiographic axial spondyloarthritis (nr-axSpA), and are reasonable when TNFi have failed or are not tolerated, or when patients prefer to avoid infusions/injections [18, 67–69]. Both are effective in PsA, AS, and nr-axSpA (appearing comparable to TNFi in PsA/nr-axSpA, with limited head-to-head data) and have efficacy in IBD; in UC, trial and network meta-analytic data suggest upadacitinib induces remission ~2.5× more often than tofacitinib in outpatients with moderate-severe disease [70, 71]. Selection should be coordinated with the prescribing dermatologist/rheumatologist and tailored to the primary IMID and IBD phenotype. Use caution in patients with multiple cardiovascular risk factors; prior venous thromboembolism (VTE)/PE; malignancy; severe cytopenias (e.g., absolute neutrophil count < 100 cells/mm3 or absolute lymphocyte count < 500 cells/mm3); significant hepatic/renal impairment; or pregnancy/planned pregnancy [67]. Published reports in IL-17i-associated IBD primarily describe successful use of tofacitinib (often after TNFi failure) to control IL-17i-associated IBD and facilitate steroid taper; we found no phenotype-specific reports with upadacitinib to date [8, 10].
Although neither agent is FDA-approved for inpatient induction, select hospitalized patients with severe disease may benefit given the rapid onset of action, particularly those with prior biologic failure (e.g., infliximab), recent outpatient failure of high-dose prednisone (≥ 40 mg), or failure of IV corticosteroids at another facility; a high CRP-to-ALB ratio may further support consideration of a JAKi [24, 67]. When pursuing this path, involve an IBD specialist and consider a protocol similar to the University of Michigan Acute Severe UC, generally reserved for patients at high risk of failing 72 hours of IV corticosteroids; case series and case-control data suggest clinical benefit when JAKi are initiated concurrently with corticosteroids in this high-risk group [24, 53, 54, 72, 73]. Data specific to accelerated inpatient JAKi use for IL-17i-associated IBD are currently limited. If a JAKi is planned, the first dose of recombinant adjuvanted herpes zoster vaccine should ideally be administered before starting therapy, followed by the second dose two months later [67].
In cases of partial advanced therapy remission, we recommend continuing the current therapy with close monitoring under gastroenterological guidance. For refractory disease, a thorough reevaluation for infectious etiologies, particularly C. difficile and CMV, is crucial, along with surgical consultation in severe refractory disease. This approach is illustrated by a reported case of IL-17i–induced UC that was refractory to PO mesalamine, IV corticosteroids, and infliximab. Although an initial concomitant C. difficile infection was treated, symptoms recurred, ultimately uncovering active CMV infection that required antiviral therapy for disease improvement [53]. Additionally, case reports have documented patients requiring colectomy for therapy-refractory severe colitis and perforation [9, 38, 44], including the case described here.
Acute abdominal pain is common during inflammatory episodes in patients with IBD and typically improves with appropriate medical therapy. Narcotics should be avoided in severe IBD flares because they can worsen inflammation, increase infection risk, induce constipation (potentially leading to toxic megacolon), mask critical symptoms, and worsen outcomes [24, 74, 75]. Optimal pain management focuses on controlling the underlying inflammation, judicious use of non-opioid agents (e.g., IV acetaminophen), and nonpharmacologic measures (e.g., dietary modifications such as a low fiber diet, if applicable).
Antibiotics should be used selectively in IL-17i-associated IBD, primarily in cases of confirmed or strongly suspected bacterial infection, or complications such as perforations, abscesses, fistulas, or perianal disease, which have been documented in case reports of IL-17i-associated IBD [7–11, 76]. Routine antibiotic use in the absence of clear evidence of infection is discouraged, as it poses risks of gut microbiome disruption, limited efficacy in modulating the underlying inflammatory process, and potential adverse outcomes, including an elevated risk of C. difficile infection. When indicated, antibiotic therapy should ideally be guided by culture and sensitivity data to ensure targeted treatment and to minimize unnecessary alterations to the gut microbiota [77].
There are no retrospective meta-analyses or prospective studies of VTE prophylaxis specific to IL-17i-associated IBD. Because IBD in general confers > 2× the VTE risk (similar in UC and CD), we recommend managing presumed risk equivalently. A notable example describes a patient on IXE for PsO and PsA who reported several months of diarrhea and was found to have colitis and developed a popliteal DVT and bilateral pulmonary emboli requiring enoxaparin [78]. Risk is highest with active disease, IV corticosteroids, hospitalization, and surgery; additional contributors include age > 65, estrogen therapy, obesity, and concomitant autoimmune disease. Unless experiencing acute, hemodynamically unstable gastrointestinal bleeding, we recommend pharmacologic prophylaxis (low-molecular-weight heparin or fondaparinux) for all hospitalized patients with active IBD and for those undergoing major surgery, with continuation for ≥ 3 weeks after discharge in surgical cases. The role of extended or ambulatory prophylaxis during severe flares remains uncertain in IBD and warrants further study, particularly in IL-17i-associated IBD [79].
Enteral nutrition should be preferred whenever the gut is functional. Initiate tube feeding when PO intake is inadequate, especially with actual or anticipated malnutrition, as enteral routes preserve mucosal integrity, maintain gut-associated lymphoid tissue, reduce bacterial translocation, support immune function and motility, and carry fewer complications than TPN. Use short-term TPN preoperatively when abscesses or phlegmon limit intake, providing bowel rest and a bridge to surgery or interventional radiology; reserve longer-term TPN for high-output fistulas, prolonged ileus, or severe malnutrition when enteral feeding is not feasible. All inpatients with complicated IBD or significant nutritional risk should be co-managed with a registered dietitian to tailor enteral/parenteral strategies and optimize perioperative outcomes [75].
In patients with iron deficiency and active inflammation, PO iron is often ineffective because inflammation impairs gastrointestinal absorption, PO formulations can provoke gastrointestinal adverse effects, and there is concern for aggravating mucosal inflammation and disrupting the gut microbiota. For individuals with active IBD in particular, IV iron circumvents the gut and delivers iron directly into the bloodstream, enabling faster and more reliable repletion; accordingly, IV iron is the preferred approach when absorption is compromised, or PO iron is poorly tolerated [80].
Before discharge, patients should have < 5 non-watery, minimally bloody stools/day, tolerate a full diet, need no IV fluids or significant electrolyte repletion, and ambulate independently. After IV corticosteroids, observe for 24 hours following a switch to PO therapy. Unless response has been exceptionally rapid, discharge most patients on 40–60 mg PO prednisone with a complete taper provided at discharge to prevent interruptions. Suggested taper: if total steroid exposure < 30 days, reduce by 10 mg/week daily; if ≥ 30 days, reduce by 5 mg/week, avoiding abrupt cessation to prevent adrenal insufficiency, particularly in those taking ≥ 20 mg/day for ≥ 3 weeks [24]. Arrange a close follow-up with gastroenterology, dermatology/rheumatology, and nutrition. A proposed management algorithm is presented in Figure 14.
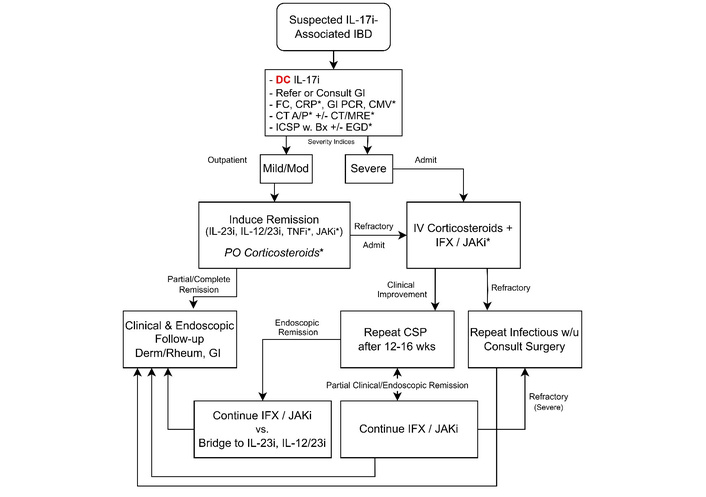
Recommendations for management of IL-17i-associated IBD. A/P: abdomen and pelvis; BX: biopsy; CMV: cytomegalovirus; CRP: C-reactive protein; CSP: colonoscopy; CT: computed tomography; DC: discharge; EGD: esophagogastroduodenoscopy; FC: fecal calprotectin; GI: gastroenterology; GI PCR: gastrointestinal polymerase chain reaction; IBD: inflammatory bowel disease; ICSP: ileocolonoscopy; IFX: infliximab; IL-12/23i: interleukin-12/23 inhibitor; IL-17i: interleukin-17 inhibitor; IL-23i: interleukin-23 inhibitor; IV: intravenous; JAKi: Janus kinase inhibitor; MRE: magnetic resonance enterography; PO: oral; TNFi: tumor necrosis factor inhibitor; W: with; W/U: work-up. *: Items marked with an asterisk are discussed in detail in the main text, which outlines specific caveats and clinical contexts in which they should or should not be considered.
Across published reports, symptom improvement after stopping the IL-17i and starting appropriate therapy occurs within 2 days to 3 months, with a median time to clinical remission of 4 weeks [7–11, 38]. Over half of patients remit with a single agent (corticosteroids or advanced therapy), and about one-third require combination therapy. Advanced therapy (alone or with corticosteroids) generally outperforms corticosteroids alone; topical 5-ASA adds little, likely reflecting disease severity and limited efficacy in CD [7–11, 20, 38]. Many are stabilized with corticosteroids or infliximab and then transitioned to an advanced agent that better controls both the primary IMID and the IBD, especially if the IMID has flared [7–11, 55]. A minority, including our patient, achieves clinical and endoscopic remission after drug withdrawal without long-term immunosuppression, supporting a drug-induced etiology [38, 45]. Relapse is uncommon but reported months later [9, 65]. Because rechallenge data are lacking and recurrent disease has been described after re-exposure (e.g., recurrent IXE-induced UC requiring repeat corticosteroid induction and transition to guselkumab) [81], we consider IL-17i contraindicated thereafter.
There are no formal data to guide surveillance timing in IL-17i-associated IBD; case series report follow-up endoscopy from 3 weeks to 12 months after clinical remission [7–11, 38, 54, 55, 65, 76]. We recommend performing ileocolonoscopy 12–16 weeks after starting therapy to evaluate disease status and mucosal healing. In those demonstrating significant improvement, continuation of infliximab or JAKi, especially for severe or refractory disease, is advisable unless the primary IMID remains uncontrolled or flares. As noted above, transitioning to a safer long-term option, such as an IL-23i or ustekinumab, has been reported in several cases, particularly when infliximab fails to sustain remission of the primary IMID [7–11, 55]. This transition should occur under the guidance of the patient’s dermatologist/rheumatologist, in close collaboration with an IBD specialist, to ensure optimal management and safety.
This case illustrates a plausible instance of de novo, severe CD precipitated by IXE in a patient without a definitive IMID. The patient had no antecedent gastrointestinal symptoms, repeatedly normal biomarkers, and a normal colonoscopy within 2 years of IL-17i initiation; symptoms escalated within 16 weeks; histopathology was consistent with CD; and sustained clinical, biochemical, and endoscopic remission was achieved after drug withdrawal without maintenance immunosuppression and has persisted for 16 months thus far. Taken together, the timing, severe phenotype, biopsy confirmation, and durable remission support a drug-induced etiology rather than unmasking of latent disease.
This clinical course underscores preventable harms that arise when complex IBD is managed without specialty involvement in resource-limited settings: delayed diagnosis; absence of a multidisciplinary team; use of constipating agents, including opioids and antimotility agents; reliance on ineffective therapy for severe disease (5-ASA, budesonide); supratherapeutic immunosuppression; failure to obtain follow-up imaging as disease worsens; underutilization of nutrition support; and delayed surgical intervention. Collectively, these system- and practice-level factors increased the risk of perforation and postoperative complications and compromised safe, effective care.
In the absence of formal guidelines, our experience supports pragmatic safeguards when prescribing IL-17i. To help close the guidance gap, we offer a stepwise, evidence-informed framework that non-IBD clinicians can use in everyday practice, including in resource-limited settings. Broader pharmacovigilance and collaborative research are needed to define mechanisms of IL-17-mediated gut injury, identify patients at risk, and establish effective acute and maintenance treatment pathways. Equally important is understanding how healthcare disparities affect recognition, access, and outcomes outside of academic centers. Until stronger data emerge, sharing real-world, case-based insights such as this report remains essential to improving patient safety and preparedness.
This is a single-case report; it cannot establish causality and demonstrates only a temporal association between IL-17 inhibition and IBD. The findings should not be generalized to all IL-17i (e.g., BKZ, BRO, SEC), as agent- and patient-specific responses and adverse events may differ. Early clinical decisions were made by outside clinicians, and our reconstruction of their diagnostic rationale, therapeutic choices, and consent discussions relies solely on available records. Undocumented elements of history, labs, imaging, or procedures may have influenced management; therefore, some questions about the initial care remain unanswered. Feasibility may limit the application of our proposed framework in rural or resource-constrained settings, where colonoscopy or advanced imaging is not readily available. In such contexts, early telemedicine consultation with tertiary centers, use of validated simplified activity indices, selective basic laboratory markers, and expedited transfer are practical interim strategies. Finally, although the patient has sustained clinical, biochemical, and endoscopic remission for 16 months, the lack of multi-year follow-up precludes conclusions about long-term relapse risk or delayed complications.
ALB: albumin
AS: ankylosing spondylitis
BKZ: bimekizumab
BRO: brodalumab
CD: Crohn’s disease
CMV: cytomegalovirus
CRP: C-reactive protein
CT: computed tomography
CTE: computed tomography enterography
ESR: erythrocyte sedimentation rate
FC: fecal calprotectin
HGB: hemoglobin
IBD: inflammatory bowel disease
ICIs: immune checkpoint inhibitors
IL-17i: interleukin-17 inhibitors
IMIDs: immune-mediated inflammatory diseases
IV: intravenous
IXE: ixekizumab
JAKi: Janus kinase inhibitors
nr-axSpA: nonradiographic axial spondyloarthritis
NSAIDs: nonsteroidal anti-inflammatory drugs
PLT: platelet
PO: oral
PsA: psoriatic arthritis
PsO: psoriasis
SEC: secukinumab
SN: sensitivity
Th17: T-helper 17
TNFi: tumor necrosis factor inhibitor
TPN: total parenteral nutrition
UC: ulcerative colitis
VTE: venous thromboembolism
WBC: white blood cell
The view(s) expressed herein are those of the author(s) and do not reflect the official policy or position of Joint Base Elmendorf-Richardson Medical Center, Brooke Army Medical Center, the U.S. Air Force and Army Medical Departments, the U.S. Air Force Office of the Surgeon General, the Department of the Air Force, the Department of the Army, Department of Defense or the U.S. Government.
During the preparation of this work, authors used ChatGPT to improve the readability and language. After using ChatGPT, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
TLS: Conceptualization, Data curation, Writing—original draft. TAS: Investigation, Conceptualization, Writing—review & editing, Supervision. AP: Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
This case report was conducted in accordance with the ethical standards set forth in the Declaration of Helsinki, as revised by the World Medical Association at its 75th General Assembly, Helsinki, Finland, in October 2024. Institutional Review Board (IRB) approval was waived by the Brooke Army Medical Center Human Research Protections Office (BAMC HRPO) / Regional Health Command-Central (RHC-C) Institutional Review Board and Ethics Committee, as the study involved only retrospective analysis of a single patient’s anonymized clinical data obtained during standard care, which is exempt from formal ethics review per institutional policy. Written informed consent was obtained from the patient for the endoscopic procedure, participation in this report, and publication of associated images and clinical details. All efforts were made to ensure patient confidentiality, including removal or generalization of all direct identifiers (e.g., name, date of birth, contact information, geographic details smaller than a state) and indirect identifiers (e.g., unique dates, rare disease descriptors) in compliance with HIPAA Privacy Rule standards (45 CFR §164.514) and institutional policy, ensuring that no information could reasonably be used to identify the individual.
Written informed consent to participate in the manuscript was obtained from the relevant patient.
Written informed consent to publication was obtained from the relevant patient.
This is a case report involving confidential patient data that cannot be publicly shared for privacy protection, despite informed consent for publication.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.